
Interactions between Growth of Muscle and Stature: Mechanisms Involved and Their Nutritional Sensitivity to Dietary Protein: The Protein-Stat Revisited


Abstract
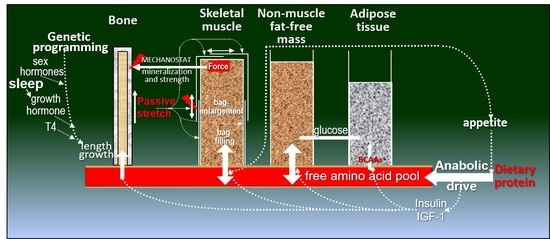
1. Introduction
2. Coordination of Growth of Muscle and Stature
2.1. Genetics and Programming
2.2. Muscle and Bone Growth Interrelationships: The Protein-Stat and the Mechanostat
2.3. Muscle and Bone Growth Interrelationships during Saltatory Growth
3. Endocrine and Cellular Mechanisms of Growth in Stature during Development
3.1. Endocrine Regulation of Statural Growth
3.2. Endochondral Ossification
4. Cellular Mechanisms of Muscle Growth during Development
4.1. Myofibre Hypertrophy within the Extracellular Matrix: The Nature of the Problem
4.2. Role of Satellite and Other Cells in Myofibre Hypertrophy
4.3. The Mechanotransduction of Satellite Cell-Induced Muscle Growth
4.3.1. Myofibre, Satellite Cell and ECM Linkages
4.3.2. Molecular Mechanisms
4.3.3. Role of mTORC1
4.3.4. Hippo Pathway and Notch Signalling
4.3.5. WNT Signalling
4.3.6. Free Calcium
4.3.7. Metalloproteinases
4.4. Myofibre Hypertrophy and Its Implications for Protein Turnover within the Myonuclear Domain
4.5. Implications of Changes in Total Muscle Nuclear Domain Number and Size for ECM Remodelling during Postnatal Growth
5. Regulation of Protein Deposition in Muscle: Experimental Studies in Animals and Human Adults
5.1. Animal Studies of Regulation of Protein Deposition in Muscle: Dietary Protein, Insulin, Amino Acids and Thyroid Hormones
5.2. Animal Studies of Muscle and Bone Growth Interactions
5.3. Regulation of “Bag-Filling” and the “Bag-Full” Signal in Human Muscle
5.3.1. Amino Acids and MPS
5.3.2. Insulin and MPB
5.3.3. Muscle Volume Changes and Regulation
6. Nutritional Sensitivity of Growth of Muscle and Stature to Dietary Protein in Children
6.1. The Breastfed Child as the Normative Growth Model
6.2. The “Early Protein Hypothesis.”
6.3. IGF-1 and Length/Height Growth in a Paediatric Context
6.4. Low Protein Intakes of the Breastfed Infant and Length Growth
6.5. Height and Muscle Mass Growth of Human Children Is Limited to a Genotypic Maximum
6.6. Length/Height Growth and Dietary Protein in Older Children and Adolescents—The Issue of Milk Intakes
6.7. Protein Deficiency and Stunting
6.8. Can Muscle Growth in Children Occur Independently from Length Growth?
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tanner, J. Growth as a measure of the nutritional and hygienic status of a population. Horm. Res. 1992, 38, 106–115. [Google Scholar] [CrossRef] [PubMed]
- NCD Risk Factor Collaboration. A century of trends in adult human height. Elife 2016, 5, 1–29. [Google Scholar] [CrossRef]
- Orsso, C.E.; Tibaes, J.R.B.; Oliveira, C.L.P.; Rubin, D.A.; Field, C.J.; Heymsfield, S.B.; Prado, C.M.; Haqq, A.M. Low muscle mass and strength in pediatrics patients: Why should we care? Clin. Nutr. 2019, 38, 2002–2015. [Google Scholar] [CrossRef] [PubMed]
- Timpka, S.; Petersson, I.F.; Zhou, C.; Englund, M. Muscle strength in adolescent men and risk of cardiovascular disease events and mortality in middle age: A prospective cohort study. BMC Med. 2014, 12, 1–8. [Google Scholar] [CrossRef]
- Lee, D.H.; Keum, N.N.; Hu, F.B.; Orav, E.J.; Rimm, E.B.; Willett, W.C.; Giovannucci, E.L. Predicted lean body mass, fat mass, and all cause and cause specific mortality in men: Prospective US cohort study. BMJ 2018, 362, k2575. [Google Scholar] [CrossRef]
- Srikanthan, P.; Karlamangla, A.S. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the Third National Health and Nutrition Examination Survey. J. Clin. Endocrinol. Metab. 2011, 96, 2898–2903. [Google Scholar] [CrossRef]
- Millward, D.J. A Protein-Stat Mechanism for Regulation of Growth and Maintenance of the Lean Body Mass. Nutr. Res. Rev. 1995, 8, 93–120. [Google Scholar] [CrossRef] [PubMed]
- Tanner, J. Growth at Adolescence, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1962. [Google Scholar]
- Millward, D.J.; Rivers, J.P.W. The need for indispensable amino acids: The concept of the anabolic drive. Diabetes Metab. Rev. 1989, 5, 191–211. [Google Scholar] [CrossRef]
- Villemure, I.; Stokes, I.A.F. Growth Plate Mechanics and Mechanobiology. A Survey of Present Understanding. J. Biomech. 2009, 42, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Karlberg, J. On the modelling of human growth. Stat. Med. 1987, 6, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Golden, M.H. Is complete catch-up possible for stunted malnourished children? Eur. J. Clin. Nutr. 1994, 48, S58–S70. [Google Scholar] [PubMed]
- Yengo, L.; Sidorenko, J.; Kemper, K.E.; Zheng, Z.; Wood, A.R.; Weedon, M.N.; Frayling, T.M.; Hirschhorn, J.; Yang, J.; Visscher, P.M. Meta-analysis of genome-wide association studies for height and body mass index in ~700,000 individuals of European ancestry. Hum. Mol. Genet. 2018, 27, 3641–3649. [Google Scholar] [CrossRef] [PubMed]
- Lui, J.C.; Baron, J. Mechanisms limiting body growth in mammals. Endocr. Rev. 2011, 32, 422–440. [Google Scholar] [CrossRef]
- Chomtho, S.; Wells, J.C.K.; Williams, J.E.; Lucas, A.; Fewtrell, M.S. Associations between birth weight and later body composition: Evidence from the 4-component model. Am. J. Clin. Nutr. 2008, 88, 1040–1048. [Google Scholar] [CrossRef]
- Karasik, D. How pleiotropic genetics of the musculoskeletal system can inform genomics and phenomics of aging. Age 2011, 33, 49–62. [Google Scholar] [CrossRef]
- Karasik, D.; Zhou, Y.; Adrienne Cupples, L.; Hannan, M.T.; Kiel, D.P.; Demissie, S. Bivariate genome-wide linkage analysis of femoral bone traits and leg lean mass: Framingham study. J. Bone Miner. Res. 2009, 24, 710–718. [Google Scholar] [CrossRef]
- Liu, J.X.; Yeung, S.L.A.; He, B.; Kwok, M.K.; Leung, J.Y.Y.; Schooling, C.M. The effect of birth weight on body composition: Evidence from a birth cohort and a Mendelian randomization study. PLoS ONE 2019, 14, e0222141. [Google Scholar] [CrossRef]
- Wang, Z.M.; Visser, M.; Ma, R.; Baumgartner, R.N.; Kotler, D.; Gallagher, D.; Heymsfield, S.B. Skeletal muscle mass: Evaluation of neutron activation and dual-energy X-ray absorptiometry methods. J. Appl. Physiol. 1996, 80, 824–831. [Google Scholar] [CrossRef]
- McCarthy, H.D.; Samani-Radia, D.; Jebb, S.A.; Prentice, A.M. Skeletal muscle mass reference curves for children and adolescents. Pediatr. Obes. 2014, 9, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Dodds, R.; Denison, H.J.; Ntani, G.; Cooper, R.; Cooper, C.; Sayer, A.A.; Baird, J. Birth weight and muscle strength: A systematic review and meta-analysis. J. Nutr. Health Aging 2012, 16, 609–615. [Google Scholar] [CrossRef]
- Barr, J.G.; Veena, S.R.; Kiran, K.N.; Wills, A.K.; Winder, N.R.; Kehoe, S.; Fall, C.H.D.; Sayer, A.A.; Krishnaveni, G.V. The relationship of birthweight, muscle size at birth and post-natal growth to grip strength in 9-year-old Indian children: Findings from the Mysore Parthenon study. J. Dev. Orig. Health Dis. 2010, 1, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, R.T.; Bravenboer, N. Biochemical interaction between muscle and bone: A physiological reality? Clin. Rev. Bone Miner. Metab. 2014, 12, 27–43. [Google Scholar] [CrossRef]
- Schoenau, E.; Fricke, O. Interaction between muscle and bone. Horm. Res. 2007, 66, 73–78. [Google Scholar] [CrossRef]
- Tagliaferri, C.; Wittrant, Y.; Davicco, M.J.; Walrand, S.; Coxam, V. Muscle and bone, two interconnected tissues. Ageing Res. Rev. 2015, 21, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Bakker, A.D.; Wu, G.; Klein-Nulend, J.; Jaspers, R.T. Physicochemical Niche Conditions and Mechanosensing by Osteocytes and Myocytes. Curr. Osteoporos. Rep. 2019, 17, 235–249. [Google Scholar] [CrossRef]
- Frost, H.M. Bone’s Mechanostat: A 2003 Update. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2003, 275, 1081–1101. [Google Scholar] [CrossRef] [PubMed]
- Farr, J.N.; Laddu, D.R.; Blew, R.M.; Lee, V.R.; Going, S.B. Effects of physical activity and muscle quality on bone development in girls. Med. Sci. Sports Exerc. 2013, 45, 2332–2340. [Google Scholar] [CrossRef] [PubMed]
- Libuda, L.; Wudy, S.A.; Schoenau, E.; Remer, T. Comparison of the effects of dietary protein, androstenediol and forearm muscle area on radial bone variables in healthy prepubertal children. Br. J. Nutr. 2011, 105, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Wu, Q.; Gong, J.; Xiao, Z.; Tang, Y.; Shang, J.; Cheng, Y.; Xu, H. Relationships between the lean mass index and bone mass and reference values of muscular status in healthy Chinese children and adolescents. J. Bone Miner. Metab. 2016, 34, 703–713. [Google Scholar] [CrossRef]
- Nelson, D.A.; Simpson, P.M.; Johnson, C.C.; Barondess, D.A.; Kleerekoper, M. The accumulation of whole body skeletal mass in third- and fourth-grade children: Effects of age, gender, ethnicity, and body composition. Bone 1997, 20, 73–78. [Google Scholar] [CrossRef]
- Iuliano-Burns, S.; Mirwald, R.L.; Bailey, D.A. Timing and magnitude of peak height velocity and peak tissue velocities for early, average, and late maturing boys and girls. Am. J. Hum. Biol. 2001, 13, 1–8. [Google Scholar] [CrossRef]
- Rauch, F.; Bailey, D.A.; Baxter-Jones, A.; Mirwald, R.; Faulkner, R. The ’muscle-bone unit’ during the pubertal growth spurt. Bone 2004, 34, 771–775. [Google Scholar] [CrossRef]
- Vandewalle, S.; Taes, Y.; Fiers, T.; Toye, K.; Van Caenegem, E.; Roggen, I.; De Schepper, J.; Kaufman, J.M. Associations of sex steroids with bone maturation, bone mineral density, bone geometry, and body composition: A cross-sectional study in healthy male adolescents. J. Clin. Endocrinol. Metab. 2014, 99, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Völgyi, E.; Tylavsky, F.A.; Lyytikäinen, A.; Törmäkangas, T.; Xu, L.; Cheng, S.M.; Kröger, H.; Alèn, M.; Kujala, U.M. Trait-specific tracking and determinants of body composition: A 7-year follow-up study of pubertal growth in girls. BMC Med. 2009, 7, 1–12. [Google Scholar] [CrossRef][Green Version]
- Lang, T.F. The Bone-Muscle Relationship in Men and Women. J. Osteoporos. 2011, 2011, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Eftestøl, E.; Egner, I.M.; Lunde, I.G.; Ellefsen, S.; Andersen, T.; Sjåland, C.; Gundersen, K.; Bruusgaard, J.C. Increased hypertrophic response with increased mechanical load in skeletal muscles receiving identical activity patterns. Am. J. Physiol. Cell Physiol. 2016, 311, C616–C629. [Google Scholar] [CrossRef]
- Boers, H.E.; Haroon, M.; Le Grand, F.; Bakker, A.D.; Klein-Nulend, J.; Jaspers, R.T. Mechanosensitivity of aged muscle stem cells. J. Orthop. Res. 2018, 36, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Juffer, P.; Bakker, A.D.; Klein-Nulend, J.; Jaspers, R.T. Mechanical Loading by Fluid Shear Stress of Myotube Glycocalyx Stimulates Growth Factor Expression and Nitric Oxide Production. Cell Biochem. Biophys. 2014, 69, 411–419. [Google Scholar] [CrossRef]
- Zöllner, A.M.; Abilez, O.J.; Böl, M.; Kuhl, E. Stretching Skeletal Muscle: Chronic Muscle Lengthening through Sarcomerogenesis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Parker, D.F.; Round, J.M.; Sacco, P.; Jones, D.A. A cross-sectional survey of upper and lower limb strength in boys and girls during childhood and adolescence. Ann. Hum. Biol. 1990, 17, 199–211. [Google Scholar] [CrossRef]
- Pařízková, J. Growth and growth velocity of lean body mass and fat in adolescent boys. Pediatr. Res. 1976, 10, 647–650. [Google Scholar] [CrossRef]
- Xu, L.; Nicholson, P.; Wang, Q.; Alén, M.; Cheng, S. Bone and muscle development during puberty in girls: A seven-year longitudinal study. J. Bone Miner. Res. 2009, 24, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Hogrel, J.Y.; Decostre, V.; Alberti, C.; Canal, A.; Ollivier, G.; Josserand, E.; Taouil, I.; Simon, D. Stature is an essential predictor of muscle strength in children. BMC Musculoskelet. Disord. 2012, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.G.; Pearn, J.; Barnes, A.; Young, C.M.; Kehoe, M.; Newman, J. Norms for hand grip strength. Arch. Dis. Child. 1984, 59, 453–459. [Google Scholar] [CrossRef]
- Eek, M.N.; Kroksmark, A.K.; Beckung, E. Isometric Muscle Torque in Children 5 to 15 Years of Age: Normative Data. Arch. Phys. Med. Rehabil. 2006, 87, 1091–1099. [Google Scholar] [CrossRef]
- Maes, C.; Kronenberg, H.M. Bone Development and Remodeling, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 1, ISBN 9780323189071. [Google Scholar]
- Hallett, S.A.; Ono, W.; Ono, N. Growth plate chondrocytes: Skeletal development, growth and beyond. Int. J. Mol. Sci. 2019, 20, 1–17. [Google Scholar] [CrossRef]
- Millward, D.J. Undernutrition, Infection, and Poor Growth in Infants and Children. In Nutrition, Immunity, and Infection; Calder, P.C., Kulkarni, A.D., Eds.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: Boca Raton, FL, USA, 2017; pp. 83–109. ISBN 13:978-1-4822-5397-9. [Google Scholar]
- Nimmanon, T.; Ziliotto, S.; Morris, S.; Flanagan, L.; Taylor, K.M. Phosphorylation of zinc channel ZIP7 drives MAPK, PI3K and mTOR growth and proliferation signalling. Metallomics 2017, 9, 471–481. [Google Scholar] [CrossRef]
- Sabatini, D.M. Twenty-five years of mTOR: Uncovering the link from nutrients to growth. Proc. Natl. Acad. Sci. USA 2017, 114, 11818–11825. [Google Scholar] [CrossRef]
- Hozain, S.; Hernandez, A.; Fuller, J.; Sharp, G.; Cottrell, J. Zinc chloride affects chondrogenesis via VEGF signaling. Exp. Cell Res. 2021, 399. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.; Iversen, T.; Cottrell, J. Zinc Chloride Treatment in ATDC5 cells Induces Chondrocyte Maturation. Int. J. Regen. Med. Sci. Repos. 2018, 1, 1–11. [Google Scholar] [CrossRef]
- Mammoto, A.; Mammoto, T.; Ingber, D.E. Mechanosensitive mechanisms in transcriptional regulation. J. Cell Sci. 2012, 125, 3061–3073. [Google Scholar] [CrossRef] [PubMed]
- Hornberger, T.A. Mechanotransduction and the regulation of mTORC1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 2011, 43, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Jansen, K.A.; Atherton, P.; Ballestrem, C. Mechanotransduction at the cell-matrix interface. Semin. Cell Dev. Biol. 2017, 71, 75–83. [Google Scholar] [CrossRef]
- Stutchbury, B.; Atherton, P.; Tsang, R.; Wang, D.Y.; Ballestrem, C. Distinct focal adhesion protein modules control different aspects of mechanotransduction. J. Cell Sci. 2017, 130, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, V.C.; McCarthy, J.J. Regulation of ribosome biogenesis in skeletal muscle hypertrophy. Physiology 2019, 34, 30–42. [Google Scholar] [CrossRef]
- Laurent, G.; Sparrow, M.; Millward, D. Turnover of Muscle Protein in the Fowl Changes in rates of protein synthesis and breakdown during hypertrophy of the anterior and posterior latissimus dorsi muscles. Biochem. J. 1978, 176, 407–417. [Google Scholar] [CrossRef]
- Kimball, S.R. Integration of signals generated by nutrients, hormones, and exercise in skeletal muscle. Am. J. Clin. Nutr. 2014, 99, 237–242. [Google Scholar] [CrossRef]
- Laurent, G.; Sparrow, M.; Bates, P.; Millward, D. Turnover of Muscle Protein in the Fowl. Collagen content and turnover in cardiac and skeletal muscles of the adult fowl and the changes during stretch-induced growth. Biochem. J. 1978, 176, 419–427. [Google Scholar] [CrossRef]
- Miller, B.F.; Olesen, J.L.; Hansen, M.; Døssing, S.; Crameri, R.M.; Welling, R.J.; Langberg, H.; Flyvbjerg, A.; Kjaer, M.; Babraj, J.A.; et al. Coordinated collagen and muscle protein synthesis in human patella tendon and quadriceps muscle after exercise. J. Physiol. 2005, 567, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.R.; Phillips, S.M.; Babraj, J.A.; Smith, K.; Rennie, M.J. Myofibrillar and collagen protein synthesis in human skeletal muscle in young men after maximal shortening and lengthening contractions. Am. J. Physiol. Endocrinol. Metab. 2005, 288, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Laurent, G.; Millward, D. Protein Turnover during Skeletal Muscle Hypertrophy. Fed. Proc. 1980, 39, 42–47. [Google Scholar]
- Boersma, B.; Wit, J.M. Catch-up Growth. Endocr. Rev. 1997, 18, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Hermanussen, M.; Largo, R.H.; Molinari, L. Canalisation in human growth: A widely accepted concept reconsidered. Eur. J. Pediatr. 2001, 160, 163–167. [Google Scholar] [CrossRef]
- Sanders, J.O.; Qiu, X.; Lu, X.; Duren, D.L.; Liu, R.W.; Dang, D.; Menendez, M.E.; Hans, S.D.; Weber, D.R.; Cooperman, D.R. The Uniform Pattern of Growth and Skeletal Maturation during the Human Adolescent Growth Spurt. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Butler, G.E.; Mckie, M.; Ratcliffe, S.G. The cyclical nature of prepubertal growth. Ann. Hum. Biol. 1990, 17, 177–198. [Google Scholar] [CrossRef]
- Molinari, L.; Butler, G.E.; Ratcliffe, S.G. Letters to the editor. Ann. Hum. Biol. 1992, 19, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Lampl, M.; Veldhuis, J.D.; Johnson, M.L. Saltation and Stasis: A Model of Human Growth. Science 1992, 258, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Lampl, M. Saltation and Stasis. In Human Growth and Development; Cameron, N., Schell, L., Eds.; Academic press: Cambridge, MA, USA, 2012; pp. 415–434. ISBN 9780123838827. [Google Scholar]
- Lampl, M.; Johnson, M.L. Infant growth in length follows prolonged sleep and increased naps. Sleep 2011, 34, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Lampl, M.; Thompson, A.L.; Frongillo, E.A. Sex differences in the relationships among weight gain, subcutaneous skinfold tissue and saltatory length growth spurts in infancy. Pediatr. Res. 2005, 58, 1238–1242. [Google Scholar] [CrossRef]
- Thalange, N.K.S.; Foster, P.J.; Gill, M.S.; Price, D.A.; Clayton, P.E. Model of normal prepubertal growth. Arch. Dis. Child. 1996, 75, 427–431. [Google Scholar] [CrossRef]
- Tillmann, V.; Thalange, N.K.S.; Foster, P.J.; Gill, M.S.; Price, D.A.; Clayton, P.E. The relationship between stature, growth, and short-term changes in height and weight in normal prepubertal children. Pediatr. Res. 1998, 44, 882–886. [Google Scholar] [CrossRef]
- Gill, M.S.; Tillmann, V.; Veldhuis, J.D.; Clayton, P.E. Patterns of GH output and their synchrony with short-term height increments influence stature and growth performance in normal children. J. Clin. Endocrinol. Metab. 2001, 86, 5860–5863. [Google Scholar] [CrossRef]
- Lehman, P.J.; Carl, R.L. Growing Pains: When to Be Concerned. Sports Health 2017, 9, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Millward, D. Catch-up Growth in Children. Nutr. Rev. 1986, 44, 57–163. [Google Scholar] [CrossRef] [PubMed]
- Golden, M.H.N.; Golden, B.E. Effect of zinc supplementation on the dietary intake, rate of weight gain, and energy cost of tissue deposition in children recovering from severe malnutrition. Am. J. Clin. Nutr. 1981, 34, 900–908. [Google Scholar] [CrossRef]
- De Wit, C.C.; Sas, T.C.J.; Wit, J.M.; Cutfield, W.S. Patterns of catch-up growth. J. Pediatr. 2013, 162, 415–420. [Google Scholar] [CrossRef]
- Wit, J.M.; Camacho-Hübner, C. Endocrine Regulation of Longitudinal Bone Growth. Endocr. Dev. 2011, 21, 30–41. [Google Scholar] [CrossRef]
- Lui, J.C.; Nilsson, O.; Baron, J. Recent insights into the regulation of the growth plate. J. Mol. Endocrinol. 2014, 53. [Google Scholar] [CrossRef] [PubMed]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bang, P. Pediatric Implications of Normal Insulin-GH-IGF-Axis Physiology; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2019. [Google Scholar]
- Giustina, A.; Mazziotti, G.; Canalis, E. Growth hormone, insulin-like growth factors, and the skeleton. Endocr. Rev. 2008, 29, 535–559. [Google Scholar] [CrossRef]
- Racine, H.L.; Serrat, M.A. The Actions of IGF-1 in the Growth Plate and Its Role in Postnatal Bone Elongation. Curr. Osteoporos. Rep. 2020, 18, 210–227. [Google Scholar] [CrossRef]
- Karlberg, J.; Jalil, F.; Lam, B.; Low, L.; Yeung, C.Y. Linear growth retardation in relation to the three phases of growth. Eur. J. Clin. Nutr. 1994, 48, 25–44. [Google Scholar]
- Low, L.C.K.; Tam, S.Y.M.; Kwan, E.Y.W.; Tsang, A.M.C.; Karlberg, J.; Centre, C.T. Onset of Significant GH Dependence of Serum IGF-I and IGF-Binding Protein 3 Concentrations in Early Life. Pediatr. Res. 2001, 50, 737–742. [Google Scholar] [CrossRef]
- Zhang, F.; He, Q.; Tsang, W.P.; Garvey, W.T.; Chan, W.Y.; Wan, C. Insulin exerts direct, IGF-1 independent actions in growth plate chondrocytes. Bone Res. 2014, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Guntur, A.R.; Rosen, C.J. IGF-1 regulation of key signaling pathways in bone. BoneKEy Rep. 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Siddle, K. Signalling by insulin and IGF receptors: Supporting acts and new players. J. Mol. Endocrinol. 2011, 47, R1–R10. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Wooh, J.W.; Tunny, K.A.; Waters, M.J. Growth hormone receptor; mechanism of action. Int. J. Biochem. Cell Biol. 2008, 40, 1984–1989. [Google Scholar] [CrossRef]
- Carter-Su, C.; Schwartz, J.; Argetsinger, L.S. Growth Hormone & IGF Research Growth hormone signaling pathways. Growth Horm. IGF Res. 2016, 28, 11–15. [Google Scholar] [CrossRef]
- Parker, E.A.; Hegde, A.; Buckley, M.; Barnes, K.M.; Baron, J.; Nilsson, O. Spatial and temporal regulation of GH—IGF-related gene expression in growth plate cartilage. J. Endocrinol. 2007, 194, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, O.; Marino, R.; De Luca, F.; Phillip, M.; Baron, J. Endocrine Regulation of the Growth Plate. Horm. Res. 2005, 64, 157–165. [Google Scholar] [CrossRef]
- Elis, S.; Courtland, H.; Wu, Y.; Rosen, C.J.; Sun, H.; Jepsen, K.J.; Majeska, R.J.; Yakar, S. Elevated Serum Levels of IGF-1 Are Sufficient to Establish Normal Body Size and Skeletal Properties Even in the Absence of Tissue IGF-1. J. Bone Miner. Res. 2010, 25, 1257–1266. [Google Scholar] [CrossRef]
- Hellström, A.; Ley, D.; Hansen-Pupp, I.; Hallberg, B.; Löfqvist, C.; Van Marter, L.; Van Weissenbruch, M.; Ramenghi, L.A.; Beardsall, K.; Dunger, D.; et al. Insulin-like growth factor 1 has multisystem effects on foetal and preterm infant development. Acta Paediatr. Int. J. Paediatr. 2016, 105, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.; Doyle, N.; Ballesteros, M.; Waters, M.J.; Ho, K.K.Y. Insulin regulation of human hepatic growth hormone receptors: Divergent effects on biosynthesis and surface translocation. J. Clin. Endocrinol. Metab. 2000, 85, 4712–4720. [Google Scholar] [CrossRef]
- Hawkes, C.P.; Grimberg, A. Measuring Growth Hormone and Insulin-like Growth Factor-I in Infants: What is Normal? Pediatr. Endocrinol. Rev. 2013, 11, 126–146. [Google Scholar]
- Ogilvy-Stuart, A.L.; Hands, S.J.; Adcock, C.J.; Holly, J.M.P.; Matthews, D.R.; Mohamed-Ali, V.; Yudkin, J.S.; Wilkinson, A.R.; Dunger, D.B. Insulin, Insulin-Like Growth Factor I (IGF-I), IGF- Binding Protein-1, Growth Hormone, and Feeding in the Newborn. J. Clin. Endocrinol. Metab. 1998, 83, 3550–3557. [Google Scholar] [CrossRef] [PubMed]
- Tse, W.Y.; Hindmarsh, P.C.; Brook, C.G. The Infancy-Childhood-Puberty Model of Growth: Clinical Aspects. Acta Paediatr. Scand. Suppl. 1989, 356, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Socha, P.; Grote, V.; Gruszfeld, D.; Janas, R.; Demmelmair, H.; Closa-Monasterolo, R.; Subías, J.E.; Scaglioni, S.; Verduci, E.; Dain, E.; et al. Milk protein intake, the metabolic-endocrine response, and growth in infancy: Data from a randomized clinical trial. Am. J. Clin. Nutr. 2011, 94, 1776–1784. [Google Scholar] [CrossRef]
- Madsen, A.L.; Larnkjær, A.; Mølgaard, C.; Michaelsen, K.F. Determinants of blood glucose and insulin in healthy 9-month-old term Danish infants; the SKOT cohort. Diabet. Med. 2010, 27, 1350–1357. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Juul, A.; Michaelsen, K.F. High intakes of skimmed milk, but not meat, increase serum IGF-I and IGFBP-3 in eight-year-old boys. Eur. J. Clin. Nutr. 2004, 58, 1211–1216. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Mølgaard, C.; Vaag, A.; Barkholt, V.; Michaelsen, K.F. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur. J. Clin. Nutr. 2005, 59, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Mullur, R.; Liu, Y.Y.; Brent, G.A. Thyroid hormone regulation of metabolism. Physiol. Rev. 2014, 94, 355–382. [Google Scholar] [CrossRef]
- Duncan Bassett, J.H.; Williams, G.R. Role of thyroid hormones in skeletal development and bone maintenance. Endocr. Rev. 2016, 37, 135–187. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.R.; Robson, H.; Shalet, S.M. Thyroid hormone actions on cartilage and bone: Interactions with other hormones at the epiphyseal plate and effects on linear growth. J. Endocrinol. 1998, 157, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Styne, D.M. The regulation of pubertal growth. Horm. Res. 2003, 60, 22–26. [Google Scholar] [CrossRef]
- Soliman, A.T.; De Sanctis, V.; Elalaily, R.; Bedair, S. Advances in pubertal growth and factors influencing it: Can we increase pubertal growth? Indian J. Endocrinol. Metab. 2014, 18, S53–S62. [Google Scholar] [CrossRef]
- Van Cauter, E.; Plat, L. Physiology of growth hormone secretion during sleep. J. Pediatr. 1996, 128, s32–s37. [Google Scholar] [CrossRef]
- Yakar, S.; Boisclair, Y.R.; Leroith, D.; Yakar, S.; Rosen, C.J.; Beamer, W.G.; Ackert-bicknell, C.L.; Wu, Y.; Liu, J.; Ooi, G.T.; et al. Circulating levels of IGF-1 directly regulate bone growth and density. J. Clin. Investig. 2002, 110, 771–781. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.-L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; Leroith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef] [PubMed]
- Domené, H.M.; Argente, J.; Wit, M.; Camacho-Hübner, C.; Jasper, H.G.; Pozo, J.; van Duyvenvoorde, H.A.; Yakar, S.; Fofanova-Gambetti, O.V.; Rosenfeld, R.G. Human Acid-Labile Subunit Deficiency: Clinical, Endocrine and Metabolic Consequences. Horm. Res. 2009, 1425, 129–141. [Google Scholar] [CrossRef]
- Murray, P.G.; Clayton, P.E. Endocrine Control of Growth. Am. J. Med. Genet. Part C 2013, 163, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Juul, A.; Holm, K.; Kastrup, K.W.; Pedersen, S.A.; Michaelsen, K.F.; Scheike, T.; Rasmussen, S.; Müller, J.; Skakkebæk, N.E. Free insulin-like growth factor I serum levels in 1430 healthy children and adults, and its diagnostic value in patients suspected of growth hormone deficiency. J. Clin. Endocrinol. Metab. 1997, 82, 2497–2502. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Prein, C.; Beier, F. ECM signaling in cartilage development and endochondral ossification. Curr. Top. Dev. Biol. 2019, 133, 25–47. [Google Scholar] [CrossRef]
- Samsa, W.E.; Zhou, X.; Zhou, G. Signaling pathways regulating cartilage growth plate formation and activity. Semin. Cell Dev. Biol. 2017, 62, 3–15. [Google Scholar] [CrossRef]
- Millward, D.J. Nutrition, infection and stunting: The roles of deficiencies of individual nutrients and foods, and of inflammation, as determinants of reduced linear growth of children. Nutr. Res. Rev. 2017, 30, 50–72. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Chin, J.E.; Schalk, E.M.; Wuthier, R.E. Effect of amino acid levels on matrix vesicle formation by epiphyseal growth plate chondrocytes in primary culturen. J. Cell. Physiol. 1986, 126, 399–406. [Google Scholar] [CrossRef]
- Phornphutkul, C.; Lee, M.; Voigt, C.; Wu, K.Y.; Ehrlich, M.G.; Gruppuso, P.A.; Chen, Q. The effect of rapamycin on bone growth in rabbits. J. Orthop. Res. 2009, 27, 1157–1161. [Google Scholar] [CrossRef]
- Phornphutkul, C.; Wu, K.Y.; Auyeung, V.; Chen, Q.; Gruppuso, P.A. mTOR signaling contributes to chondrocyte differentiation. Dev. Dyn. 2008, 237, 702–712. [Google Scholar] [CrossRef]
- Chen, J.; Long, F. mTORC1 signaling controls mammalian skeletal growth through stimulation of protein synthesis. Development 2014, 141, 2848–2854. [Google Scholar] [CrossRef] [PubMed]
- Adegoke, O.A.J.; Beatty, B.E.; Kimball, S.R.; Wing, S.S. Interactions of the super complexes: When mTORC1 meets the proteasome. Int. J. Biochem. Cell Biol. 2019, 117, 105638. [Google Scholar] [CrossRef] [PubMed]
- Newton, P.T.; Vuppalapati, K.K.; Bouderlique, T.; Chagin, A.S. Pharmacological inhibition of lysosomes activates the MTORC1 signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy 2015, 11, 1594–1607. [Google Scholar] [CrossRef]
- White, R.B.; Biérinx, A.S.; Gnocchi, V.F.; Zammit, P.S. Dynamics of muscle fibre growth during postnatal mouse development. BMC Dev. Biol. 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Gillies, A.R.; Lieber, R.L. Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 2011, 44, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Delhaas, T.; Van der Meer, S.F.T.; Schaart, G.; Degens, H.; Drost, M.R. Steep Increase in Myonuclear Domain Size During Infancy. Anat. Rec. 2013, 296, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Jorgenson, K.W.; Phillips, S.M.; Hornberger, T.A. Identifying the Structural Adaptations that Drive the Mechanical Load-Induced Growth of Skeletal Muscle: A Scoping Review. Cells 2020, 9, 1658. [Google Scholar] [CrossRef]
- Dumont, N.A.; Wang, Y.X.; Rudnicki, M.A. Intrinsic and extrinsic mechanisms regulating satellite cell function. Development 2015, 142, 1572–1581. [Google Scholar] [CrossRef]
- Betts, G.; Kelly, A.; Wise, J.; Johnson, E.; Poe, B.; Kruse, D.; Korol, O.; Johnson, J.; Womble, M.; DeSaix, P. Anatomy and Physiology; OpenStax: Houston, TX, USA, 2013. [Google Scholar]
- 10.2 Skeletal Muscle. Available online: openstax.org/books/anatomy-and-physiology/pages/10-2-skeletal-muscle (accessed on 6 January 2021).
- Millward, D.J. The Endocrine Response to Dietary Protein: The Anabolic Drive on Growth. In Milk Proteins: Nutritional, Clinical, Functional and Technological Aspects; Barth, C.A., Schlimme, E., Eds.; Steinkopff: Darmstadt, Germany, 1988; pp. 49–61. ISBN 13:978-3-642-85375-3. [Google Scholar]
- Coutinho, E.L.; Gomes, A.R.S.; França, C.N.; Oishi, J.; Salvini, T.F. Effect of passive stretching on the immobilized soleus muscle fiber morphology. Braz. J. Med. Biol. Res. 2004, 37, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Latroche, C.; Gitiaux, C.; Chrétien, F.; Desguerre, I.; Mounie, R.; Chazaud, B. Skeletal muscle microvasculature: A highly dynamic lifeline. Physiology 2015, 30, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Giordani, L.; He, G.J.; Negroni, E.; Sakai, H.; Law, J.Y.C.; Siu, M.M.; Wan, R.; Corneau, A.; Tajbakhsh, S.; Cheung, T.H.; et al. High-Dimensional Single-Cell Cartography Reveals Novel Skeletal Muscle-Resident Cell Populations. Mol. Cell 2019, 74, 609–621. [Google Scholar] [CrossRef]
- Mierzejewski, B.; Archacka, K.; Grabowska, I.; Florkowska, A.; Ciemerych, M.A.; Brzoska, E. Human and mouse skeletal muscle stem and progenitor cells in health and disease. Semin. Cell Dev. Biol. 2020, 104, 93–104. [Google Scholar] [CrossRef]
- Evano, B.; Tajbakhsh, S. Skeletal muscle stem cells in comfort and stress. npj Regen. Med. 2018, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dellavalle, A.; Maroli, G.; Covarello, D.; Azzoni, E.; Innocenzi, A.; Perani, L.; Antonini, S.; Sambasivan, R.; Brunelli, S.; Tajbakhsh, S.; et al. Pericytes resident in postnatal skeletal muscle differentiate into muscle fibres and generate satellite cells. Nat. Commun. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Harrell, C.R.; Simovic Markovic, B.; Fellabaum, C.; Arsenijevic, A.; Djonov, V.; Volarevic, V. Molecular mechanisms underlying therapeutic potential of pericytes. J. Biomed. Sci. 2018, 25, 1–12. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Pannérec, A.; Cadot, B.; Parlakian, A.; Besson, V.; Gomes, E.R.; Marazzi, G.; Sassoon, D.A. Identification and characterization of a non-satellite cell muscle resident progenitor during postnatal development. Nat. Cell Biol. 2010, 12, 257–266. [Google Scholar] [CrossRef]
- Liu, N.; Garry, G.A.; Li, S.; Bezprozvannaya, S.; Sanchez-Ortiz, E.; Chen, B.; Shelton, J.M.; Jaichander, P.; Bassel-Duby, R.; Olson, E.N. A Twist2-dependent progenitor cell contributes to adult skeletal muscle. Nat. Cell Biol. 2017, 19, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro–Adipogenic Progenitors Cross-Talk in Skeletal Muscle: The Social Network. Front. Physiol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wosczyna, M.N.; Rando, T.A. A Muscle Stem Cell Support Group: Coordinated Cellular Responses in Muscle Regeneration. Dev. Cell 2018, 46, 135–143. [Google Scholar] [CrossRef]
- Bachman, J.F.; Klose, A.; Liu, W.; Paris, N.D.; Blanc, R.S.; Schmalz, M.; Knapp, E.; Chakkalakal, J.V. Prepubertal skeletal muscle growth requires Pax7-expressing satellite cell-derived myonuclear contribution. Development 2018, 145, dev167197. [Google Scholar] [CrossRef]
- Ohlstein, B.; Kai, T.; Decotto, E.; Spradling, A. The stem cell niche: Theme and variations. Curr. Opin. Cell Biol. 2004, 16, 693–699. [Google Scholar] [CrossRef]
- Yin, H.; Price, F.; Rudnicki, M.A. Satellite cells and the muscle stem cell niche. Physiol. Rev. 2013, 93, 23–67. [Google Scholar] [CrossRef]
- Feige, P.; Rudnicki, M.A. Muscle stem cells. Curr. Biol. 2018, 28, R589–R590. [Google Scholar] [CrossRef]
- Mashinchian, O.; Pisconti, A.; Le Moal, E.; Bentzinger, C.F. The Muscle Stem Cell Niche in Health and Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018; Volume 126. [Google Scholar]
- Christov, C.; Chrétien, F.; Abou-Khalil, R.; Bassez, G.; Vallet, G.; Authier, F.-J.; Bassaglia, Y.; Shinin, V.; Tajbakhsh, S.; Chazaud, B.; et al. Muscle Satellite Cells and Endothelial Cells: Close Neighbors and Privileged Partners. Mol. Biol. Cell 2007, 18, 1397–1409. [Google Scholar] [CrossRef]
- Collins, B.C.; Kardon, G. Won’t You Be My Neighbor? Muscle Stem Cells Recruit Endothelial Cells to Their Niche. Cell Stem Cell 2018, 23, 455–456. [Google Scholar] [CrossRef]
- Verma, M.; Asakura, Y.; Murakonda, B.S.R.; Pengo, T.; Latroche, C.; Chazaud, B.; McLoon, L.K.; Asakura, A. Muscle Satellite Cell Cross-Talk with a Vascular Niche Maintains Quiescence via VEGF and Notch Signaling. Cell Stem Cell 2018, 23, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, J.T.; King, K.Y.; Brett, J.O.; Cromie, M.J.; Charville, G.W.; Maguire, K.K.; Brunson, C.; Mastey, N.; Liu, L.; Tsai, C.R.; et al. MTORC1 controls the adaptive transition of quiescent stem cells from G 0 to GAlert. Nature 2014, 510, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Schüler, S.C.; Hüttner, S.S.; von Eyss, B.; von Maltzahn, J. Adult stem cells at work: Regenerating skeletal muscle. Cell. Mol. Life Sci. 2019, 76, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Shea, K.L.; Xiang, W.; LaPorta, V.S.; Licht, J.D.; Keller, C.; Basson, M.A.; Brack, A.S. Sprouty1 Regulates Reversible Quiescence of a Self-Renewing Adult Muscle Stem Cell Pool during Regeneration. Cell Stem Cell 2010, 6, 117–129. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Jones, A.E.; Parks, R.J.; Rudnicki, M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA 2013, 110, 16474–16479. [Google Scholar] [CrossRef] [PubMed]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell. Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Asfour, H.A.; Allouh, M.Z.; Said, R.S. Myogenic regulatory factors: The orchestrators of myogenesis after 30 years of discovery. Exp. Biol. Med. 2018, 243, 118–128. [Google Scholar] [CrossRef]
- Zammit, P.S. Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin. Cell Dev. Biol. 2017, 72, 19–32. [Google Scholar] [CrossRef]
- Hernández-Hernández, J.M.; García-González, E.G.; Brun, C.E.; Rudnicki, M.A. The myogenic regulatory factors, determinants of muscle development, cell identity and regeneration. Semin. Cell Dev. Biol. 2017, 72, 10–18. [Google Scholar] [CrossRef]
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Epigenetic control of adult skeletal muscle stem cell functions. FEBS J. 2015, 282, 1571–1588. [Google Scholar] [CrossRef] [PubMed]
- Barruet, E.; Garcia, S.M.; Striedinger, K.; Wu, J.; Lee, S.; Byrnes, L.; Wong, A.; Xuefeng, S.; Tamaki, S.; Brack, A.S.; et al. Functionally heterogeneous human satellite cells identified by single cell RNA sequencing. Elife 2020, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.V.; Hughes, S.M. Mef2 and the skeletal muscle differentiation program. Semin. Cell Dev. Biol. 2017, 72, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Moretti, I.; Ciciliot, S.; Dyar, K.A.; Abraham, R.; Murgia, M.; Agatea, L.; Akimoto, T.; Bicciato, S.; Forcato, M.; Pierre, P.; et al. MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Dyar, K.A.; Calabria, E. Skeletal muscle mass is controlled by the MRF4-MEF2 axis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 164–167. [Google Scholar] [CrossRef]
- Buckingham, M.; Rigby, P.W.J. Gene Regulatory Networks and Transcriptional Mechanisms that Control Myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Li, M.; He, J.; Huang, Z. Regulation of skeletal myogenesis by microRNAs. J. Cell. Physiol. 2020, 235, 87–104. [Google Scholar] [CrossRef]
- Fry, C.S.; Kirby, T.J.; Kosmac, K.; McCarthy, J.J.; Peterson, C.A. Myogenic Progenitor Cells Control Extracellular Matrix Production by Fibroblasts during Skeletal Muscle Hypertrophy. Cell Stem Cell 2017, 20, 56–69. [Google Scholar] [CrossRef]
- Collard, L.; Herledan, G.; Pincini, A.; Guerci, A.; Randrianarison-Huetz, V.; Sotiropoulos, A. Nuclear actin and myocardin-related transcription factors control disuse muscle atrophy through regulation of Srf activity. J. Cell Sci. 2014, 127, 5157–5163. [Google Scholar] [CrossRef]
- Gauthier-Rouviere, C.; Vandromme, M.; Tuil, D.; Lautredou, N.; Morris, M.; Soulez, M.; Kahn, A.; Fernandez, A.; Lamb, N. Expression and activity of serum response factor is required for expression of the muscle-determining factor MyoD in both dividing and differentiating mouse C2C12 myoblasts. Mol. Biol. Cell 1996, 7, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Randrianarison-Huetz, V.; Papaefthymiou, A.; Herledan, G.; Noviello, C.; Faradova, U.; Collard, L.; Pincini, A.; Schol, E.; Decaux, J.F.; Maire, P.; et al. Srf controls satellite cell fusion through the maintenance of actin architecture. J. Cell Biol. 2018, 217, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Leikina, E.; Gamage, D.G.; Prasad, V.; Goykhberg, J.; Crowe, M.; Diao, J.; Kozlov, M.M.; Chernomordik, L.V.; Millay, D.P. Myomaker and Myomerger Work Independently to Control Distinct Steps of Membrane Remodeling during Myoblast Fusion. Dev. Cell 2018, 46, 767–780. [Google Scholar] [CrossRef]
- Heredia, J.E.; Mukundan, L.; Chen, F.M.; Mueller, A.A.; Deo, R.C.; Locksley, R.M.; Rando, T.A.; Chawla, A. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 2013, 153, 376–388. [Google Scholar] [CrossRef]
- Murphy, M.M.; Lawson, J.A.; Mathew, S.J.; Hutcheson, D.A.; Kardon, G. Satellite cells, connective tissue fibroblasts and their interactions are crucial for muscle regeneration. Development 2011, 138, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Forcina, L.; Miano, C.; Scicchitano, B.; Musarò, A. Signals from the Niche: Insights into the Role of IGF-1 and IL-6 in Modulating Skeletal Muscle Fibrosis. Cells 2019, 8, 232. [Google Scholar] [CrossRef]
- Fiore, D.; Judson, R.N.; Low, M.; Lee, S.; Zhang, E.; Hopkins, C.; Xu, P.; Lenzi, A.; Rossi, F.M.V.; Lemos, D.R. Pharmacological blockage of fibro/adipogenic progenitor expansion and suppression of regenerative fibrogenesis is associated with impaired skeletal muscle regeneration. Stem Cell Res. 2016, 17, 161–169. [Google Scholar] [CrossRef]
- Winchester, P.K.; Davis, M.E.; Alway, S.E.; Gonyea, W.J. Satellite cell activation in the stretch-enlarged anterior latissimus dorsi muscle of the adult quail. Am. J. Physiol. Cell Physiol. 1991, 260. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, R.; Liu, X.; Pulido, A.; Morales, M.; Sakata, T.; Dial, S.; Hattori, A.; Ikeuchi, Y.; Allen, R.E. Satellite cell activation in stretched skeletal muscle and the role of nitric oxide and hepatocyte growth factor. Am. J. Physiol. Cell Physiol. 2006, 290, 1487–1494. [Google Scholar] [CrossRef]
- Tatsumi, R.; Sheehan, S.M.; Iwasaki, H.; Hattori, A.; Allen, R.E. Mechanical stretch induces activation of skeletal muscle satellite cells in vitro. Exp. Cell Res. 2001, 267, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.S.; Kim, J.J.; Kim, H.W.; Lewis, M.P.; Wall, I. Impact of mechanical stretch on the cell behaviors of bone and surrounding tissues. J. Tissue Eng. 2016, 7, 1–24. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Etlinger, J.D.; Goldspink, D.F.; Jablecki, C. Mechanism of work-induced hypertrophy of skeletal muscle. Med. Sci Sport. 1975, 7, 185–198. [Google Scholar]
- Goldberg, A.L. Protein synthesis during work-induced growth of skeletal muscle. J. Cell Biol. 1968, 36, 653–658. [Google Scholar] [CrossRef]
- Kjaer, M.; Jørgensen, N.R.; Heinemeier, K.; Magnusson, S.P. Exercise and Regulation of Bone and Collagen Tissue Biology. Prog. Mol. Biol. Transl. Sci. 2015, 135, 259–291. [Google Scholar] [CrossRef]
- Jaka, O.; Casas, L.; Lopez-de-munain, A. Costamere proteins and their involvement in myopathic processes. Expert Rev. Mol. Med. 2015, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Henderson, C.A.; Gomez, C.G.; Novak, S.M.; Mi-Mi, L.; Gregorio, C.C. Overview of the muscle cytoskeleton. Compr. Physiol. 2017, 7, 891–944. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Yan, Y.; Li, S.; Tong, H. SPARCL1 Influences Bovine Skeletal Muscle-Derived. Animals 2020, 10, 1361. [Google Scholar] [CrossRef] [PubMed]
- Sanes, J.R. The basement membrane/basal lamina of skeletal muscle. J. Biol. Chem. 2003, 278, 12601–12604. [Google Scholar] [CrossRef]
- Lund, D.K.; Cornelison, D.D.W. Enter the matrix: Shape, signal and superhighway. FEBS J. 2013, 280, 4089–4099. [Google Scholar] [CrossRef]
- Tierney, M.T.; Gromova, A.; Sesillo, F.B.; Sala, D.; Spenlé, C.; Orend, G.; Sacco, A. Autonomous Extracellular Matrix Remodeling Controls a Progressive Adaptation in Muscle Stem Cell Regenerative Capacity during Development. Cell Rep. 2016, 14, 1940–1952. [Google Scholar] [CrossRef] [PubMed]
- Grzelkowska-Kowalczyk, K. The Importance of Extracellular Matrix in Skeletal Muscle Development and Function. In Composition and Function of the Extracellular Matrix in the Human Body; InTech: Francesco, UK, 2016. [Google Scholar] [CrossRef]
- Murach, K.A.; Fry, C.S.; Kirby, T.J.; Jackson, J.R.; Lee, J.D.; White, S.H.; Dupont-Versteegden, E.E.; McCarthy, J.J.; Peterson, C.A. Starring or supporting role? Satellite cells and skeletal muscle fiber size regulation. Physiology 2018, 33, 26–38. [Google Scholar] [CrossRef]
- Ingber, D.E.; Wang, N.; Stamenović, D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep. Prog. Phys. 2014, 77. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, S.V.; Waterman, C.M. Guiding cell migration by tugging. Curr. Opin. Cell Biol. 2013, 25, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Tytell, J.D.; Ingber, D.E. Mechanotransduction at a distance: Mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Olsen, L.A.; Nicoll, J.X.; Fry, A.C. The skeletal muscle fiber: A mechanically sensitive cell. Eur. J. Appl. Physiol. 2019, 119, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; McNally, E.M. The Dystrophin Complex: Structure, function and implications for therapy. Compr. Physiol. 2015, 5, 1223–1239. [Google Scholar] [CrossRef]
- Chang, N.C.; Sincennes, M.C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Goel, A.J.; Rieder, M.K.; Arnold, H.H.; Radice, G.L.; Krauss, R.S. Niche Cadherins Control the Quiescence-to-Activation Transition in Muscle Stem Cells. Cell Rep. 2017, 21, 2236–2250. [Google Scholar] [CrossRef] [PubMed]
- Gattazzo, F.; Urciuolo, A.; Bonaldo, P. Extracellular matrix: A dynamic microenvironment for stem cell niche. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2506–2519. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.D.; Coon, B.G.; Yun, S.; Baeyens, N.; Tanaka, K.; Ouyang, M.; Schwartz, M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013, 25, 613–618. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Thorsteinsdottir, S.; Deries, M.; Cachaço, A.S.; Bajanca, F. The extracellular matrix dimension of skeletal muscle development. Dev. Biol. 2011, 354, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Boppart, M.D.; Mahmassani, Z.S. Integrin signaling: Linking mechanical stimulation to skeletal muscle hypertrophy. Am. J. Physiol. Cell Physiol. 2019, 317, C629–C641. [Google Scholar] [CrossRef]
- Bachir, A.I.; Horwitz, A.R.; Nelson, W.J.; Bianchini, J.M. Actin-based adhesion modules mediate cell interactions with the extracellular matrix and neighboring cells. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.T.; Navajas, P.L.; Lietha, D. FAK structure and regulation by membrane interactions and force in focal adhesions. Biomolecules 2020, 10, 179. [Google Scholar] [CrossRef]
- Niessen, C.M.; Leckband, D.; Yap, A.S. Tissue organization by cadherin adhesion molecules: Dynamic Molecular and Cellular Mechanisms of Morphogenetic Regulation. Physiol. Rev. 2011, 91, 691–731. [Google Scholar] [CrossRef] [PubMed]
- Leckband, D.E.; de Rooij, J. Cadherin adhesion and mechanotransduction. Annu. Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef] [PubMed]
- DeMali, K.A.; Sun, X.; Bui, G.A. Force transmission at cell-cell and cell-matrix adhesions. Biochemistry 2014, 53, 7706–7717. [Google Scholar] [CrossRef]
- Ladoux, B.; Nelson, W.J.; Yan, J.; Mège, R.M. The mechanotransduction machinery at work at adherens junctions. Integr. Biol. 2015, 7, 1109–1119. [Google Scholar] [CrossRef]
- Cho, S.; Irianto, J.; Discher, D.E. Mechanosensing by the nucleus: From pathways to scaling relationships. J. Cell Biol. 2017, 216, 305–315. [Google Scholar] [CrossRef]
- Case, L.B.; Waterman, C.M. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol. 2015, 17, 955–963. [Google Scholar] [CrossRef]
- Karaman, R.; Halder, G. Cell junctions in Hippo signaling. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Luo, L.; Chen, J. Roles of mTOR Signaling in Tissue Regeneration. Cells 2019, 8, 1075. [Google Scholar] [CrossRef] [PubMed]
- Risson, V.; Mazelin, L.; Roceri, M.; Sanchez, H.; Moncollin, V.; Corneloup, C.; Richard-Bulteau, H.; Vignaud, A.; Baas, D.; Defour, A.; et al. Muscle inactivation of mTOR causes metabolic and dystrophin defects leading to severe myopathy. J. Cell Biol. 2009, 187, 859–874. [Google Scholar] [CrossRef]
- Bentzinger, C.F.; Romanino, K.; Cloëtta, D.; Lin, S.; Mascarenhas, J.B.; Oliveri, F.; Xia, J.; Casanova, E.; Costa, C.F.; Brink, M.; et al. Skeletal Muscle-Specific Ablation of raptor, but Not of rictor, Causes Metabolic Changes and Results in Muscle Dystrophy. Cell Metab. 2008, 8, 411–424. [Google Scholar] [CrossRef]
- Rion, N.; Castets, P.; Lin, S.; Enderle, L.; Reinhard, J.R.; Eickhorst, C.; Ru, M.A. mTOR controls embryonic and adult myogenesis via mTORC1. Development 2019, 146, 1–15. [Google Scholar] [CrossRef]
- Kleinert, M.; Parker, B.L.; Chaudhuri, R.; Fazakerley, D.J.; Serup, A.; Thomas, K.C.; Krycer, J.R.; Sylow, L.; Fritzen, A.M.; Hoffman, N.J.; et al. mTORC2 and AMPK differentially regulate muscle triglyceride content via Perilipin 3. Mol. Metab. 2016, 5, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Adams, G.R. Autocrine/paracrine IGF-I and skeletal muscle adaptation. J. Appl. Physiol. 2002, 93, 1159–1167. [Google Scholar] [CrossRef]
- Goodman, C.A. The Role of mTORC1 in Regulating Protein Synthesis and Skeletal Muscle Mass in Response to Various Mechanical Stimuli. Rev. Physiol. Biochem. Pharmacol. 2014, 1–56. [Google Scholar] [CrossRef]
- Zou, Y.; Dong, Y.; Meng, Q.; Zhao, Y.; Li, N. Incorporation of a skeletal muscle-specific enhancer in the regulatory region of Igf1 upregulates IGF1 expression and induces skeletal muscle hypertrophy. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Shavlakadze, T.; Chai, J.; Maley, K.; Cozens, G.; Grounds, G.; Winn, N.; Rosenthal, N.; Grounds, M.D. A growth stimulus is needed for IGF-1 to induce skeletal muscle hypertrophy in vivo. J. Cell Sci. 2010, 123, 960–971. [Google Scholar] [CrossRef]
- Spangenburg, E.E.; Le Roith, D.; Ward, C.W.; Bodine, S.C. A functional insulin-like growth factor receptor is not necessary for load-induced skeletal muscle hypertrophy. J. Physiol. 2008, 586, 283–291. [Google Scholar] [CrossRef]
- Hamilton, D.L.; Baar, K. Muscle growth: No IGFs, ands, or buts. J. Physiol. 2008, 586, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, F.Y.; Zhen, Y.Y.; Yuen, C.M.; Fan, R.; Chen, Y.T.; Sheu, J.J.; Chen, Y.L.; Wang, C.J.; Sun, C.K.; Yip, H.K. The mTOR-FAK mechanotransduction signaling axis for focal adhesion maturation and cell proliferation. Am. J. Transl. Res. 2017, 9, 1603–1617. [Google Scholar] [PubMed]
- Kikani, C.K.; Wu, X.; Paul, L.; Sabic, H.; Shen, Z.; Shakya, A.; Keefe, A.; Villanueva, C.; Kardon, G.; Graves, B.; et al. Pask integrates hormonal signaling with histone modification via Wdr5 phosphorylation to drive myogenesis. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kikani, C.K.; Wu, X.; Fogarty, S.; Kang, S.A.W.; Dephoure, N.; Gygi, S.P.; Sabatini, D.M.; Rutter, J. Activation of PASK by mTORC1 is required for the onset of the terminal differentiation program. Proc. Natl. Acad. Sci. USA 2019, 116, 10382–10391. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Rikeit, P.; Knaus, P.; Coirault, C. YAP-mediated mechanotransduction in skeletal muscle. Front. Physiol. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Pocaterra, A.; Romani, P.; Dupont, S. YAP/TAZ functions and their regulation at a glance. J. Cell Sci. 2020, 133, 1–9. [Google Scholar] [CrossRef]
- Goodman, C.A.; Dietz, J.M.; Jacobs, B.L.; McNally, R.M.; You, J.S.; Hornberger, T.A. Yes-Associated Protein is up-regulated by mechanical overload and is sufficient to induce skeletal muscle hypertrophy. FEBS Lett. 2015, 589, 1491–1497. [Google Scholar] [CrossRef]
- Gnimassou, O.; Francaux, M.; Deldicque, L. Hippo pathway and skeletal muscle mass regulation in mammals: A controversial relationship. Front. Physiol. 2017, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; De Mello, V.; Mohamed, A.; Ortuste Quiroga, H.P.; Garcia-Munoz, A.; Al Bloshi, A.; Tremblay, A.M.; von Kriegsheim, A.; Collie-Duguid, E.; Vargesson, N.; et al. Common and Distinctive Functions of the Hippo Effectors Taz and Yap in Skeletal Muscle Stem Cell Function. Stem Cells 2017, 35, 1958–1972. [Google Scholar] [CrossRef] [PubMed]
- Abuammah, A.; Maimari, N.; Towhidi, L.; Frueh, J.; Chooi, K.Y.; Warboys, C.; Krams, R. New developments in mechanotransduction: Cross talk of the Wnt, TGF-β and Notch signalling pathways in reaction to shear stress. Curr. Opin. Biomed. Eng. 2018, 5, 96–104. [Google Scholar] [CrossRef]
- Totaro, A.; Castellan, M.; Di Biagio, D.; Piccolo, S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol. 2018, 28, 560–573. [Google Scholar] [CrossRef]
- Mourikis, P.; Tajbakhsh, S. Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev. Biol. 2014, 14, 1–8. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Von Maltzahn, J.; Chang, N.C.; Bentzinger, C.F.; Rudnicki, M.A. Wnt signaling in myogenesis. Trends Cell Biol. 2012, 22, 602–609. [Google Scholar] [CrossRef]
- Le Grand, F.; Jones, A.E.; Seale, V.; Scimè, A.; Rudnicki, M.A. Wnt7a Activates the Planar Cell Polarity Pathway to Drive the Symmetric Expansion of Satellite Stem Cells. Cell Stem Cell 2009, 4, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; Wang, Y.X.; Von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Poncet, N.; Halley, P.A.; Lipina, C.; Gierliński, M.; Dady, A.; Singer, G.A.; Febrer, M.; Shi, Y.; Yamaguchi, T.P.; Taylor, P.M.; et al. Wnt regulates amino acid transporter Slc7a5 and so constrains the integrated stress response in mouse embryos. EMBO Rep. 2020, 21, 1–20. [Google Scholar] [CrossRef]
- Tatsumi, R. Mechano-biology of skeletal muscle hypertrophy and regeneration: Possible mechanism of stretch-induced activation of resident myogenic stem cells. Anim. Sci. J. 2010, 81, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Benavides Damm, T.; Egli, M. Calcium’s role in mechanotransduction during muscle development. Cell. Physiol. Biochem. 2014, 33, 249–272. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.K.; Levin, J.B.; Hamilton, A.M.; Borodinsky, L.N. Calcium signaling in skeletal muscle development, maintenance and regeneration. Cell Calcium 2016, 59, 91–97. [Google Scholar] [CrossRef]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Perdiguero, E.; Muñoz-Cánoves, P. Regulation of muscle stem cell functions: A focus on the p38 MAPK signaling pathway. Front. Cell Dev. Biol. 2016, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alameddine, H.S.; Morgan, J.E. Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteinases in Inflammation and Fibrosis of Skeletal Muscles. J. Neuromuscul. Dis. 2016, 3, 455–473. [Google Scholar] [CrossRef]
- Smith, L.R.; Kok, H.J.; Zhang, B.; Chung, D.; Spradlin, R.A.; Rakoczy, K.D.; Lei, H.; Boesze-Battaglia, K.; Barton, E.R. Matrix metalloproteinase 13 from satellite cells is required for efficient muscle growth and regeneration. Cell. Physiol. Biochem. 2020, 54, 333–353. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Garlick, P.J.; James, W.P.T.; Nnanyelugo, D.O.; Ryatt, J.S. Relationship between protein synthesis and RNA content in skeletal muscle. Nature 1973, 241, 204–205. [Google Scholar] [CrossRef]
- Brook, M.S.; Wilkinson, D.J.; Smith, K.; Atherton, P.J. It’s not just about protein turnover: The role of ribosomal biogenesis and satellite cells in the regulation of skeletal muscle hypertrophy. Eur. J. Sport Sci. 2019, 19, 952–963. [Google Scholar] [CrossRef]
- Waterlow, J.; Garlick, P.; Millward, D. Protein Turnover in Mammalian Tissues and in the Whole Body; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1978; ISBN 0444800212. [Google Scholar]
- Enesco, M.; Puddy, D. Increase in the number of nuclei and weight in skeletal muscle of rats of various ages. Am. J. Anat. 1964, 114, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Garlick, P.J.; Stewart, R.J.C.; Nnanyelugo, D.O.; Waterlow, J.C. Skeletal muscle growth and protein turnover. Biochem. J. 1975, 150, 235–243. [Google Scholar] [CrossRef]
- Stewart, R.J.C.; Preece, R.F.; Sheppard, H.G. Twelve generations of marginal protein deficiency. Br. J. Nutr. 1975, 33, 233–253. [Google Scholar] [CrossRef]
- Laurent, G.; Sparrow, M.; Bates, P.; Millward, D. Turnover of muscle protein in the fowl (Gallus domesticus). Rates of protein synthesis in fast and slow skeletal, cardiac and smooth muscle of the adult fowl. Biochem. J. 1978, 176, 393–401. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Hennebry, A.; Oldham, J.; Shavlakadze, T.; Grounds, M.D.; Sheard, P.; Fiorotto, M.L.; Falconer, S.; Smith, H.K.; Berry, C.; Jeanplong, F.; et al. IGF1 stimulates greater muscle hypertrophy in the absence of myostatin in male mice. J. Endocrinol. 2017, 234, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Garlick, P.J.; Nnanyelugo, D.; Waterlow, J.C. The Relative Importance of Muscle Protein Synthesis and Breakdown in the Regulation of Muscle Mass. Biochem. J. 1976, 156, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Rudar, M.; Fiorotto, M.L.; Davis, T.A. Regulation of Muscle Growth in Early Postnatal Life in a Swine Model. Annu. Rev. Anim. Biosci. 2019, 7, 309–335. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Odedra, B.; Bates, P.C. The role of insulin, corticosterone and other factors in the acute recovery of muscle protein synthesis on refeeding food-deprived rats. Biochem. J. 1983, 216, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Millward, D.J.; Nnanyelugo, D.O.; James, W.P.T.; Garlick, P.J. Protein metabolism in skeletal muscle: The effect of feeding and fasting on muscle RNA, free amino acids and plasma insulin concentrations. Br. J. Nutr. 1974, 32, 127–142. [Google Scholar] [CrossRef]
- Jepson, M.M.; Bates, P.C.; Millward, D.J. The role of insulin and thyroid hormones in the regulation of muscle growth and protein turnover in response to dietary protein in the rat. Br. J. Nutr. 1988, 59, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.C.; Lang, C.H.; Crozier, S.J.; Anthony, T.G.; MacLean, D.A.; Kimball, S.R.; Jefferson, L.S. Contribution of insulin to the translational control of protein synthesis in skeletal muscle by leucine. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E1092–E1101. [Google Scholar] [CrossRef]
- Jefferson, L.; Li, J.B.; Rannels, S.R. Regulation by Insulin of Amino Acid Release and Protein Turnover in the Perfused Rat Hemicorpus. J. Biol. Chem. 1977, 252, 1476–1483. [Google Scholar] [CrossRef]
- Davis, T.A.; Fiorotto, M.L.; Burrin, D.G.; Reeds, P.J.; Nguyen, H.V.; Beckett, P.R.; Vann, R.C.; O’Connor, P.M.J. Stimulation of protein synthesis by both insulin and amino acids is unique to skeletal muscle in neonatal pigs. Am. J. Physiol. Endocrinol. Metab. 2002, 282, 880–890. [Google Scholar] [CrossRef]
- Atherton, P.J.; Wilkinson, D.J.; Smith, K. Feeding Modulation of Amino Acid Utilization: Role of Insulin and Amino Acids in Skeletal Muscle. In The Molecular Nutrition of Amino Acids and Proteins; Academic Press: Amsterdam, The Netherlands, 2016; pp. 109–124. ISBN 9780128021675. [Google Scholar] [CrossRef]
- Millward, D.J.; Smith, K. The application of stable-isotope tracers to study human musculoskeletal protein turnover: A tale of bag filling and bag enlargement. J. Physiol. 2019, 597, 1235–1249. [Google Scholar] [CrossRef] [PubMed]
- Prod’homme, M.; Balage, M.; Debras, E.; Farges, M.C.; Kimball, S.; Jefferson, L.; Grizard, J. Differential effects of insulin and dietary amino acids on muscle protein synthesis in adult and old rats. J. Physiol. 2005, 563, 235–248. [Google Scholar] [CrossRef]
- Anthony, J.C.; Reiter, A.K.; Anthony, T.G.; Crozier, S.J.; Lang, C.H.; MacLean, D.A.; Kimball, S.R.; Jefferson, L.S. Orally administered leucine enhances protein synthesis in skeletal muscle of diabetic rats in the absence of increases in 4E-BP1 or S6K1 phosphorylation. Diabetes 2002, 51, 928–936. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.M.J.; Kimball, S.R.; Suryawan, A.; Bush, J.A.; Nguyen, H.V.; Jefferson, L.S.; Davis, T.A. Regulation of translation initiation by insulin and amino acids in skeletal muscle of neonatal pigs. Am. J. Physiol. Endocrinol. Metab. 2003, 285, 40–53. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Xu, D.; Shimkus, K.L.; Lacko, H.A.; Kutzler, L.; Jefferson, L.S.; Kimball, S.R. Evidence for a role for sestrin1 in mediating leucine-induced activation of mtorc1 in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E817–E828. [Google Scholar] [CrossRef] [PubMed]
- Kusnadi, E.P.; Hannan, K.M.; Hicks, R.J.; Hannan, R.D.; Pearson, R.B.; Kang, J. Regulation of rDNA transcription in response to growth factors, nutrients and energy. Gene 2015, 556, 27–34. [Google Scholar] [CrossRef]
- Chaillou, T.; Kirby, T.J.; Mccarthy, J.J. Ribosome Biogenesis: Emerging Evidence for a Central Role in the Regulation of Skeletal Muscle Mass. J. Cell. Physiol. 2014, 229, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Control of the translational machinery by amino acids. Am. J. Clin. Nutr. 2014, 99, 231–236. [Google Scholar] [CrossRef]
- Antonetti, D.A.; Kimball, S.R.; Horetsky, R.L.; Jefferson, L.S. Regulation of rDNA transcription by insulin in primary cultures of rat hepatocytes. J. Biol. Chem. 1993, 268, 25277–25284. [Google Scholar] [CrossRef]
- Hannan, K.M.; Rothblum, L.I.; Jefferson, L.S. Regulation of ribosomal DNA transcription by insulin. Am. J. Physiol. 1998, 275, 130–138. [Google Scholar] [CrossRef]
- Flaim, K.E.; Li, J.B.; Jefferson, L.S. Effects of thyroxine on protein turnover in rat skeletal muscle. Am. J. Physiol. Endocrinol. Metab. Gastrointest. Physiol. 1978, 4, E231–E237. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.G.; Bates, P.C.; Holliday, M.A.; Millward, D.J. Thyroid hormones and muscle protein turnover. The effect of thyroid-hormone deficiency and replacement in thryoidectomized and hypophysectomized rats. Biochem. J. 1981, 194, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.G.; Van Bueren, J.; Millward, D.J. The effect of tri-iodothyronine administration on protein synthesis in the diabetic rat. Biochem. J. 1983, 214, 637–640. [Google Scholar] [CrossRef]
- Brown, J.G.; Millward, D.J. Dose response of protein turnover in rat skeletal muscle to triiodothyronine treatment. BBA Gen. Subj. 1983, 757, 182–190. [Google Scholar] [CrossRef]
- Clément, K.; Viguerie, N.; Diehn, M.; Alizadeh, A.; Barbe, P.; Thalamas, C.; Storey, J.D.; Brown, P.O.; Barsh, G.S.; Langin, D. In vivo regulation of human skeletal muscle gene expression by thyroid hormone. Genome Res. 2002, 12, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, A.; Ojamaa, K. Thyroid hormone stimulates protein synthesis in the cardiomyocyte by activating the Akt-mTOR and p70S6K pathways. J. Biol. Chem. 2006, 281, 20666–20672. [Google Scholar] [CrossRef]
- Waterlow, J.C. Protein-Energy Malnutrition; Smith-Gorden: New Barnet, UK, 2006; ISBN 1-85463-231-0. [Google Scholar]
- Chauvin, C.; Koka, V.; Nouschi, A.; Mieulet, V.; Hoareau-Aveilla, C.; Dreazen, A.; Cagnard, N.; Carpentier, W.; Kiss, T.; Meyuhas, O.; et al. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014, 33, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.J.; Bishop, D.J.; Bartlett, J.D.; Hanson, E.D.; Anderson, M.J.; Garnham, A.P.; Stepto, N.K. Enhanced skeletal muscle ribosome biogenesis, yet attenuated mTORC1 and ribosome biogenesis-related signalling, following short-term concurrent versus single-mode resistance training. Sci. Rep. 2018, 8, 1–21. [Google Scholar] [CrossRef]
- Yahya, Z.A.H.; Millward, D.J. Dietary protein and the regulation of long-bone and muscle growth in the rat. Clin. Sci. 1994, 87, 213–224. [Google Scholar] [CrossRef]
- Tirapegui, J.; Yayha, Z.A.H.; Bates, P.C.; Millward, D.J. Energy, Glucocorticoids and the Regulation of Long Bone and muscle growth. Clin. Sci. 1994, 87, 599–606. [Google Scholar] [CrossRef]
- Yahya, Z.A.H.; Tirapegui, J.O.; Bates, P.C.; Millward, D.J. Influence of dietary protein, energy and corticosteroids on protein turnover, proteoglycan sulphation and growth of long bone and skeletal muscle in the rat. Clin. Sci. 1994, 87, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Yahya, Z.A.H.; Bates, P.C.; Tirapegui, J.O.; Morrell, D.; Buchanan, C.; Millward, D.J. IGF-1 concentrations in protein deficient rat plasma and tissues in relation to protein and proteoglycan synthesis rates. Biochem. Soc. Trans. 1988, 16, 624–625. [Google Scholar] [CrossRef]
- Yahya, Z.A.H.; Bates, P.C.; Millward, D.J. Responses to protein deficiency of plasma and tissue insulin-like growth factor-I levels and proteoglycan synthesis rates in rat skeletal muscle and bone. J. Endocrinol. 1990, 127, 56–79. [Google Scholar] [CrossRef] [PubMed]
- Davis, T.A.; Suryawan, A.; Wilson, F.A.; Fiorotto, M.L.; Gazzaneo, M.C.; Orellana, R.A.; Burrin, D.G. Prolonged stimulation of protein synthesis by leucine is dependent on amino acid availability. EAAP Sci. Ser. 2010, 127, 253–254. [Google Scholar] [CrossRef]
- Wilson, G.J.; Layman, D.K.; Moulton, C.J.; Norton, L.E.; Anthony, T.G.; Proud, C.G.; Indu Rupassara, S.; Garlick, P.J. Leucine or carbohydrate supplementation reduces AMPK and eEF2 phosphorylation and extends postprandial muscle protein synthesis in rats. Am. J. Physiol. Endocrinol. Metab. 2011, 301, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.J.; Moulton, C.J.; Garlick, P.J.; Anthony, T.G.; Layman, D.K. Post-meal responses of elongation factor 2 (eEF2) and adenosine monophosphate-activated protein kinase (AMPK) to leucine and carbohydrate supplements for regulating protein synthesis duration and energy homeostasis in rat skeletal muscle. Nutrients 2012, 4, 1723–1739. [Google Scholar] [CrossRef]
- Abdulla, H.; Smith, K.; Atherton, P.J.; Idris, I. Role of insulin in the regulation of human skeletal muscle protein synthesis and breakdown: A systematic review and meta-analysis. Diabetologia 2016, 59, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, W.K.; Wilkinson, D.J.; Phillips, B.E.; Lund, J.N.; Smith, K.; Atherton, P.J. Human Skeletal Muscle Protein Metabolism Responses to Amino Acid Nutrition. Adv. Nutr. 2016, 7, 828S–838S. [Google Scholar] [CrossRef] [PubMed]
- Deane, C.S.; Ely, I.A.; Wilkinson, D.J.; Smith, K.; Phillips, B.E.; Atherton, P.J. Dietary protein, exercise, ageing and physical inactivity: Interactive influences on skeletal muscle proteostasis. Proc. Nutr. Soc. 2020. [Google Scholar] [CrossRef]
- Atherton, P.J.; Smith, K.; Etheridge, T.; Rankin, D.; Rennie, M.J. Distinct anabolic signalling responses to amino acids in C2C12 skeletal muscle cells. Amino Acids 2010, 38, 1533–1539. [Google Scholar] [CrossRef]
- Wilkinson, D.J.; Bukhari, S.S.I.; Phillips, B.E.; Limb, M.C.; Cegielski, J.; Brook, M.S.; Rankin, D.; Mitchell, W.K.; Kobayashi, H.; Williams, J.P.; et al. Effects of leucine-enriched essential amino acid and whey protein bolus dosing upon skeletal muscle protein synthesis at rest and after exercise in older women. Clin. Nutr. 2018, 37, 2011–2021. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.S.; Albright, R.C.; Bigelow, M.L.; Toffolo, G.; Cobelli, C.; Nair, K.S. Mechanism of insulin’s anabolic effect on muscle: Measurements of muscle protein synthesis and breakdown using aminoacyl-tRNA and other surrogate measures. Am. J. Physiol. Endocrinol. Metab. 2006, 291, 729–736. [Google Scholar] [CrossRef]
- Greenhaff, P.L.; Karagounis, L.G.; Peirce, N.; Simpson, E.J.; Hazell, M.; Layfield, R.; Wackerhage, H.; Smith, K.; Atherton, P.; Selby, A.; et al. Disassociation between the effects of amino acids and insulin on signaling, ubiquitin ligases, and protein turnover in human muscle. Am. J. Physiol. Endocrinol. Metab. 2008, 295, 595–604. [Google Scholar] [CrossRef]
- Hillier, T.A.; Fryburg, D.A.; Jahn, L.A.; Barrett, E.J. Extreme hyperinsulinemia unmasks insulin’s effect to stimulate protein synthesis in the human forearm. Am. J. Physiol. Endocrinol. Metab. 1998, 274. [Google Scholar] [CrossRef]
- Wilkes, E.A.; Selby, A.L.; Atherton, P.J.; Patel, R.; Rankin, D.; Smith, K.; Rennie, M.J. Blunting of insulin inhibition of proteolysis in legs of older subjects may contribute to age-related sarcopenia. Am. J. Clin. Nutr. 2009, 90, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Takahara, T.; Amemiya, Y.; Sugiyama, R.; Maki, M.; Shibata, H. Amino acid-dependent control of mTORC1 signaling: A variety of regulatory modes. J. Biomed. Sci. 2020, 27, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Meng, D.; Yang, Q.; Wang, H.; Melick, C.H.; Navlani, R.; Frank, A.R.; Jewell, J.L. Glutamine and asparagine activate mTORC1 independently of Rag GTPases. J. Biol. Chem. 2020, 295, 2890–2899. [Google Scholar] [CrossRef]
- Proud, C.G. Phosphorylation and signal transduction pathways in translational control. Cold Spring Harb. Perspect. Biol. 2019, 11. [Google Scholar] [CrossRef]
- Pinilla, J.; Aledo, J.C.; Cwiklinski, E.; Hyde, R.; Taylor, P.M.; Hundal, H.S. SNAT2 transceptor signalling via mTOR: A role in cell growth and proliferation? Front Biosci. 2011, 3E, 1289–1299. [Google Scholar] [CrossRef]
- Hundal, H.S.; Taylor, P.M. Amino acid transceptors: Gate keepers of nutrient exchange and regulators of nutrient signaling. Am. J. Physiol. Endocrinol. Metab. 2009, 296. [Google Scholar] [CrossRef]
- Taylor, P.M. Role of amino acid transporters in amino acid sensing 1—4. Am. J. Clin. Nutr. 2014, 99, 223S–230S. [Google Scholar] [CrossRef]
- Graber, T.G.; Borack, M.S.; Reidy, P.T.; Volpi, E.; Rasmussen, B.B. Essential amino acid ingestion alters expression of genes associated with amino acid sensing, transport, and mTORC1 regulation in human skeletal muscle. Nutr. Metab. 2017, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.C.; Grimble, G.K.; Sparrow, M.P.; Millward, D.J. Myofibrillar protein turnover.Synthesis of protein-bound 3-methylhistidine, actin, myosin heavy chain and aldolase in rat skeletal muscle in the fed and starved states. Biochem. J. 1983, 214, 587–592. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2014, 14, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, X.; Miereles, C.; Bailey, J.L.; Debigare, R.; Zheng, B.; Price, S.R.; Mitch, W.E. Activation of caspase-3 is an initial step triggering accelerated muscle proteolysis in catabolic conditions. J. Clin. Investig. 2004, 113, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Physiol. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef]
- Lorin, S.; Pattingre, S.; Meijer, A.J.; Codogno, P. Regulation of Macroautophagy by Nutrients and Metabolites. Mol. Nutr. Amin. Acids Proteins 2016, 181–203. [Google Scholar] [CrossRef]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef]
- Collins, G.A.; Goldberg, A.L. The Logic of the 26S Proteasome. Cell 2017, 169, 792–806. [Google Scholar] [CrossRef]
- Rousseau, A.; Bertolotti, A. Regulation of proteasome assembly and activity in health and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 697–712. [Google Scholar] [CrossRef] [PubMed]
- Chantranupong, L.; Sabatini, D. The TORC1 pathway to protein destruction. Nature 2016, 536, 156. [Google Scholar] [CrossRef][Green Version]
- Zhao, J.; Goldberg, A.L. Coordinate regulation of autophagy and the ubiquitin proteasome system by MTOR. Autophagy 2016, 12, 1967–1970. [Google Scholar] [CrossRef]
- Häussinger, D.; Gerok, W.; Roth, E.; Lang, F. Cellular hydration state: An important determinant of protein catabolism in health and disease. Lancet 1993, 341, 1330–1332. [Google Scholar] [CrossRef]
- Gosmanov, A.R.; Lindinger, M.I.; Thomason, D.B. Riding the tides: K+ concentration and volume regulation by muscle Na+-K+-2Cl− cotransport activity. News Physiol. Sci. 2003, 18, 196–200. [Google Scholar] [CrossRef]
- Schliess, F.; Reinehr, R.; Häussinger, D. Osmosensing and signaling in the regulation of mammalian cell function. FEBS J. 2007, 274, 5799–5803. [Google Scholar] [CrossRef] [PubMed]
- Schliess, F.; Richter, L.; Vom Dahl, S.; Häussinger, D. Cell hydration and mTOR-dependent signalling. Acta Physiol. 2006, 187, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Low, S.Y.; Rennie, M.J.; Taylor, P.M. Involvement of integrins and the cytoskeleton in modulation of skeletal muscle glycogen synthesis by changes in cell volume. FEBS Lett. 1997, 417, 101–103. [Google Scholar] [CrossRef]
- Low, S.Y.; Rennie, M.J.; Taylor, P.M. Signaling elements involved in amino acid transport responses to altered muscle cell volume. FASEB J. 1997, 11, 1111–1117. [Google Scholar] [CrossRef]
- Keller, U.; Szinnai, G.; Bilz, S.; Berneis, K. Effects of changes in hydration on protein, glucose and lipid metabolism in man: Impact on health. Eur. J. Clin. Nutr. 2003, 57, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Gill, G.V.; Baylis, P.H.; Flear, C.T.G.; Lawson, J.Y. Changes in plasma solutes after food. J. R. Soc. Med. 1985, 78, 1009–1013. [Google Scholar] [CrossRef]
- Vom Dahl, S.; Schliess, F.; Reissmann, R.; Görg, B.; Weiergräber, O.; Kocalkova, M.; Dombrowski, F.; Häussinger, D. Involvement of integrins in osmosensing and signaling toward autophagic proteolysis in rat liver. J. Biol. Chem. 2003, 278, 27088–27095. [Google Scholar] [CrossRef] [PubMed]
- Low, S.Y.; Taylor, P.M. Integrin and cytoskeletal involvement in signalling cell volume changes to glutamine transport in rat skeletal muscle. J. Physiol. 1998, 512, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Bohé, J.; Aili Low, J.F.; Wolfe, R.R.; Rennie, M.J. Latency and duration of stimulation of human muscle protein synthesis during continuous infusion of amino acids. J. Physiol. 2001, 532, 575–579. [Google Scholar] [CrossRef]
- Atherton, P.J.; Etheridge, T.; Watt, P.W.; Wilkinson, D.; Selby, A.; Rankin, D.; Smith, K.; Rennie, M.J. Muscle full effect after oral protein: Time-dependent concordance and discordance between human muscle protein synthesis and mTORC1 signaling. Am. J. Clin. Nutr. 2010, 92, 1080–1088. [Google Scholar] [CrossRef]
- Mitchell, W.K.; Phillips, B.E.; Hill, I.; Greenhaff, P.; Lund, J.N.; Williams, J.P.; Rankin, D.; Wilkinson, D.J.; Smith, K.; Atherton, P.J. Human skeletal muscle is refractory to the anabolic effects of leucine during the postprandial muscle-full period in older men. Clin. Sci. 2017, 131, 2643–2653. [Google Scholar] [CrossRef]
- Mitchell, W.K.; Phillips, B.E.; Williams, J.P.; Rankin, D.; Lund, J.N.; Smith, K.; Atherton, P.J. A Dose-rather than delivery profile-dependent mechanism regulates the ‘muscle-full’ effect in response to oral essential amino acid intake in young men. J. Nutr. 2015, 145, 207–214. [Google Scholar] [CrossRef]
- Kumar, A.; Xie, L.; Ta, C.M.; Hinton, A.O.; Gunasekar, S.K.; Minerath, R.A.; Shen, K.; Maurer, J.M.; Grueter, C.E.; Abel, E.D.; et al. SWELL1 regulates skeletal muscle cell size, intracellular signalling, adiposity and glucose metabolism. eLife 2020, 9, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Demian, W.L.; Persaud, A.; Jiang, C.; Kafri, R.; Raught, B.; Liu, S.; Kapus, A.; Kafri, R. The Ion Transporter NKCC1 Links Cell Volume to Cell Mass Regulation by Suppressing mTORC1 Article The Ion Transporter NKCC1 Links Cell Volume to Cell Mass Regulation by Suppressing mTORC1. Cell Rep. 2019, 27, 1886–1896. [Google Scholar] [CrossRef]
- Norton, L.E.; Layman, D.K.; Bunpo, P.; Anthony, T.G.; Brana, D.V.; Garlick, P.J. The leucine content of a complete meal directs peak activation but not duration of skeletal muscle protein synthesis and mammalian target of rapamycin signaling in rats. J. Nutr. 2009, 139, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Child Growth Standards; WHO Press: Geneva, Switzerland, 2006; ISBN 92-4-154693-X. [Google Scholar]
- World Health Organization. Multicentre Growth Reference Study Group WHO child growth standards. Acta Pædiatr. 2006, 450, 56–65. [Google Scholar] [CrossRef]
- World Health Organization. Protein and Amino Acid Requirements in Human Nutrition Report of a Joint FAO/WHO/UNU Expert Consultation (WHO Technical Report Series 935); WHO: Geneva, Switzerland, 2007; ISBN 92-4-120935-6. [Google Scholar]
- Millward, D.J. Identifying recommended dietary allowances for protein and amino acids: A critique of the 2007 WHO/FAO/UNU report. Br. J. Nutr. 2012, 108, S3–S21. [Google Scholar] [CrossRef]
- Hörnell, A.; Lagström, H.; Lande, B.; Thorsdottir, I. Protein intake from 0 to 18 years of age and its relation to health: A systematic literature review for the 5th Nordic Nutrition Recommendations. Food Nutr. Res. 2013, 57, 21083. [Google Scholar] [CrossRef] [PubMed]
- Victora, C.G.; Barros, A.J.D. Effect of breastfeeding on infant and child mortality due to infectious diseases in less developed countries: A pooled analysis. Lancet 2000, 355, 451–455. [Google Scholar] [CrossRef]
- World Health Organization. Report of the Commission on Ending Childhood Obesity; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Grummer-Strawn, L.M.; Rollins, N. Summarising the health effects of breastfeeding. Acta Paediatr. Int. J. Paediatr. 2015, 104, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qiao, Y.; Zhao, P.; Li, W.; Katzmarzyk, P.T.; Chaput, J.P.; Fogelholm, M.; Kuriyan, R.; Lambert, E.V.; Maher, C.; et al. Breastfeeding and childhood obesity: A 12-country study. Matern. Child Nutr. 2020, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, I.E.M.; Ivarsson, S.A.; Raiha, N.C.R. Protein intake in early infancy: Effects on plasma amino acid concentrations, insulin metabolism, and growth. Pediatr. Res. 1989, 26, 614–617. [Google Scholar] [CrossRef]
- Koletzko, B.; Von Kries, R.; Monasterolo, R.C.; Subías, J.E.; Scaglioni, S.; Giovannini, M.; Beyer, J.; Demmelmair, H.; Anton, B.; Gruszfeld, D.; et al. Can infant feeding choices modulate later obesity risk? Am. J. Clin. Nutr. 2009, 89, 1502–1509. [Google Scholar] [CrossRef]
- Elks, C.E.; Heude, B.; De Zegher, F.; Barton, S.J.; Clément, K.; Inskip, H.M.; Koudou, Y.; Cooper, C.; Dunger, D.B.; Ibáñez, L.; et al. Associations between genetic obesity susceptibility and early postnatal fat and lean mass: An individual participant meta-analysis. JAMA Pediatr. 2014, 168, 1122–1130. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.L.; Ahmed, M.L.; Dunger, D.B.; Emmett, P.M.; Preece, M.A. Association between postnatal catch-up growth and obesity in childhood: Prospective cohort study. Br. Med. J. 2000, 320, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.; Petry, C.J.; Emmett, P.M.; Saradhu, M.S.; Kiess, W.; Hales, C.N.; Ness, A.R.; Dunger, D.B. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia 2004, 47, 1064–1070. [Google Scholar] [CrossRef]
- Druet, C.; Stettler, N.; Sharp, S.; Simmons, R.K.; Cooper, C.; Davey Smith, G.; Ekelund, U.; Lévy-Marchal, C.; Jarvelin, M.R.; Kuh, D.; et al. Prediction of childhood obesity by infancy weight gain: An individual-level meta-analysis. Paediatr. Perinat. Epidemiol. 2012, 26, 19–26. [Google Scholar] [CrossRef]
- Patro-Gołąb, B.; Zalewski, B.M.; Kołodziej, M.; Kouwenhoven, S.; Poston, L.; Godfrey, K.M.; Koletzko, B.; van Goudoever, J.B.; Szajewska, H. Nutritional interventions or exposures in infants and children aged up to 3 years and their effects on subsequent risk of overweight, obesity and body fat: A systematic review of systematic reviews. Obes. Rev. 2016, 17, 1245–1257. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.G.; Martin, R.M.; Whincup, P.H.; Davey-Smith, G.; Gillman, M.W.; Cook, D.G. The effect of breastfeeding on mean body mass index throughout life: A quantitative review of published and unpublished observational evidence. Am. J. Clin. Nutr. 2005, 82, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Brion, M.J.A.; Lawlor, D.A.; Matijasevich, A.; Horta, B.; Anselmi, L.; Araújo, C.L.; Menezes, A.M.B.; Victora, C.G.; Smith, G.D. What are the causal effects of breastfeeding on IQ, obesity and blood pressure? Evidence from comparing high-income with middle-income cohorts. Int. J. Epidemiol. 2011, 40, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.M.; Kramer, M.S.; Patel, R.; Rifas-Shiman, S.L.; Thompson, J.; Yang, S.; Vilchuck, K.; Bogdanovich, N.; Hameza, M.; Tilling, K.; et al. Effects of promoting long-term, exclusive breastfeeding on adolescent adiposity, blood pressure, and growth trajectories: A secondary analysis of a randomized clinical trial. JAMA Pediatr. 2017, 171, e170698. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.S.; Matush, L.; Vanilovich, I.; Platt, R.W.; Bogdanovich, N.; Sevkovskaya, Z.; Dzikovich, I.; Shishko, G.; Collet, J.P.; Martin, R.M.; et al. Effects of prolonged and exclusive breastfeeding on child height, weight, adiposity, and blood pressure at age 6.5 y: Evidence from a large randomized trial. Am. J. Clin. Nutr. 2007, 86, 1717–1721. [Google Scholar] [CrossRef]
- Ruckinger, S.; Kries, R. von Breastfeeding and reduced risk of childhood obesity: Will randomized trials on breastfeeding promotion give the definite answer? Am. J. Clin. Nutr. 2009, 89, 653–655. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.S.; Platt, R.W. Reply to Rückinger and von Kries. Am. J. Clin. Nutr. 2009, 89, 655. [Google Scholar] [CrossRef][Green Version]
- Lind, M.V.; Larnkjær, A.; MØlgaard, C.; Michaelsen, K.F. Dietary protein intake and quality in early life: Impact on growth and obesity. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Rzehak, P.; Oddy, W.H.; Mearin, M.L.; Grote, V.; Mori, T.A.; Szajewska, H.; Shamir, R.; Koletzko, S.; Weber, M.; Beilin, L.J.; et al. Infant feeding and growth trajectory patterns in childhood and body composition in young adulthood. Am. J. Clin. Nutr. 2017, 106, 568–580. [Google Scholar] [CrossRef]
- Koletzko, B.; Demmelmair, H.; Grote, V.; Totzauer, M. Optimized protein intakes in term infants support physiological growth and promote long-term health. Semin. Perinatol. 2019, 43. [Google Scholar] [CrossRef]
- Clemmons, D.R. Consensus statement on the standardization and evaluation of growth hormone and insulin-like growth factor assays. Clin. Chem. 2011, 57, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Bidlingmaier, M.; Friedrich, N.; Emeny, R.T.; Spranger, J.; Wolthers, O.D.; Roswall, J.; Körner, A.; Obermayer-Pietsch, B.; Hübener, C.; Dahlgren, J.; et al. Reference intervals for insulin-like growth factor-1 (IGF-I) from birth to senescence: Results from a multicenter study using a new automated chemiluminescence IGF-I immunoassay conforming to recent international recommendations. J. Clin. Endocrinol. Metab. 2014, 99, 1712–1721. [Google Scholar] [CrossRef] [PubMed]
- Bozzola, M.; Meazza, C. Growth Velocity Curves: What They Are and How to Use Them. In Handbook of Growth and Growth Monitoring in Health and Disease; Preedy, V.R., Ed.; Springer: New York, NY, USA, 2012; pp. 2999–3011. [Google Scholar] [CrossRef]
- Hawkes, C.P.; Murray, D.M.; Kenny, L.C.; Kiely, M.; O’B Hourihane, J.; Irvine, A.D.; Wu, Z.; Argon, Y.; Reitz, R.E.; McPhaul, M.J.; et al. Correlation of Insulin-Like Growth Factor-I and -II Concentrations at Birth Measured by Mass Spectrometry and Growth from Birth to Two Months. Horm. Res. Paediatr. 2018, 89, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Juul, A.; Bang, P.; Hertel, N.T.; Main, K.; Dalgaard, P.; Jørgensen, K.; Müller, J.; Hall, K.; Skakkebæk, N.E. Serum insulin-like growth factor-I in 1030 healthy children, adolescents, and adults: Relation to age, sex, stage of puberty, testicular size, and body mass index. J. Clin. Endocrinol. Metab. 1994, 78, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.; Langkamp, M.; Ranke, M.B.; Whitehead, K.; Hughes, I.A.; Acerini, C.L.; Dunger, D.B. Insulin-like growth factor I concentrations in infancy predict differential gains in body length and adiposity: The Cambridge Baby Growth Study. Am. J. Clin. Nutr. 2009, 90, 156–161. [Google Scholar] [CrossRef]
- Madsen, A.L.; Larnkjær, A.; Mølgaard, C.; Michaelsen, K.F. IGF-I and IGFBP-3 in healthy 9month old infants from the SKOT cohort: Breastfeeding, diet, and later obesity. Growth Horm. IGF Res. 2011, 21, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Rogers, I.; Metcalfe, C.; Gunnell, D.; Emmett, P.; Dunger, D.; Holly, J. Insulin-like growth factor-I and growth in height, leg length, and trunk length between ages 5 and 10 years. J. Clin. Endocrinol. Metab. 2006, 91, 2514–2519. [Google Scholar] [CrossRef]
- Wiley, A.S.; Lubree, H.G.; Joshi, S.M.; Bhat, D.S.; Ramdas, L.V.; Rao, A.S.; Thuse, N.V.; Deshpande, V.U.; Yajnik, C.S. Cord IGF-I concentrations in Indian newborns: Associations with neonatal body composition and maternal determinants. Pediatr. Obes. 2016, 11, 151–157. [Google Scholar] [CrossRef]
- Wiley, A.S.; Joshi, S.M.; Lubree, H.G.; Bhat, D.S.; Memane, N.S.; Raut, D.A.; Yajnik, C.S. IGF-I and IGFBP-3 concentrations at 2 years: Associations with anthropometry and milk consumption in an Indian cohort. Eur. J. Clin. Nutr. 2018, 72, 564–571. [Google Scholar] [CrossRef]
- Chellakooty, M.; Juul, A.; Boisen, K.A.; Damgaard, I.N.; Kai, C.M.; Schmidt, I.M.; Petersen, J.H.; Skakkebæk, N.E.; Main, K.M. A prospective study of serum insulin-like growth factor I (IGF-I) and IGF-binding protein-3 in 942 healthy infants: Associations with birth weight, gender, growth velocity, and breastfeeding. J. Clin. Endocrinol. Metab. 2006, 91, 820–826. [Google Scholar] [CrossRef]
- Putet, G.; Labaune, J.M.; Mace, K.; Steenhout, P.; Grathwohl, D.; Raverot, V.; Morel, Y.; Picaud, J.C. Effect of dietary protein on plasma insulin-like growth factor-1, growth, and body composition in healthy term infants: A randomised, double-blind, controlled trial (Early Protein and Obesity in Childhood (EPOCH) study). Br. J. Nutr. 2016, 115, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Mølgaard, C.; Thomsen, B.L.; Juul, A.; Michaelsen, K.F. Protein intake at 9 mo of age is associated with body size but not with body fat in 10-y-old Danish children. Am. J. Clin. Nutr. 2004, 79, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, C.; Udam, T.R.; Lauritzen, L.; Mølgaard, C.; Juul, A.; Michaelsen, K.F. Animal protein intake, serum insulin-like growth factor I, and growth in healthy 2.5-y-old Danish children. Am. J. Clin. Nutr. 2004, 80, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Larnkjær, A.; Hoppe, C.; Mølgaard, C.; Michaelsen, K.F. The effects of whole milk and infant formula on growth and IGF-I in late infancy. Eur. J. Clin. Nutr. 2009, 63, 956–963. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ejlerskov, K.T.; Larnkjaer, A.; Pedersen, D.; Ritz, C.; Mølgaard, C.; Michaelsen, K.F. IGF-I at 9 and 36 months of age—Relations with body composition and diet at 3 years—The SKOT cohort. Growth Horm. IGF Res. 2014, 24, 239–244. [Google Scholar] [CrossRef]
- Hoppe, C.; Mølgaard, C.; Dalum, C.; Vaag, A.; Michaelsen, K.F. Differential effects of casein versus whey on fasting plasma levels of insulin, IGF-1 and IGF-1/IGFBP-3: Results from a randomized 7-day supplementation study in prepubertal boys. Eur. J. Clin. Nutr. 2009, 63, 1076–1083. [Google Scholar] [CrossRef]
- Kadakia, R.; Josefson, J. The relationship of insulin-like growth factor 2 to fetal growth and adiposity. Horm. Res. Paediatr. 2016, 85, 75–82. [Google Scholar] [CrossRef]
- Kineman, R.D.; del Rio-Moreno, M.; Sarmento-Cabral, A. 40 years of IGF1: Understanding the tissue-specific roles of IGF1/IGF1R in regulating metabolism using the Cre/loxP system. J. Mol. Endocrinol. 2018, 61, T187–T198. [Google Scholar] [CrossRef]
- De Onis, M.; Yip, R. The WHO growth chart: Historical considerations and current scientific issues. In Bibliotheca Nutritio et Dieta; Karger Publishers: Basel, Switzerland, 1996; pp. 74–89. [Google Scholar]
- Dewey, K.G. Growth Characteristics of Breast-Fed Compared to Formula-Fed Infants. Biol. Neonate 1998, 74, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, E.R.J.; Horta, B.L.; Loret De Mola, C.; Lisboa, B.O.; Victora, C.G. Effect of breastfeeding promotion interventions on child growth: A systematic review and meta-analysis. Acta Paediatr. Int. J. Paediatr. 2015, 104, 20–29. [Google Scholar] [CrossRef]
- Weber, M.; Grote, V.; Ricardo Closa-Monasterolo, J.E.; Langhendries, J.-P.; Dain, E.; Giovannini, M.; Verduci, E.; Gruszfeld, D.; Socha, P.; Koletzko, B. Lower protein content in infant formula reduces BMI and obesity risk at school age: Follow-up of a randomized trial. Am. J. Clin. Nutr. 2014, 99, 1041–1051. [Google Scholar] [CrossRef]
- Costa-Orvay, J.A.; Figueras-Aloy, J.; Romera, G.; Closa-Monasterolo, R.; Carbonell-Estrany, X. The effects of varying protein and energy intakes on the growth and body composition of very low birth weight infants. Nutr. J. 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Fenton, T.R.; Premji, S.S.; Al-Wassia, H.; Sauve, R.S. Higher versus lower protein intake in formula-fed low birth weight infants. Cochrane Database Syst. Rev. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Tonkin, E.L.; Collins, C.T.; Miller, J. Protein Intake and Growth in Preterm Infants. Glob. Pediatr. Health 2014, 1, 2333794X1455469. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.K.L.; Preece, M.A.; Emmett, P.M.; Ahmed, M.L.; Dunger, D.B. Size at Birth and Early Childhood Growth in Relation to Maternal Smoking, Parity and Infant Breast-Feeding: Longitudinal Birth Cohort Study and Analysis. Pediatr. Res. 2002, 52, 863–867. [Google Scholar] [CrossRef]
- Nielsen, G.A.; Thomsen, B.L.; Michaelsen, K.F. Influence of breastfeeding and complementary food on growth between 5 and 10 months. Acta Paediatr. Int. J. Paediatr. 1998, 87, 911–917. [Google Scholar] [CrossRef]
- Braun, K.V.; Erler, N.S.; Kiefte-de Jong, J.C.; Jaddoe, V.W.; van den Hooven, E.H.; Franco, O.H.; Voortman, T. Dietary Intake of Protein in Early Childhood Is Associated with Growth Trajectories between 1 and 9 Years of Age. J. Nutr. 2016, 146, 2361–2367. [Google Scholar] [CrossRef]
- Switkowski, K.M.; Jacques, P.F.; Must, A.; Fleisch, A.; Oken, E. Associations of protein intake in early childhood with body composition, height, and insulin-like growth factor i in mid-childhood and early adolescence. Am. J. Clin. Nutr. 2019, 109, 1154–1163. [Google Scholar] [CrossRef]
- Thorisdottir, B.; Gunnarsdottir, I.; Palsson, G.I.; Halldorsson, T.I.; Thorsdottir, I. Animal protein intake at 12 months is associated with growth factors at the age of six. Acta Paediatr. Int. J. Paediatr. 2014, 103, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Bogin, B. Milk and Human Development: An Essay on the Milk Hypothesis. Antropol. Port. 1998, 15, 23–36. [Google Scholar]
- Hoppe, C.; Mølgaard, C.; Michaelsen, K.F. Cow’s Milk and Linear Growth in Industrialized and Developing Countries. Annu. Rev. Nutr. 2006, 26, 131–173. [Google Scholar] [CrossRef]
- Berkey, C.S.; Colditz, G.A.; Rockett, H.R.H.; Frazier, A.L.; Willett, W.C. Dairy consumption and female height growth: Prospective cohort study. Cancer Epidemiol. Biomarkers Prev. 2009, 18, 1881–1887. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Pang, X.; Yang, Z.; Wang, J.; Jiang, S.; Bi, Y.; Wang, S.; Zhang, H.; Lai, J. Association between dairy intake and linear growth in Chinese pre-school children. Nutrients 2020, 12, 2576. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, B.; Cao, S.; Jia, C.; Yu, X.; Zhao, L.; Dellarco, M.; Duan, X. Association between milk intake and childhood growth: Results from a nationwide cross-sectional survey. Int. J. Obes. 2020, 44, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Herber, C.; Bogler, L.; Subramanian, S.V.; Vollmer, S. Association between milk consumption and child growth for children aged 6–59 months. Sci. Rep. 2020, 10, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Grasgruber, P.; Sebera, M.; Hrazdíra, E.; Cacek, J.; Kalina, T. Major correlates of male height: A study of 105 countries. Econ. Hum. Biol. 2016, 21, 172–195. [Google Scholar] [CrossRef] [PubMed]
- Ségurel, L.; Bon, C. On the Evolution of Lactase Persistence in Humans. Annu. Rev. Genomics Hum. Genet. 2017, 18, 297–319. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C.; John, S.M.; Schmitz, G. Milk is not just food but most likely a genetic transfection system activating mTORC1 signaling for postnatal growth. Nutr. J. 2013, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Melnik, B.C. Milk—A nutrient system of mammalian evolution promoting mTORC1-dependent translation. Int. J. Mol. Sci. 2015, 16, 17048–17087. [Google Scholar] [CrossRef]
- Wiley, A.S. The Evolution of Lactase Persistence: Milk Consumption, Insulin-Like Growth Factor I, and Human Life-History Parameters. Q. Rev. Biol. 2018, 93, 319–345. [Google Scholar] [CrossRef]
- De Beer, H. Dairy products and physical stature: A systematic review and meta-analysis of controlled trials. Econ. Hum. Biol. 2012, 10, 299–309. [Google Scholar] [CrossRef]
- Kouvelioti, R.; Josse, A.R.; Klentrou, P. Effects of dairy consumption on body composition and bone properties in youth: A systematic review. Curr. Dev. Nutr. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Sotunde, O.F.; Weiler, H.A. Effects of milk and milk-product consumption on growth among children and adolescents aged 6–18 years: A meta-analysis of randomized controlled trials. Adv. Nutr. 2019, 10, 250–261. [Google Scholar] [CrossRef]
- Dougkas, A.; Barr, S.; Reddy, S.; Summerbell, C.D. A critical review of the role of milk and other dairy products in the development of obesity in children and adolescents. Nutr. Res. Rev. 2019, 32, 106–127. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, J.M.; Dibley, M.J.; Sierra, J.; Wallace, B.; Marks, J.S.; Yip, R. Growth of Vegetarian Children: The farm study. Pediatrics 1989, 84, 475–481. [Google Scholar]
- Yen, C.E.; Yen, C.H.; Huang, M.C.; Cheng, C.H.; Huang, Y.C. Dietary intake and nutritional status of vegetarian and omnivorous preschool children and their parents in Taiwan. Nutr. Res. 2008, 28, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Sanders, T. Growth and development of British vegan children. Am. J. Clin. Nutr. 1988, 48, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Weder, S.; Hoffmann, M.; Becker, K.; Alexy, U.; Keller, M. Energy, macronutrient intake, and anthropometrics of vegetarian, vegan, and omnivorous children (1-3 years) in Germany (VeChi diet study). Nutrients 2019, 11, 832. [Google Scholar] [CrossRef]
- Sanders, T.A.B.; Reddy, S. Vegetarian diets and children. Am. J. Clin. Nutr. 1994, 59, 1176S–1181S. [Google Scholar] [CrossRef] [PubMed]
- Van Dusseldorp, M.; Arts, I.C.W.; Bergsma, J.S.; De Jong, N.; Dagnelie, P.C.; Van Staveren, W.A. Catch-Up Growth in Children Fed a Macrobiotic Diet in Early Childhood. J. Nutr. 1996, 126, 2977–2983. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.T.; Andrew, E.M.; Berkey, C.; Valadian, I.; Reed, R.B. Growth in ‘new’ vegetarian preschool children using the Jenns-Bayley curve fitting technique. Am. J. Clin. Nutr. 1983, 37, 815–827. [Google Scholar] [CrossRef]
- Golden, M.H. The role of individual nutrient deficiencies in growth retardation of children as exemplified by zinc and protein. In Linear Growth Retardation in Less Developed Countries; Waterlow, J.C., Ed.; Raven Press: New York, NY, USA, 1988; pp. 143–163. [Google Scholar]
- Swaminathan, S.; Vaz, M.; Kurpad, A.V. Protein intakes in India. Br. J. Nutr. 2012, 108, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Li, K.; Li, H.; Guo, M.; He, J.; Shen, X.; Song, J. Low Habitual Dietary Calcium and Linear Growth from Adolescence to Young Adulthood: Results from the China Health and Nutrition Survey. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.; Lembcke, J.; Morales, E. Quality-Protein Maize as the Sole Source of Dietary Protein. Pediatrics 1990, 85, 85–91. [Google Scholar] [PubMed]
- Gunaratna, N.S.; De Groote, H.; Nestel, P.; Pixley, K.V.; McCabe, G.P. A meta-analysis of community-based studies on quality protein maize. Food Policy 2010, 35, 202–210. [Google Scholar] [CrossRef]
- Wessells, K.R.; Brown, K.H. Estimating the Global Prevalence of Zinc Deficiency: Results Based on Zinc Availability in National Food Supplies and the Prevalence of Stunting. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Millward, D.J.; Truby, H.; Fox, K.R.; Livingstone, M.B.E.; Macdonald, I.A.; Tothill, P. Sex differences in the composition of weight gain and loss in overweight and obese adults. Br. J. Nutr. 2014, 111, 933–943. [Google Scholar] [CrossRef]
- Fotheringham, J.; Weatherley, N.; Kawar, B.; Fogarty, D.G.; Ellam, T. The body composition and excretory burden of lean, obese, and severely obese individuals has implications for the assessment of chronic kidney disease. Kidney Int. 2014, 86, 1221–1228. [Google Scholar] [CrossRef]
- Dériaz, O.; Fournier, G.; Tremblay, A.; Després, J.P.; Bouchard, C. Lean-body-mass composition and resting energy expenditure before and after long term overfeeding. Am. J. Clin. Nutr. 1992, 56, 840–847. [Google Scholar] [CrossRef]
- Murton, A.J.; Marimuthu, K.; Mallinson, J.E.; Selby, A.L.; Smith, K.; Rennie, M.J.; Greenhaff, P.L. Obesity appears to be associated with altered muscle protein synthetic and breakdown responses to increased nutrient delivery in older men, but not reduced muscle mass or contractile function. Diabetes 2015, 64, 3160–3171. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Smith, S.R.; Rood, J.; Martin, C.K.; Most, M.; Brock, C.; Mancuso, S. Effect of Dietary Protein Content on Weight Gain, Energy Expenditure and Body Composition During Overeating. JAMA 2012, 307, 47–55. [Google Scholar] [CrossRef]
- Cameron, J.D.; Sigal, R.J.; Kenny, G.P.; Alberga, A.S.; Prud’homme, D.; Phillips, P.; Doucette, S.; Goldfield, G. Body Composition and Energy Intake—Skeletal Muscle Mass is the Strongest Predictor of Food Intake in Obese Adolescents: The HEARTY Trial. Appl. Physiol. Nutr. Metab. 2016, 41, 611–617. [Google Scholar] [CrossRef]
- Jen, V.; Karagounis, L.G.; Jaddoe, V.W.V.; Franco, O.H.; Voortman, T. Dietary protein intake in school-age children and detailed measures of body composition: The Generation R Study. Int. J. Obes. 2018, 42, 1715–1723. [Google Scholar] [CrossRef]
- Van Vught, A.J.A.H.; Heitmann, B.L.; Nieuwenhuizen, A.G.; Veldhorst, M.A.B.; Andersen, L.B.; Hasselstrom, H.; Brummer, R.J.M.; Westerterp-Plantenga, M.S. Association between intake of dietary protein and 3-year-change in body growth among normal and overweight 6-year-old boys and girls (CoSCIS). Public Health Nutr. 2010, 13, 647–653. [Google Scholar] [CrossRef]
- Harris, C.; Buyken, A.; Von Berg, A.; Berdel, D.; Lehmann, I.; Hoffmann, B.; Koletzko, S.; Koletzko, B.; Heinrich, J.; Standl, M. Prospective associations of meat consumption during childhood with measures of body composition during adolescence: Results from the GINIplus and LISAplus birth cohorts. Nutr. J. 2016, 15, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Assmann, K.E.; Joslowski, G.; Buyken, A.E.; Cheng, G.; Remer, T.; Kroke, A.; Günther, A.L.B. Prospective association of protein intake during puberty with body composition in young adulthood. Obesity 2013, 21, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Albala, C.; Ebbeling, C.B.; Cifuentes, M.; Lera, L.; Bustos, N.; Ludwig, D.S. Effects of replacing the habitual consumption of sugar-sweetened beverages with milk in Chilean children. Am. J. Clin. Nutr. 2008, 88, 605–611. [Google Scholar] [CrossRef][Green Version]
- Merrilees, M.J.; Smart, E.J.; Gilchrist, N.L.; Frampton, C.; Turner, J.G.; Hooke, E.; March, R.L.; Maguire, P. Effects of dairy food supplements on bone mineral density in teenage girls. Eur. J. Nutr. 2000, 39, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Cadogan, J.; Eastell, R.; Jones, N.; Barker, M.E. Milk intake and bone mineral acquisition in adolescent girls: Randomised, controlled intervention trial. Br. Med. J. 1997, 315, 1255–1260. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
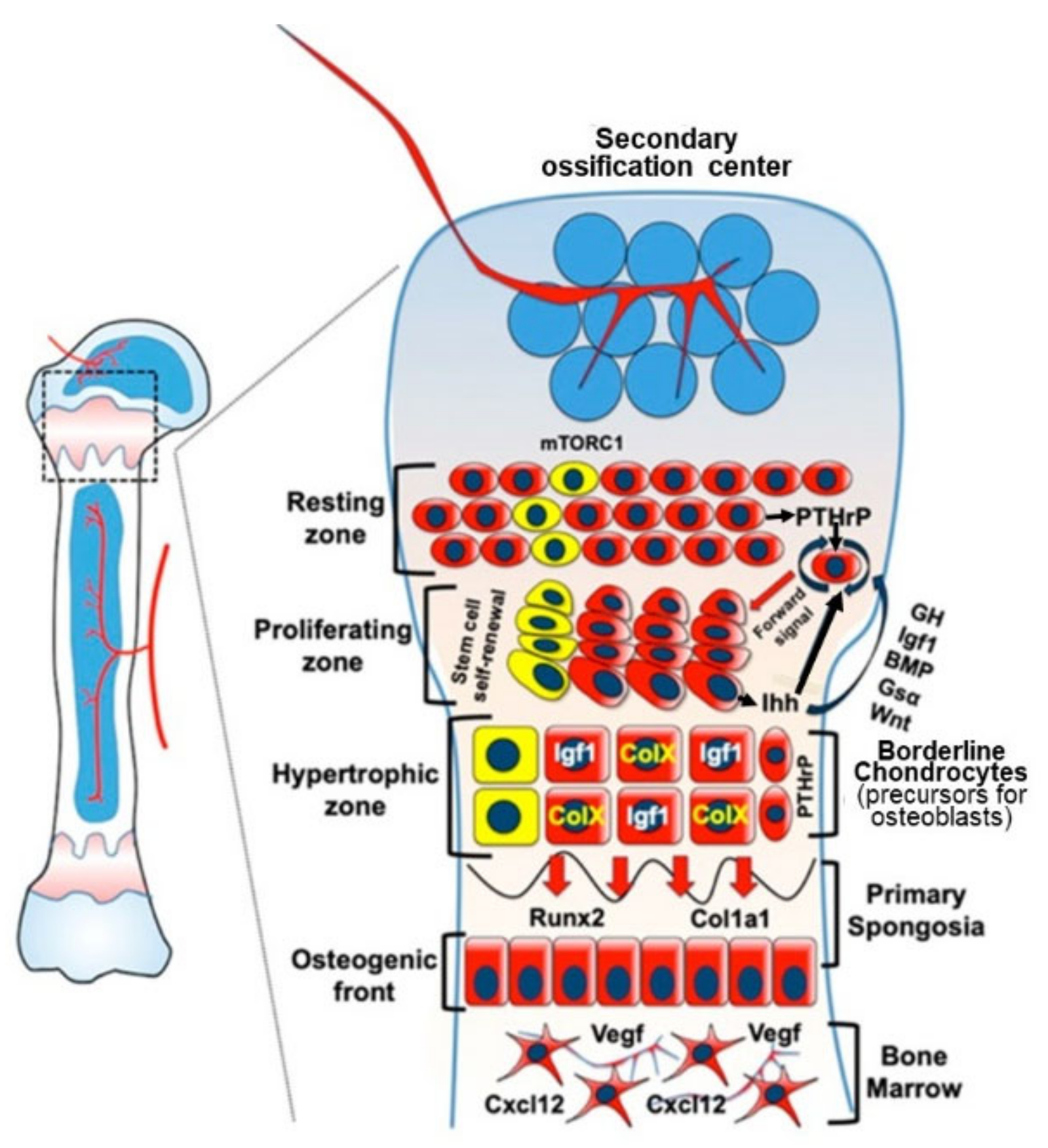{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
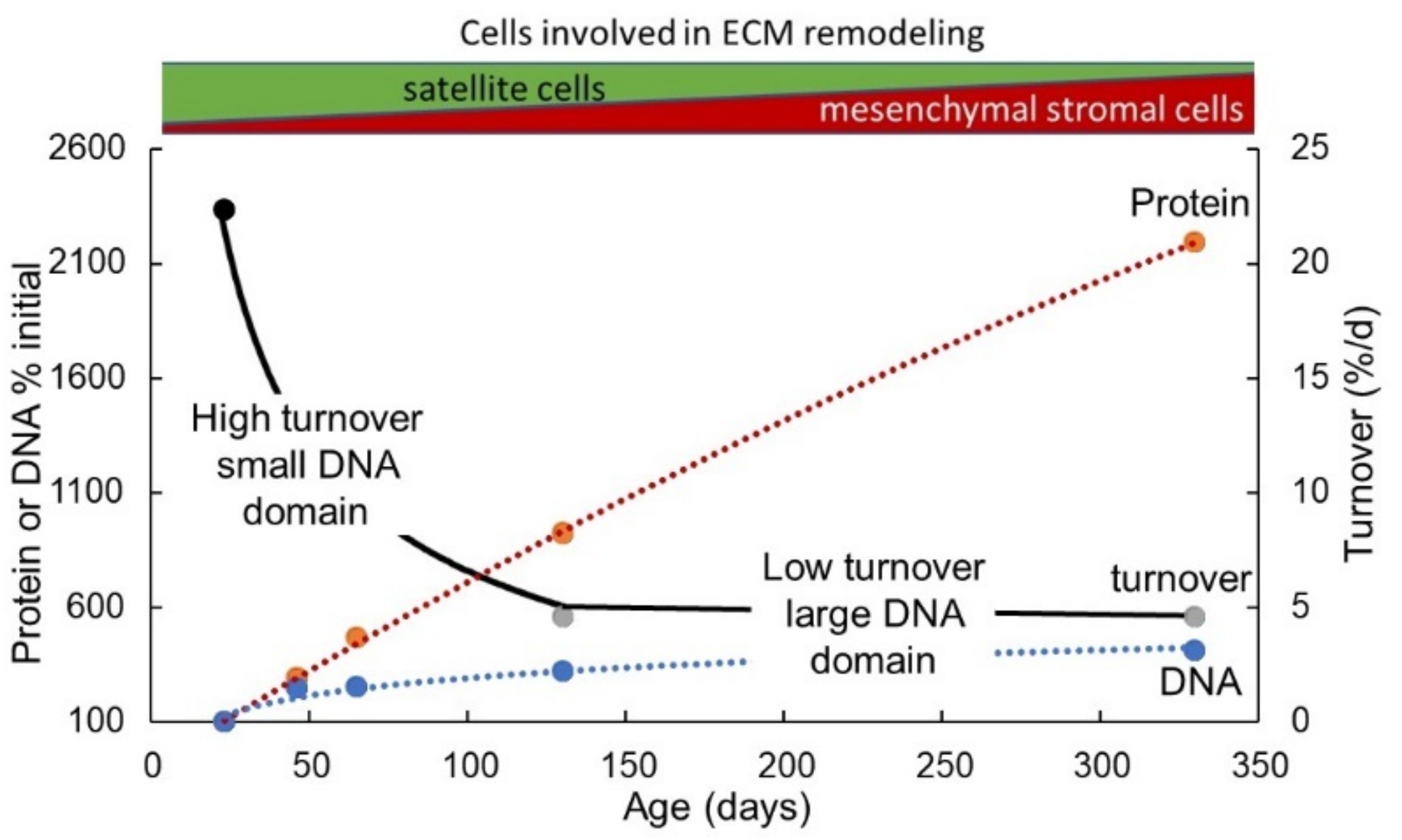{kind=link}
{kind=link}
{kind=link}
{kind=link}
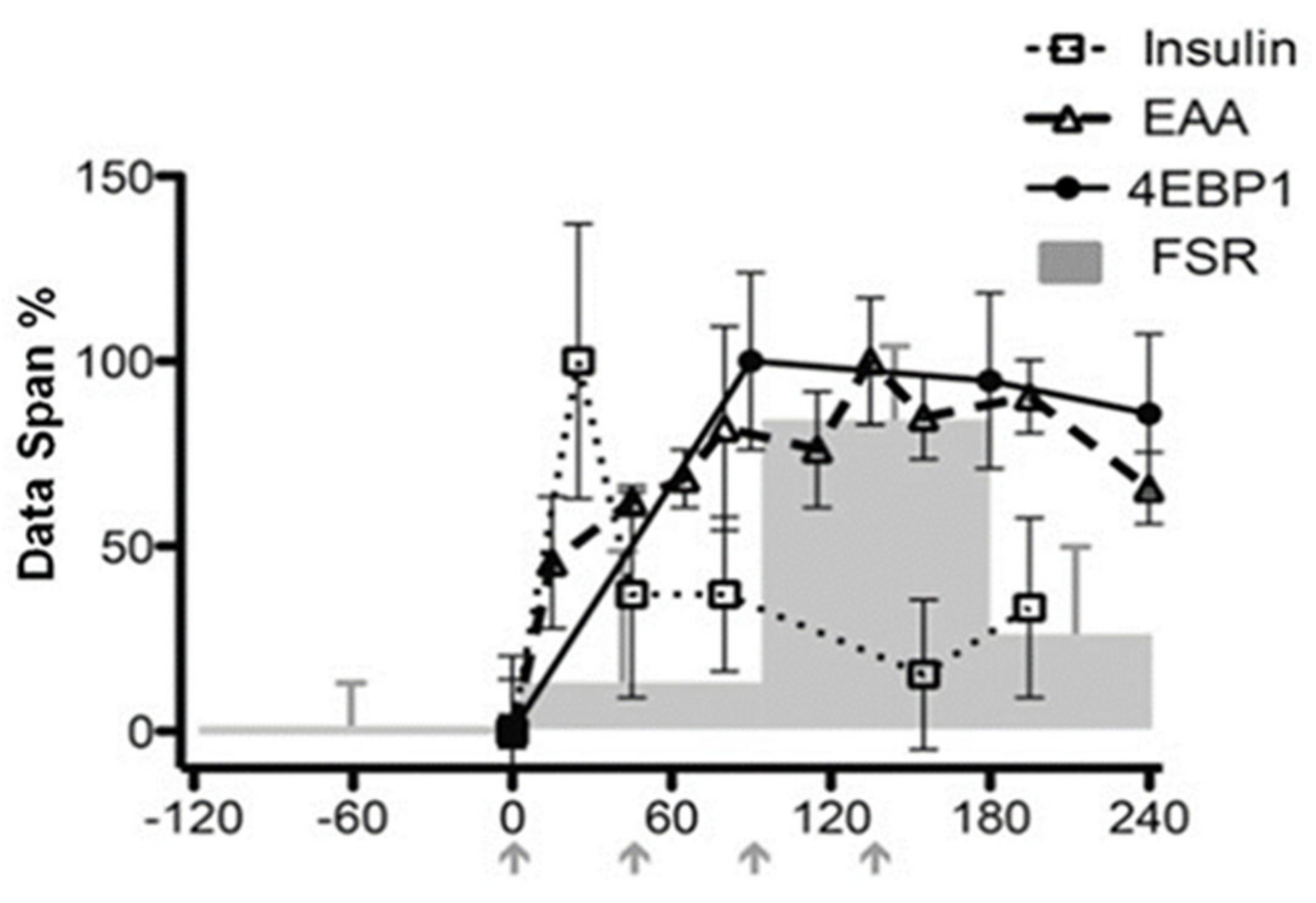{kind=link}
{kind=link}
{kind=link}
| Parameter | Peak Growth Velocity | Growth State at 18 Years |
|---|---|---|
| TRM (months) | ||
| Tibial length (mm) | −20 | complete |
| Total bone cross-sectional area (mm2) | −20 | complete |
| Muscle cross-sectional area (mm2) | −8 | incomplete |
| Cortical cross-sectional area (mm2) | −7 | incomplete |
| Total bone mineral content (mg/mm) | −5 | incomplete |
| Cortical volumetric bone mineral content (mg/cm3) | 0 | incomplete |
| A. Male rat (gastrocnemius + quadriceps muscle) [255] | ||||||||
| Well fed | ||||||||
| Age | Protein | DNA | RNA/ | Protein/ | RNA × 103 | protein synthesis | ||
| days | mg | mg | DNA | DNA | /protein | %day−1 | g.day−1gRNA−1 | g.day−1gDNA−1 |
| 23 | 49 | 0.35 | 2.1 | 141 | 14.8 | 28.6 | 19.2 | 40.3 |
| 46 | 145 | 0.87 | 1.9 | 167 | 11.3 | 16.1 | 14.2 | 27.2 |
| 65 | 230 | 0.9 | 2.1 | 255 | 8.3 | 11.5 | 13.8 | 29.4 |
| 130 | 453 | 1.12 | 2.1 | 402 | 5.2 | 5.3 | 10.1 | 21.2 |
| 330 | 1074 | 1.45 | 3.1 | 740 | 4.2 | 4.9 | 11.5 | 35.7 |
| Marginally malnourished | ||||||||
| 30 | 20 | 0.25 | 1 | 80 | 12.1 | 13.8 | 11.4 | 11.4 |
| 60 | 73 | 0.26 | 2.9 | 282 | 9.9 | 10.9 | 11 | 30.7 |
| 120 | 325 | 0.85 | 2.1 | 384 | 5.5 | 6.5 | 11.8 | 25 |
| 185 | 470 | 1.07 | 2.1 | 438 | 4.4 | 6.3 | 14.4 | 27.6 |
| 330 | 570 | 0.96 | 2.7 | 595 | 4.6 | 4.2 | 9.1 | 25 |
| B. Adult Fowl (Gallus domesticus) ** [257] | ||||||||
| Posterior latissimus dorsi | ||||||||
| 240 | 0.26 | 3.05 | 909 | 3.4 | 6.9 | 20.4 | 62.1 | |
| Anterior latissimus dorsi | ||||||||
| 124 | 0.32 | 2.7 | 384 | 7.6 | 17 | 22.4 | 60.5 | |
| C. Changes, (% initial value), during load-induced hypertrophy of ALD [59] | ||||||||
| Days | Protein | DNA | RNA/ | Protein/ | RNA × 103 | protein synthesis | ||
| mg | mg | DNA | DNA | /protein | %day−1 | g.day−1gRNA−1 | g.day−1gDNA−1 | |
| 1 | 10 | 25 | 11 | −20 | 27 | 103 | 60 | 43 |
| 3 | 38 | 90 | 92 | −23 | 153 | 122 | −14 | 58 |
| 7 | 80 | 150 | 63 | −31 | 125 | 71 | −24 | 26 |
| 28 | 113 | 90 | 56 | 7.6 | 27 | 47 | 13 | 67 |
| 58 | 128 | 130 | 65 | 16 | 11 | nsd *** | nsd | nsd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millward, D.J. Interactions between Growth of Muscle and Stature: Mechanisms Involved and Their Nutritional Sensitivity to Dietary Protein: The Protein-Stat Revisited. Nutrients 2021, 13, 729. https://doi.org/10.3390/nu13030729
Millward DJ. Interactions between Growth of Muscle and Stature: Mechanisms Involved and Their Nutritional Sensitivity to Dietary Protein: The Protein-Stat Revisited. Nutrients. 2021; 13(3):729. https://doi.org/10.3390/nu13030729
Chicago/Turabian StyleMillward, D Joe. 2021. "Interactions between Growth of Muscle and Stature: Mechanisms Involved and Their Nutritional Sensitivity to Dietary Protein: The Protein-Stat Revisited" Nutrients 13, no. 3: 729. https://doi.org/10.3390/nu13030729
APA StyleMillward, D. J. (2021). Interactions between Growth of Muscle and Stature: Mechanisms Involved and Their Nutritional Sensitivity to Dietary Protein: The Protein-Stat Revisited. Nutrients, 13(3), 729. https://doi.org/10.3390/nu13030729