
Dietary AGEs as Exogenous Boosters of Inflammation



{kind=link}
Abstract
1. Introduction
1.1. Dietary Modulation of Inflammation
1.1.1. Dietary Advanced Glycation End Products
1.1.2. The Mediterranean Diet
1.1.3. The Dietary Approaches to Stop Hypertension (DASH) Diet
1.1.4. Dietary Inflammatory Index (DII)
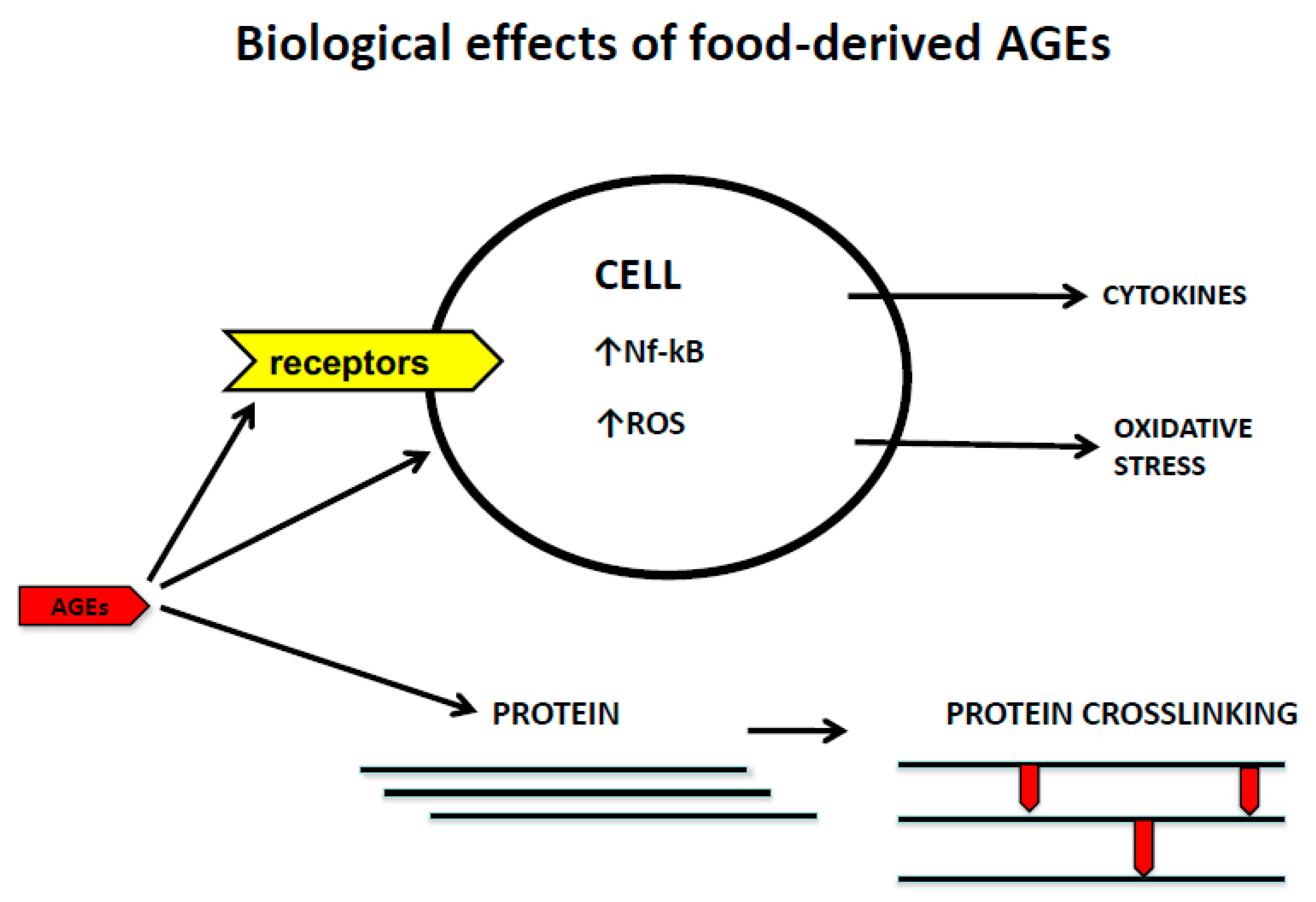
2. Mechanisms of Cell/Tissue Damage Induced by AGEs
2.1. Dietary AGEs and Extracellular Matrix Modifications
2.2. Dietary AGEs and Receptor-Mediated Mechanisms
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation-Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Doré, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Lauterbach, M.; Latz, T. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 19, 794–811. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, N.; Andreeva, V.A.; Kesse-Guyot, E.; Hercberg, E. Dietary patterns, inflammation and the metabolic síndrome. Diabetes Metab. 2013, 39, 99–110. [Google Scholar] [CrossRef]
- Garcia-Arellano, A.; Martínez-González, M.A.; Ramallal, R.; Salas-Salvadó, J.; Hébert, J.R.; Corella, D.; Shivappa, N.; Forga, L.; Schröder, H.; Muñoz-Bravo, C.; et al. Dietary inflammatory index and all-cause mortality in large cohorts: The SUN and PREDIMED studies. Clin. Nutr. 2019, 38, 1221–1231. [Google Scholar] [CrossRef]
- Mazidi, M.; Shivappa, N.; Wirth, M.D.; Hebert, J.R.; Mikhailidis, D.P.; Kengne, A.P.; Banach, M. Dietary inflammatory index and cardiometabolic risk in US adults. Atherosclerosis 2018, 276, 23–27. [Google Scholar] [CrossRef]
- Shivappa, N.; Godos, J.; Hébert, J.R.; Wirth, M.D.; Piuri, G.; Speciani, A.F.; Grosso, G. Dietary Inflammatory Index and Colorectal Cancer Risk—A Meta-Analysis. Nutrients 2017, 9, 1043. [Google Scholar] [CrossRef]
- Shivappa, N.; Godos, J.; Hébert, J.R.; Wirth, M.D.; Piuri, G.; Speciani, A.F.; Grosso, G. Dietary Inflammatory Index and Cardiovascular Risk and Mortality—A Meta-Analysis. Nutrients 2018, 10, 200. [Google Scholar] [CrossRef]
- Koschinsky, T.; He, C.J.J.; Mitsuhashi, T.; Bucala, R.; Liu, C.; Buenting, C.; Heitmann, K.; Vlassara, H. Orally absorbed reactive glycation products (glycotoxins): An environmental risk factor in diabetic nephropathy. Proc. Natl. Acad. Sci. USA 1997, 94, 6474–6479. [Google Scholar] [CrossRef] [PubMed]
- Garay-Sevilla, M.E.; Beeri, M.S.; de la Maza, M.P.; Rojas, A.; Salazar-Villanea, S.; Uribarri, J. The potential role of dietary advanced glycation end products in the development of chronic non-infectious diseases: A narrative review. Nutr. Res. Rev. 2020, 33, 298–311. [Google Scholar] [CrossRef]
- Snelson, M.; Coughlan, M.T. Dietary Advanced Glycation End Products: Digestion, Metabolism and Modulation of Gut Microbial Ecology. Nutrients 2019, 22, 215. [Google Scholar] [CrossRef]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced glycation end products in foods and a practical guide to their reduction in the diet. J. Am. Diet Assoc. 2010, 110, 911–916. [Google Scholar] [CrossRef]
- Birlouez-Aragon, I.; Saavedra, G.; Tessier, F.J.; Galinier, A.; Ait-Ameur, L.; Lacoste, F.; Niamba, C.N.; Alt, N.; Somoza, V.; Lecerf, J.M. A diet based on high-heat-treated foods promotes risk factors for diabetes mellitus and cardiovascular diseases. Am. J. Clin. Nutr. 2010, 91, 1220–1226. [Google Scholar] [CrossRef]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation end products (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Tripp, E.; Pyzik, R.; Yee, K.; Goldberg, L.; Tansman, L.; Chen, X.; Mani, V.; Fayad, Z.A.; et al. Oral AGE restriction ameliorates insulin resistance in obese individuals with the metabolic syndrome: A randomised controlled trial. Diabetologia 2016, 59, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Pyzik, R.; Goodman, S.; Chen, X.; Zhu, L.; Ramdas, M.; Striker, G.E.; Vlassara, H. Suppression of native defense mechanisms, SIRT1 and PPARγ, by dietary glycoxidants precedes disease in adult humans; relevance to lifestyle-engendered chronic diseases. Amino Acids 2014, 46, 301–309. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Woodward, M.; Tripp, E.; Goldberg, L.; Pyzik, R.; Yee, K.; Tansman, L.; Chen, X.; Mani, V.; et al. Elevated serum advanced glycation end products in obese indicate risk for the metabolic syndrome: A link between healthy and unhealthy obesity? J. Clin. Endocrinol. Metab. 2015, 100, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Scheijen, J.L.J.M.; Clevers, E.; Engelen, L.; Dagnelie, P.C.; Brouns, F.; Stehouwer, C.D.A.; Schalkwijk, C.G. Analysis of advanced glycation end products in selected food items by ultra-performance liquid chromatography tandem mass spectrometry: Presentation of a dietary AGE database. Food Chem. 2016, 1, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Baye, E.; Kiriakova, V.; Uribarri, J.; Moran, L.J.; de Courten, B. Consumption of diets with low advanced glycation end products improves cardiometabolic parameters: Meta-analysis of randomised controlled trials. Sci. Rep. 2017, 23, 2266. [Google Scholar] [CrossRef]
- Goudarzi, R.; Sedaghat, M.; Hedayati, M.; Hekmatdoost, A.; Sohrab, G. Low advanced Glycation end product diet improves the central obesity, insulin resistance and inflammatory profiles in Iranian patients with metabolic syndrome: A randomized clinical trial. J. Diabetes Metab. Disord. 2020, 26, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Keogh, J.B.; Clifton, P.M. Effects of Two Different Dietary Patterns on Inflammatory Markers, Advanced Glycation End Products and Lipids in Subjects without Type 2 Diabetes: A Randomised Crossover Study. Nutrients 2017, 9, 336. [Google Scholar] [CrossRef]
- Capurso, C.; Bellanti, F.; Lo Buglio, A.; Vendemiale, G. The Mediterranean Diet Slows Down the Progression of Aging and Helps to Prevent the Onset of Frailty: A Narrative Review. Nutrients 2019, 21, 35. [Google Scholar] [CrossRef]
- Davis, C.; Bryan, J.; Hodgson, J.; Murphy, K. Definition of the Mediterranean Diet; A Literature Review. Nutrients 2015, 7, 9139–9153. [Google Scholar] [CrossRef]
- Urquiaga, I.; Echeverría, G.; Dussaillant, C.; Rigotti, A. Origen, componentes y mechanisms of action de acciónof the Mediterranean diet. Rev. Med. Chil. 2017, 145, 85–95. [Google Scholar] [CrossRef]
- Lăcătușu, C.M.; Grigorescu, E.D.; Floria, M.; Onofriescu, A.; Mihai, B.M. The Mediterranean Diet: From an Environment-Driven Food Culture to an Emerging Medical Prescription. Int. J. Environ. Res. Public Health 2019, 16, 942. [Google Scholar] [CrossRef]
- Llorente-Cortés, V.; Estruch, R.; Mena, M.P.; Ros, E.; González, M.A.; Fitó, M.; Lamuela-Raventós, R.M.; Badimon, L. Effect of Mediterranean diet on the expression of pro-atherogenic genes in a population at high cardiovascular risk. Atherosclerosis 2010, 208, 442–450. [Google Scholar] [CrossRef]
- Martínez-González, M.A.; Salas-Salvadó, J.; Estruch, R.; Corella, D.; Fitó, M.; Ros, E.; Predimed Investigators. Benefits of the Mediterranean diet: Insights from the PREDIMED Study. Prog. Cardiovasc. Dis. 2015, 58, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Widmer, R.J.; Flammer, A.J.; Lerman, L.O.; Lerman, A. The Mediterranean diet, its components, and cardiovascular disease. Am. J. Med. 2015, 128, 229–238. [Google Scholar] [CrossRef]
- Salas-Salvadó, J.; Fernández-Ballart, J.; Ros, E.; Martínez-González, M.A.; Fitó, M.; Estruch, R.; Corella, D.; Fiol, M.; Gómez-Gracia, E.; Arós, F.; et al. Effect of a Mediterranean diet supplemented with nuts on metabolic syndrome status: One-year results of the PREDIMED randomized trial. Arch. Intern. Med. 2008, 168, 2449–2458. [Google Scholar] [CrossRef] [PubMed]
- Salas-Salvadó, J.; Bulló, M.; Babio, N.; Martínez-González, M.Á.; Ibarrola-Jurado, N.; Basora, J.; Estruch, R.; Covas, M.I.; Corella, D.; Arós, F.; et al. Reduction in the incidence of type 2 diabetes with the Mediterranean diet: Results of the PREDIMED-Reus nutrition intervention randomized trial. Diabetes Care 2011, 34, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Camargo, A.; Jimenez-Lucena, R.; Delgado-Lista, J.; Marin, C.; Tinahones, F.J.; Striker, G.E.; Roche, H.M.; Perez-Martinez, P.; et al. Dietary fat quantity and quality modifies advanced glycation end products metabolism in patients with metabolic syndrome. Mol. Nutr. Food Res. 2017, 61, 1601029. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Moreno, J.; Quintana-Navarro, G.M.; Delgado-Lista, J.; Garcia-Rios, A.; Delgado-Casado, N.; Camargo, A.; Perez-Martinez, P.; Striker, G.E.; Tinahones, F.J.; Perez-Jimenez, F.; et al. Mediterranean Diet Reduces Serum Advanced Glycation End Products and Increases Antioxidant Defenses in Elderly Adults: A Randomized Controlled Trial. J. Am. Geriatr. Soc. 2016, 64, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R., 3rd; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef]
- Siervo, M.; Lara, J.; Chowdhury, S.; Ashor, A.; Oggioni, C.; Mathers, J.C. Effects of the Dietary Approach to Stop Hypertension (DASH) diet on cardiovascular risk factors: A systematic review and meta-analysis. Br. J. Nutr. 2015, 113, 1–15. [Google Scholar] [CrossRef]
- Jannasch, F.; Kroger, J.; Schulze, M.B. Dietary Patterns and Type 2 Diabetes: A Systematic Literature Review and Meta-Analysis of Prospective Studies. J. Nutr. 2017, 147, 1174–1182. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Diet quality as assessed by the Healthy Eating Index, the Alternate Healthy Eating Index, the Dietary Approaches to Stop Hypertension score, and health outcomes: A systematic review and meta-analysis of cohort studies. J. Acad. Nutr. Diet. 2015, 115, 780–800. [Google Scholar] [CrossRef]
- Chiavaroli, L.; Viguiliouk, E.; Nishi, S.K.; Blanco Mejia, S.; Rahelić, D.; Kahleová, H.; Salas-Salvadó, J.; Kendall, C.W.; Sievenpiper, J.L. DASH Dietary Pattern and Cardiometabolic Outcomes: An Umbrella Review of Systematic Reviews and Meta-Analyses. Nutrients 2019, 11, 338. [Google Scholar] [CrossRef]
- Soltani, S.; Chitsazi, M.J.; Salehi-Abargouei, A. The effect of dietary approaches to stop hypertension (DASH) on serum inflammatory markers: A systematic review and meta-analysis of randomized trials. Clin. Nutr. 2018, 37, 542–550. [Google Scholar] [CrossRef]
- Cavicchia, P.P.; Steck, S.E.; Hurley, T.G.; Hussey, J.R.; Ma, Y.; Ockene, I.S.; Hébert, J.R. A new dietary inflammatory index predicts interval changes in serum high-sensitivity C-reactive protein. J. Nutr. 2009, 139, 2365–2372. [Google Scholar] [CrossRef]
- Shivappa, N.; Steck, S.E.; Hurley, T.G.; Hussey, J.R.; Hébert, J.R. Designing and developing a literature-derived, population-based dietary inflammatory index. Public Health Nutr. 2014, 17, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Chen, L.W.; Heude, B.; Bernard, J.Y.; Harvey, N.C.; Duijts, L.; Mensink-Bout, S.M.; Polanska, K.; Mancano, G.; Suderman, M.; et al. Dietary Inflammatory Index and Non-Communicable Disease Risk: A Narrative Review. Nutrients 2019, 11, 1873. [Google Scholar] [CrossRef] [PubMed]
- Nikniaz, L.; Nikniaz, Z.; Shivappa, N.; Hébert, J.R. The association between dietary inflammatory index and metabolic syndrome components in Iranian adults. Prim. Care Diabetes 2018, 5, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.M.; Shivappa, N.; Hébert, J.R.; Perry, I.J. Dietary Inflammatory Index and Biomarkers of Lipoprotein Metabolism, Inflammation and Glucose Homeostasis in Adults. Nutrients 2018, 10, 1033. [Google Scholar] [CrossRef] [PubMed]
- Pimenta, A.M.; Toledo, E.; Rodriguez-Diez, M.C.; Gea, A.; Lopez-Iracheta, R.; Shivappa, N.; Hébert, J.R.; Martinez-Gonzalez, M.A. Dietary indexes, food patterns and incidence of metabolic syndrome in a Mediterranean cohort: The SUN project. Clin. Nutr. 2015, 34, 508–514. [Google Scholar] [CrossRef]
- Ruiz-Canela, M.; Zazpe, I.; Shivappa, N.; Hébert, J.R.; Sánchez-Tainta, A.; Corella, D.; Salas-Salvadó, J.; Fitó, M.; Lamuela-Raventós, R.M.; Rekondo, J.; et al. Dietary inflammatory index and anthropometric measures of obesity in a population sample at high cardiovascular risk from the PREDIMED (PREvención con DIeta MEDiterránea) trial. Br. J. Nutr. 2015, 113, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.A.; Silva, A.A.M.; Assunção, M.C.F.; Fonseca, P.C.A.; Barbieri, M.A.; Bettiol, H.; Shivappa, N.; Hébert, J.R. The dietary inflammatory index and insulin resistance or metabolic syndrome in young adults. Nutrition 2019, 58, 187–193. [Google Scholar] [CrossRef]
- Ren, Z.; Zhao, A.; Wang, Y.; Meng, L.; Szeto, I.M.; Li, T.; Gong, H.; Tian, Z.; Zhang, Y.; Wang, P. Association between Dietary Inflammatory Index, C-Reactive Protein and Metabolic Syndrome: A Cross-Sectional Study. Nutrients 2018, 10, 831. [Google Scholar] [CrossRef]
- Monnier, V.M.; Sell, D.R.; Nagaraj, R.H.; Miyata, S.; Grandhee, S.; Odetti, P.; Ibrahim, S.A. Maillard reaction-mediated molecular damage to extracellular matrix and other tissue proteins in diabetes, aging, and uremia. Diabetes 1992, 41 (Suppl. 2), 36–41. [Google Scholar] [CrossRef]
- Bucala, R.; Mitchell, R.; Arnold, K.; Innerarity, T.; Vlassara, H.; Cerami, A. Identification of the major site of apolipoprotein B modification by advanced glycosylation end products blocking uptake by the low density lipoprotein receptor. J. Biol. Chem. 1995, 270, 10828–10832. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs. Cell. Mol. Biol. 1998, 44, 1013–1023. [Google Scholar]
- Chakravarthy, U.; Hayes, R.G.; Stitt, A.W.; McAuley, E.; Archer, D.B. Constitutive nitric oxide synthase expression in retinal vascular endothelial cells is suppressed by high glucose and advanced glycation end products. Diabetes 1998, 47, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Saremi, A.; Howell, S.; Schwenke, D.C.; Bahn, G.; Beisswenger, P.J.; Reaven, P.D.; VADT Investigators. Advanced Glycation End Products, Oxidation Products, and the Extent of Atherosclerosis during the VA Diabetes Trial and Follow-up Study. Diabetes Care 2017, 40, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Beisswenger, P.J.; Makita, Z.; Curphey, T.J.; Moore, L.L.; Jean, S.; Brinck-Johnsen, T.; Bucala, R.; Vlassara, H. Formation of immunochemical advanced glycosylation end products precedes and correlates with early manifestations of renal and retinal disease in diabetes. Diabetes 1995, 44, 824–829. [Google Scholar] [CrossRef]
- Tsai, T.T.; Ho, N.Y.; Lin, Y.T.; Lai, P.L.; Fu, T.S.; Niu, C.C.; Chen, L.H.; Chen, W.J.; Pang, J.H. Advanced glycation end products in degenerative nucleus pulposus with diabetes. J. Orthop. Res. 2014, 32, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Fields, A.J.; Berg-Johansen, B.; Metz, L.N.; Miller, S.; La, B.; Liebenberg, E.C.; Coughlin, D.G.; Graham, J.L.; Stanhope, K.L.; Havel, P.J.; et al. Alterations in intervertebral disc composition, matrix homeostasis and biomechanical behavior in the UCD-T2DM rat model of type 2 diabetes. J. Orthop. Res. 2015, 33, 738–746. [Google Scholar] [CrossRef]
- Illien-Junger, S.; Grosjean, F.; Laudier, D.M.; Vlassara, H.; Striker, G.E.; Iatridis, J.C. Combined anti-inflammatory and anti-AGE drug treatments have a protective effect on intervertebral discs in mice with diabetes. PLoS ONE 2013, 8, e64302. [Google Scholar] [CrossRef]
- Cannizzaro, L.; Rossoni, G.; Savi, F.; Altomare, A.; Marinello, C.; Saethang, T.; Carini, M.; Payne, D.M.; Pisitkun, T.; Aldini, G.; et al. Regulatory landscape of AGE-RAGE-oxidative stress axis and its modulation by PPARγ activation in high fructose diet-induced metabolic syndrome. Nutr. Metab. 2017, 14, 1–13. [Google Scholar] [CrossRef]
- Uribarri, J.; Peppa, M.; Cai, W.; Goldberg, T.; Lu, M.; He, C.; Vlassara, H. Restriction of dietary glycotoxins reduces excessive advanced glycation end products in renal failure patients. J. Am. Soc. Nephrol. 2003, 14, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Tessier, F.J.; Niquet-Léridon, C.; Jacolot, P.; Jouquand, C.; Genin, M.; Schmidt, A.M.; Grossin, N.; Boulanger, E. Quantitative assessment of organ distribution of dietary protein-bound 13C-labeled Nɛ-carboxymethyllysine after a chronic oral exposure in mice. Mol. Nutr. Food Res. 2016, 60, 2446–2456. [Google Scholar] [CrossRef]
- He, C.; Sabol, J.; Mitsuhashi, T.; Vlassara, H. Dietary glycotoxins: Inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999, 48, 1308–1315. [Google Scholar] [CrossRef]
- Illien-Jünger, S.; Lu, Y.; Qureshi, S.A.; Hecht, A.C.; Cai, W.; Vlassara, H.; Striker, G.E.; Iatridis, J.C. Chronic ingestion of advanced glycation end products induces degenerative spinal changes and hypertrophy in aging pre-diabetic mice. PLoS ONE 2015, 10, e0116625. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Illien-Jünger, S.; Palacio-Mancheno, P.; Kindschuh, W.F.; Chen, X.; Sroga, G.E.; Vashishth, D.; Iatridis, J.C. Dietary Advanced Glycation End Products Have Sex- and Age-Dependent Effects on Vertebral Bone Microstructure and Mechanical Function in Mice. J. Bone Miner. Res. 2018, 33, 437–448. [Google Scholar] [CrossRef]
- Krishnamoorthy, D.; Hoy, R.C.; Natelson, D.M.; Torre, O.M.; Laudier, D.M.; Iatridis, J.C.; Illien-Jünger, S. Dietary advanced glycation end-product consumption leads to mechanical stiffening of murine intervertebral discs. Dis. Model Mech. 2018, 11, dmm036012. [Google Scholar] [CrossRef]
- Sivan, S.S.; Tsitron, E.; Wachtel, E.; Roughley, P.; Sakkee, N.; van der Ham, F.; Degroot, J.; Maroudas, A. Age-related accumulation of pentosidine in aggrecan and collagen from normal and degenerate human intervertebral discs. Biochem. J. 2006, 399, 29–35. [Google Scholar] [CrossRef]
- Verzijl, N.; DeGroot, J.; Ben, Z.C.; Brau-Benjamin, O.; Maroudas, A.; Bank, R.A.; Mizrahi, J.; Schalkwijk, C.G.; Thorpe, S.R.; Baynes, J.W.; et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: A possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 2002, 46, 114–123. [Google Scholar] [CrossRef]
- Vashishth, D.; Gibson, G.J.; Khoury, J.I.; Schaffler, M.B.; Kimura, J.; Fyhrie, D.P. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 2001, 28, 195–201. [Google Scholar] [CrossRef]
- Pokharna, H.K.; Phillips, F.M. Collagen crosslinks in human lumbar intervertebral disc aging. Spine 1998, 23, 1645–1648. [Google Scholar] [CrossRef]
- Wagner, D.R.; Reiser, K.M.; Lotz, J.C. Glycation increases human annulus fibrosus stiffness in both experimental measurements and theoretical predictions. J. Biomech. 2006, 39, 1021–1029. [Google Scholar] [CrossRef]
- Reiser, K.; McCormick, R.J.; Rucker, R.B. Enzymatic and nonenzymatic cross-linking of collagen and elastin. FASEB J. 1992, 6, 2439–2449. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.Y.; Ghazaryan, A.A.; Lo, W.; Chen, Y.F.; Hovhannisyan, V.; Chen, S.J.; Tan, H.Y.; Dong, C.Y. Multiphoton spectral microscopy for imaging and quantification of tissue glycation. Biomed. Opt. Express. 2010, 2, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.M.; Eichler, J.; Reiser, K.M.; Rubenchik, A.M.; Da Silva, L.B. Collagen structure and nonlinear susceptibility: Effects of heat, glycation, and enzymatic cleavage on second harmonic signal intensity. Lasers Surg. Med. 2000, 27, 329–335. [Google Scholar] [CrossRef]
- Bai, P.; Phua, K.; Hardt, T.; Cernadas, M.; Brodsky, B. Glycation alters collagen fibril organization. Connect. Tissue Res. 1992, 28, 1–12. [Google Scholar] [CrossRef]
- Tanaka, S.; Avigad, G.; Brodsky, B.; Eikenberry, E.F. Glycation induces expansion of the molecular packing of collagen. J. Mol. Biol. 1988, 203, 495–505. [Google Scholar] [CrossRef]
- Bourne, J.W.; Lippell, J.M.; Torzilli, P.A. Glycation cross-linking induced mechanical-enzymatic cleavage of microscale tendon fibers. Matrix Biol. 2014, 34, 179–184. [Google Scholar] [CrossRef][Green Version]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell. Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef]
- Ott, C.; Jacobs, K.; Haucke, E.; Santos, A.N.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef]
- Buetler, T.M.; Leclerc, E.; Baumeyer, A.; Latado, H.; Newell, J.; Adolfsson, O.; Parisod, V.; Richoz, J.; Maurer, S.; Foata, F.; et al. Nε-carboxymethyllysine-modified proteins are unable to bind to RAGE and activate an inflammatory response. Mol. Nutr. Food Res. 2008, 52, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, A.; Niquet-Leridon, C.; Boulanger, E.; Tessier, F.J. How Can Diet Affect the Accumulation of Advanced Glycation End-Products in the Human Body? Foods 2016, 6, 84. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, Y.; Fu, L. Dietary advanced glycation end-products: Perspectives linking food processing with health implications. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2559–2587. [Google Scholar] [CrossRef]
- Roncero-Ramos, I.; Delgado-Andrade, C.; Haro, A.; Ruiz-Roca, B.; Morales, F.J.; Navarro, M.P. Effects of dietary bread crust Maillard reaction products on calcium and bone metabolism in rats. Amino Acids 2013, 44, 1409–1418. [Google Scholar] [CrossRef] [PubMed]
- Roncero-Ramos, I.; Delgado-Andrade, C.; Tessier, F.J.; Niquet-Léridon, C.; Strauch, C.; Monnier, V.M.; Navarro, M.P. Metabolic transit of N(ε)-carboxymethyl-lysine after consumption of AGEs from bread crust. Food Funct. 2013, 4, 1032–1039. [Google Scholar] [CrossRef]
- Grossin, N.; Auger, F.; Niquet-Leridon, C.; Durieux, N.; Montaigne, D.; Schmidt, A.M.; Susen, S.; Jacolot, P.; Beuscart, J.B.; Tessier, F.J.; et al. Dietary CML-enriched protein induces functional arterial aging in a RAGE-dependent manner in mice. Mol. Nutr. Food Res. 2015, 59, 927–938. [Google Scholar] [CrossRef]
- Kratochvilová, M.; Zakiyanov, O.; Kalousová, M.; Kříha, V.; Zima, T.; Tesař, V. Associations of serum levels of advanced glycation end products with nutrition markers and anemia in patients with chronic kidney disease. Ren. Fail. 2011, 33, 131–137. [Google Scholar] [CrossRef]
- Odetti, P.; Fogarty, J.; Sell, D.R.; Monnier, V.M. Chromatographic quantitation of plasma and erythrocyte pentosidine in diabetic and uremic subjects. Diabetes 1992, 41, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Henle, T.; Miyata, T. Advanced glycation end products in uremia. Adv. Ren. Replace Ther. 2003, 10, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J.; Ferrucci, L.; Cai, W.; Torreggiani, M.; Post, J.B.; Zheng, F.; Striker, G.E. Identifying advanced glycation end products as a major source of oxidants in aging: Implications for the management and/or prevention of reduced renal function in elderly persons. Semin. Nephrol. 2009, 29, 594–603. [Google Scholar] [CrossRef]
- McIntyre, N.J.; Fluck, R.J.; McIntyre, C.W.; Taal, M.W. Skin autofluorescence and the association with renal and cardiovascular risk factors in chronic kidney disease stage 3. Clin. J. Am. Soc. Nephrol. 2011, 6, 2356–2363. [Google Scholar] [CrossRef]
- Uribarri, J.; del Castillo, M.D.; de la Maza, M.P.; Filip, R.; Gugliucci, A.; Luevano-Contreras, C.; Macías-Cervantes, M.H.; Markowicz Bastos, D.H.; Medrano, A.; Menini, T.; et al. Dietary advanced glycation end products and their role in health and disease. Adv. Nutr. 2015, 6, 461–473. [Google Scholar] [CrossRef] [PubMed]
- West, R.K.; Moshier, E.; Lubitz, I.; Schmeidler, J.; Godbold, J.; Cai, W.; Uribarri, J.; Vlassara, H.; Silverman, J.M.; Beeri, M.S. Dietary advanced glycation end products are associated with decline in memory in young elderly. Mech. Ageing Dev. 2014, 140, 10–12. [Google Scholar] [CrossRef]
- Chilelli, N.C.; Burlina, S.; Lapolla, A. AGEs, rather than hyperglycemia, are responsible for microvascular complications in diabetes: A “glycoxidation-centric” point of view. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 913–919. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyl stress, protein glycation and the unfolded protein response. Glycoconj. J. 2021, 38, 331–340. [Google Scholar] [CrossRef]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 27, 7889. [Google Scholar] [CrossRef]
- Snelson, M.; Tan, S.M.; Clarke, R.E.; de Pasquale, C.; Thallas-Bonke, V.; Nguyen, T.V.; Penfold, S.A.; Harcourt, B.E.; Sourris, K.C.; Lindblom, R.S.; et al. Processed foods drive intestinal barrier permeability and microvascular diseases. Sci. Adv. 2021, 7, eabe4841. [Google Scholar] [CrossRef]
- Rajan, B.S.; Manivasagam, S.; Dhanusu, S.; Chandrasekar, N.; Krishna, K.; Kalaiarasu, L.P.; Babu, A.A.; Vellaichamy, E. Diet with high content of advanced glycation end products induces systemic inflammation and weight gain in experimental mice: Protective role of curcumin and gallic acid. Food Chem. Toxicol. 2018, 114, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Chikazawa, M.; Shibata, T.; Hatasa, Y.; Hirose, S.; Otaki, N.; Nakashima, F.; Ito, M.; Machida, S.; Maruyama, S.; Uchida, K. Identification of C1q as a Binding Protein for Advanced Glycation End Products. Biochemistry 2016, 55, 435–446. [Google Scholar] [CrossRef]
- Tian, Y.; Kijlstra, A.; van der Veen, R.L.; Makridaki, M.; Murray, I.J.; Berendschot, T.T. The effect of lutein supplementation on blood plasma levels of complement factor D, C5a and C3d. PLoS ONE 2013, 8, e73387. [Google Scholar]
- Muramatsu, D.; Wakabayashi, Y.; Usui, Y.; Okunuki, Y.; Kezuka, T.; Goto, H. Correlation of complement fragment C5a with inflammatory cytokines in the vitreous of patients with proliferative diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2013, 251, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; A randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE signaling in inflammatory disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.K.; Ramasamy, R.; Schmidt, A.M. The receptor for advanced glycation end-products (RAGE) and DIAPH1: Implications for vascular and neuroinflammatory dysfunction in disorders of the central nervous system. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay Between Glycation, Inflammation, and Hallmarks of Cancer-a Review. Horm. Cancer 2018, 5, 295–325. [Google Scholar] [CrossRef]
- Rojas, A.; Añazco, C.; Gonzalez, I. AGEs clearance mechanisms. In Dietary AGEs and their Roles in Health and Disease; Uribarri, J., Ed.; CRC Press: New York, NY, USA, 2017; pp. 37–49. [Google Scholar]
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural basis for ligand recognition and activation of RAGE. Structure 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M. Soluble RAGEs—Prospects for treating & tracking metabolic and inflammatory disease. Vascul. Pharmacol. 2015, 72, 1–8. [Google Scholar]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2008, 22, 1572–1580. [Google Scholar] [CrossRef] [PubMed]
- Raucci, A.; Cugusi, S.; Antonelli, A.; Barabino, S.M.; Monti, L.; Bierhaus, A.; Reiss, K.; Saftig, P.; Bianchi, M.E. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J. 2008, 22, 3716–3727. [Google Scholar] [CrossRef]
- Rojas, A.; Morales, M.; Gonzalez, I.; Araya, P. Inhibition of RAGE axis signaling: A pharmacological challenge. Curr. Drug Targets 2019, 20, 340–346. [Google Scholar] [CrossRef]
- González, I.; Romero, J.; Rodríguez, B.L.; Pérez-Castro, R.; Rojas, A. The immunobiology of the receptor of advanced glycation end-products: Trends and challenges. Immunobiology 2013, 218, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Kalea, A.Z.; Del Mar Arriero, M.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.H. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef]
- Slowik, A.; Merres, J.; Elfgen, A.; Jansen, S.; Mohr, F.; Wruck, C.J.; Pufe, T.; Brandenburg, L.O. Involvement of formyl peptide receptors in receptor for advanced glycation end products (RAGE)—And amyloid beta 1-42-induced signal transduction in glial cells. Mol. Neurodegener. 2012, 7, 1–18. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Kinoshita, R.; Putranto, E.W.; Ruma, I.M.W.; Sumardika, I.W.; Youyi, C.; Tomonobu, N.; Yamamoto, K.I.; Murata, H. Signal diversity of receptor for advanced glycation end products. Acta Med. Okayama 2017, 71, 459–465. [Google Scholar]
- Pickering, R.J.; Tikellis, C.; Rosado, C.J.; Tsorotes, D.; Dimitropoulos, A.; Smith, M.; Huet, O.; Seeber, R.M.; Abhayawardana, R.; Johnstone, E.K.; et al. Transactivation of RAGE mediates angiotensin-induced inflammation and atherogenesis. J. Clin. Investig. 2019, 129, 406–421. [Google Scholar] [CrossRef] [PubMed]
- Sitkiewicz, E.; Tarnowski, K.; Poznański, J.; Kulma, M.; Dadlez, M. Oligomerization interface of RAGE receptor revealed by MS-monitored hydrogen deuterium exchange. PLoS ONE 2013, 8, e76353. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the receptor for advanced glycation end-products (RAGE): A Medicinal chemistry perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE ligands, and their role in cancer and inflammation. J. Transl. Med. 2009, 7, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Rodríguez, M.E.; Uribarri, J.; Solorio-Meza, S.E.; Fajardo-Araujo, M.E.; Cai, W.; Torres-Graciano, S.; Rangel-Salazar, R.; Wrobel, K.; Garay-Sevilla, M.E. The AGE-RAGE axis and its relationship to markers of cardiovascular disease in newly diagnosed diabetes patients. PLoS ONE 2016, 11, e0159175. [Google Scholar] [CrossRef]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef]
- Akirav, E.M.; Preston-Hurlburt, P.; Garyu, J.; Henegariu, O.; Clynes, R.; Schmidt, A.M.; Herold, K.C. RAGE expression in human T cells: A link between environmental factors and adaptive immune responses. PLoS ONE 2012, 7, e34698. [Google Scholar] [CrossRef]
- Akira, S.; Takeda, K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.J.; Yang, H.Y.; Pai, M.H.; Wu, C.H.; Chen, J.R. Long-term administration of advanced glycation end-product stimulates the activation of NLRP3 inflammasome and sparking the development of renal injury. J. Nutr. Biochem. 2017, 39, 68–76. [Google Scholar] [CrossRef]
- Seno, K.; Sase, S.; Ozeki, A.; Takahashi, H.; Ohkuchi, A.; Suzuki, H.; Matsubara, S.; Iwata, H.; Kuwayama, T.; Shirasuna, K. Advanced glycation end-products regulate interleukin-1β production in human placenta. J. Reprod. Dev. 2017, 63, 401–408. [Google Scholar] [CrossRef]
- Gao, F.; Zhang, S. Loratadine alleviates advanced glycation end product-induced activation of NLRP3 inflammasome in human chondrocytes. Drug Des. Devel. Ther. 2020, 14, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Meng, X.F.; Zhang, C. Inflammasome activation in podocytes: A new mechanism of glomerular diseases. Inflamm. Res. 2020, 69, 731–743. [Google Scholar] [CrossRef]
- Son, S.; Hwang, I.; Han, S.H.; Shin, J.S.; Shin, O.S.; Yu, J.W. Advanced glycation end-products impair NLRP3 inflammasome-mediated innate immune responses in macrophages. J. Biol. Chem. 2017, 292, 20437–20448. [Google Scholar] [CrossRef]
- Son, S.; Hwang, I.; Yu, J.W. Modulation of inflammasome signaling by advanced glycation end-products (AGEs). FASEB J. 2017, 31, lb194. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garay-Sevilla, M.E.; Rojas, A.; Portero-Otin, M.; Uribarri, J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients 2021, 13, 2802. https://doi.org/10.3390/nu13082802
Garay-Sevilla ME, Rojas A, Portero-Otin M, Uribarri J. Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients. 2021; 13(8):2802. https://doi.org/10.3390/nu13082802
Chicago/Turabian StyleGaray-Sevilla, Ma. Eugenia, Armando Rojas, Manuel Portero-Otin, and Jaime Uribarri. 2021. "Dietary AGEs as Exogenous Boosters of Inflammation" Nutrients 13, no. 8: 2802. https://doi.org/10.3390/nu13082802
APA StyleGaray-Sevilla, M. E., Rojas, A., Portero-Otin, M., & Uribarri, J. (2021). Dietary AGEs as Exogenous Boosters of Inflammation. Nutrients, 13(8), 2802. https://doi.org/10.3390/nu13082802