
The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. PMA Treatment
2.3. DNA Extraction
2.4. PacBio Sequencing
2.5. Sequence Processing
2.6. Data Availability
2.7. Statistical Analysis
2.7.1. DNA Quantification
2.7.2. Alpha Diversity
2.7.3. Beta Diversity
2.7.4. Comparison of OTU Relative Abundance
3. Results
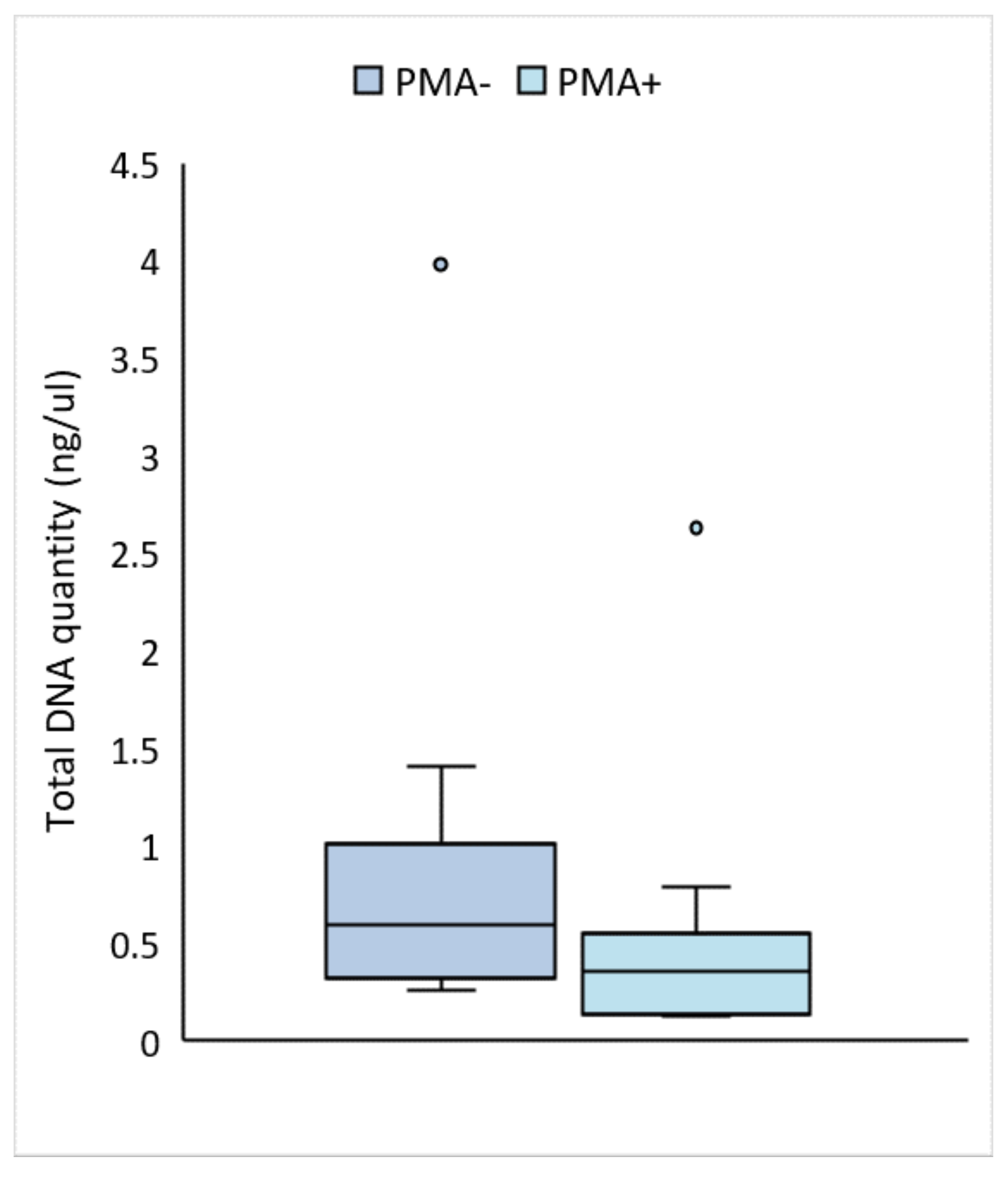
3.1. PMA-Treated Milk Contains a Significantly Lower Concentration of DNA
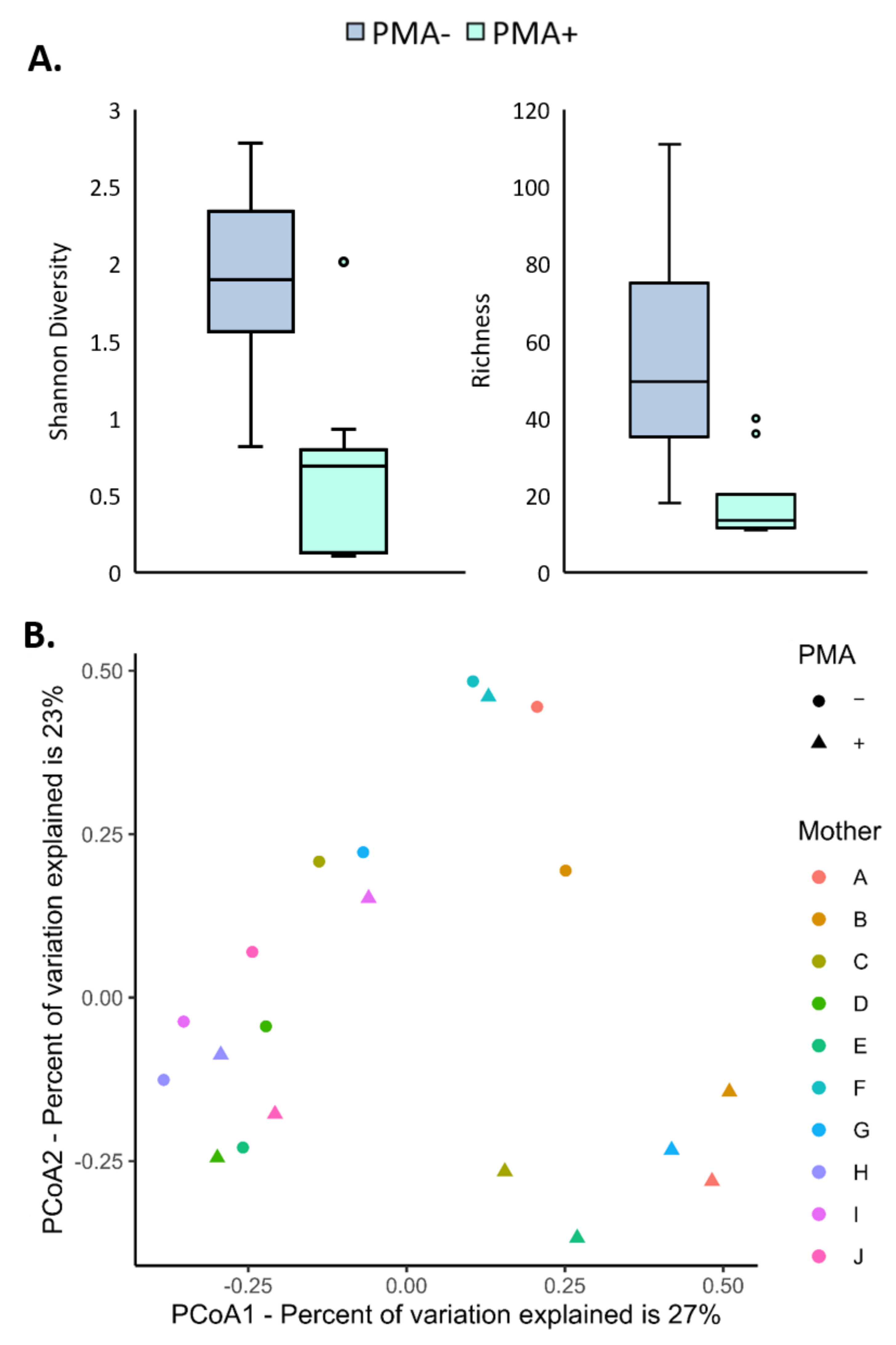
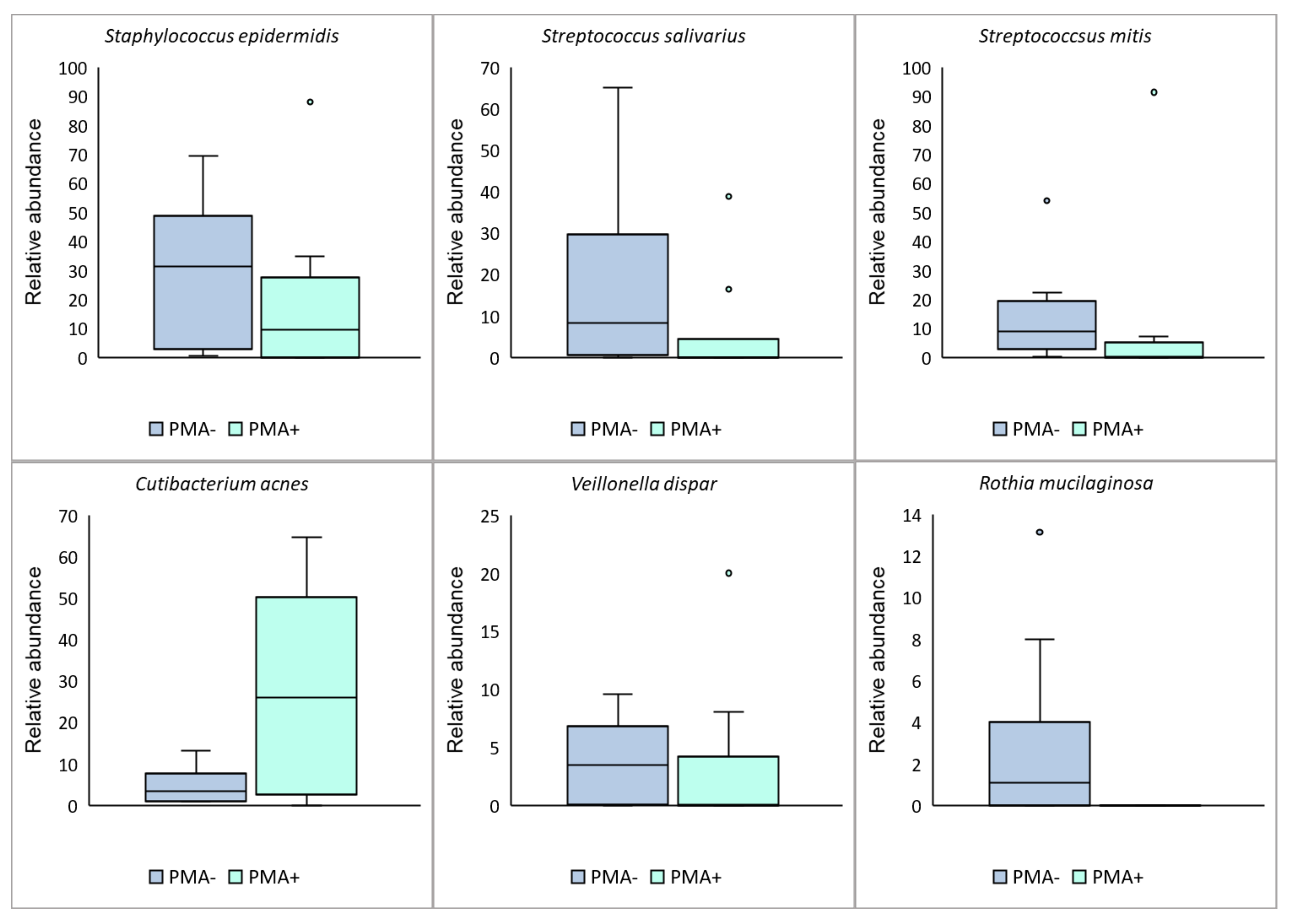
3.2. The Viable Microbiome Differs from the Total Bacterial DNA Profile in Human Milk
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stinson, L.F.; Sindi, A.S.M.; Cheema, A.S.; Lai, C.T.; Muhlhausler, B.S.; Wlodek, M.E.; Payne, M.S.; Geddes, D.T. The human milk microbiome: Who, what, when, where, why, and how? Nutr. Rev. 2021, 79, 529–543. [Google Scholar] [CrossRef]
- Li, L.; Mendis, N.; Trigui, H.; Oliver, J.D.; Faucher, S.P. The importance of the viable but non-culturable state in human bacterial pathogens. Front. Microbiol. 2014, 5, 258. [Google Scholar] [CrossRef] [Green Version]
- Kordy, K.; Gaufin, T.; Mwangi, M.; Li, F.; Cerini, C.; Lee, D.J.; Adisetiyo, H.; Woodward, C.; Pannaraj, P.S.; Tobin, N.H.; et al. Contributions to human breast milk microbiome and enteromammary transfer of Bifidobacterium breve. PLoS ONE 2020, 15, e0219633. [Google Scholar] [CrossRef] [Green Version]
- Moossavi, S.; Sepehri, S.; Robertson, B.; Bode, L.; Goruk, S.; Field, C.J.; Lix, L.M.; de Souza, R.J.; Becker, A.B.; Mandhane, P.J.; et al. Composition and Variation of the Human Milk Microbiota Are Influenced by Maternal and Early-Life Factors. Cell Host Microbe 2019, 25, 324–335.e4. [Google Scholar] [CrossRef] [Green Version]
- Lackey, K.A.; Williams, J.E.; Meehan, C.L.; Zachek, J.A.; Benda, E.D.; Price, W.J.; Foster, J.A.; Sellen, D.W.; Kamau-Mbuthia, E.W.; Kamundia, E.W.; et al. What’s Normal? Microbiomes in Human Milk and Infant Feces Are Related to Each Other but Vary Geographically: The INSPIRE Study. Front. Nutr. 2019, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Igumbor, E.O.; Mukura, R.D.; Makandiramba, B.; Chihota, V. Storage of breast milk: Effect of temperature and storage duration on microbial growth. Cent. Afr. J. Med. 2000, 46, 247–251. [Google Scholar] [CrossRef]
- Sosa, R.; Barness, L. Bacterial growth in refrigerated human milk. Am. J. Dis. Child. 1987, 141, 111–112. [Google Scholar] [CrossRef]
- Rodriguez-Cruz, M.; Alba, C.; Aparicio, M.; Checa, M.A.; Fernandez, L.; Rodriguez, J.M. Effect of Sample Collection (Manual Expression vs. Pumping) and Skimming on the Microbial Profile of Human Milk Using Culture Techniques and Metataxonomic Analysis. Microorganisms 2020, 8, 1278. [Google Scholar] [CrossRef] [PubMed]
- Stinson, L.F.; Ma, J.; Rea, A.; Dymock, M.; Geddes, D.T. Centrifugation does not remove bacteria from the fat fraction of human milk. Sci. Rep. 2021, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Cheema, A.S.; Stinson, L.F.; Lai, C.T.; Geddes, D.T.; Payne, M.S. DNA extraction method influences human milk bacterial profiles. J. Appl. Microbiol. 2021, 130, 142–156. [Google Scholar] [CrossRef] [PubMed]
- Douglas, C.A.; Ivey, K.L.; Papanicolas, L.E.; Best, K.P.; Muhlhausler, B.S.; Rogers, G.B. DNA extraction approaches substantially influence the assessment of the human breast milk microbiome. Sci. Rep. 2020, 10, 123. [Google Scholar] [CrossRef]
- Perrella, S.; Gridneva, Z.; Lai, C.T.; Stinson, L.; George, A.; Bilston-John, S.; Geddes, D. Human milk composition promotes optimal infant growth, development and health. Semin. Perinatol. 2021, 45, 151380. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.L.; Craft, K.M.; Doster, R.S.; Weitkamp, J.H.; Aronoff, D.M.; Gaddy, J.A.; Townsend, S.D. Antimicrobial and Antibiofilm Activity of Human Milk Oligosaccharides against Streptococcus agalactiae, Staphylococcus aureus, and Acinetobacter baumannii. ACS Infect. Dis. 2018, 4, 315–324. [Google Scholar] [CrossRef]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef] [Green Version]
- Nocker, A.; Sossa-Fernandez, P.; Burr, M.D.; Camper, A.K. Use of propidium monoazide for live/dead distinction in microbial ecology. Appl. Environ. Microbiol. 2007, 73, 5111–5117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Boyce, M.C.; Payne, M.S.; Keelan, J.A. The Not-so-Sterile Womb: Evidence That the Human Fetus Is Exposed to Bacteria Prior to Birth. Front. Microbiol. 2019, 10, 1124. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Ho, N.T.; Li, F.; Wang, S.; Kuhn, L. metamicrobiomeR: An R package for analysis of microbiome relative abundance data using zero-inflated beta GAMLSS and meta-analysis across studies using random effects models. BMC Bioinform. 2019, 20, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stinson, L.F.; Keelan, J.A.; Payne, M.S. Characterization of the bacterial microbiome in first-pass meconium using propidium monoazide (PMA) to exclude nonviable bacterial DNA. Lett. Appl. Microbiol. 2019, 68, 378–385. [Google Scholar] [CrossRef]
- Young, G.R.; Smith, D.L.; Embleton, N.D.; Berrington, J.E.; Schwalbe, E.C.; Cummings, S.P.; van der Gast, C.J.; Lanyon, C. Reducing Viability Bias in Analysis of Gut Microbiota in Preterm Infants at Risk of NEC and Sepsis. Front. Cell. Infect. Microbiol. 2017, 7, 237. [Google Scholar] [CrossRef]
- Weinmaier, T.; Probst, A.J.; La Duc, M.T.; Ciobanu, D.; Cheng, J.F.; Ivanova, N.; Rattei, T.; Vaishampayan, P. A viability-linked metagenomic analysis of cleanroom environments: Eukarya, prokaryotes, and viruses. Microbiome 2015, 3, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishampayan, P.; Probst, A.J.; La Duc, M.T.; Bargoma, E.; Benardini, J.N.; Andersen, G.L.; Venkateswaran, K. New perspectives on viable microbial communities in low-biomass cleanroom environments. ISME J. 2013, 7, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checinska, A.; Probst, A.J.; Vaishampayan, P.; White, J.R.; Kumar, D.; Stepanov, V.G.; Fox, G.E.; Nilsson, H.R.; Pierson, D.L.; Perry, J.; et al. Microbiomes of the dust particles collected from the International Space Station and Spacecraft Assembly Facilities. Microbiome 2015, 3, 50. [Google Scholar] [CrossRef] [Green Version]
- Cheema, A.S.; Lai, C.T.; Dymock, M.; Rae, A.; Geddes, D.T.; Payne, M.S.; Stinson, L.F. Impact of expression mode and timing of sample collection, relative to milk ejection, on human milk bacterial DNA profiles. J. Appl. Microbiol. 2021, 131, 988–995. [Google Scholar] [CrossRef]
- Biagi, E.; Aceti, A.; Quercia, S.; Beghetti, I.; Rampelli, S.; Turroni, S.; Soverini, M.; Zambrini, A.V.; Faldella, G.; Candela, M.; et al. Microbial Community Dynamics in Mother’s Milk and Infant’s Mouth and Gut in Moderately Preterm Infants. Front. Microbiol. 2018, 9, 2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, J.B.; Adams, R.I.; Roman, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Schrodinger’s microbes: Tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 2017, 5, 86. [Google Scholar] [CrossRef] [PubMed]
- Ben-Amor, K.; Heilig, H.; Smidt, H.; Vaughan, E.E.; Abee, T.; de Vos, W.M. Genetic diversity of viable, injured, and dead fecal bacteria assessed by fluorescence-activated cell sorting and 16S rRNA gene analysis. Appl. Environ. Microbiol. 2005, 71, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Witkowska-Zimny, M.; Kaminska-El-Hassan, E. Cells of human breast milk. Cell. Mol. Biol. Lett. 2017, 22, 11. [Google Scholar] [CrossRef]
- Cabrera-Rubio, R.; Collado, M.C.; Laitinen, K.; Salminen, S.; Isolauri, E.; Mira, A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am. J. Clin. Nutr. 2012, 96, 544–551. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, E.; Brereton, N.J.B.; Li, C.; Lopez Leyva, L.; Solomons, N.W.; Agellon, L.B.; Scott, M.E.; Koski, K.G. Distinct Changes Occur in the Human Breast Milk Microbiome Between Early and Established Lactation in Breastfeeding Guatemalan Mothers. Front. Microbiol. 2021, 12, 557180. [Google Scholar] [CrossRef]
- Khodayar-Pardo, P.; Mira-Pascual, L.; Collado, M.C.; Martinez-Costa, C. Impact of lactation stage, gestational age and mode of delivery on breast milk microbiota. J. Perinatol. 2014, 34, 599–605. [Google Scholar] [CrossRef]
- Milani, C.; Mancabelli, L.; Lugli, G.A.; Duranti, S.; Turroni, F.; Ferrario, C.; Mangifesta, M.; Viappiani, A.; Ferretti, P.; Gorfer, V.; et al. Exploring Vertical Transmission of Bifidobacteria from Mother to Child. Appl. Environ. Microbiol. 2015, 81, 7078–7087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asnicar, F.; Manara, S.; Zolfo, M.; Truong, D.T.; Scholz, M.; Armanini, F.; Ferretti, P.; Gorfer, V.; Pedrotti, A.; Tett, A.; et al. Studying Vertical Microbiome Transmission from Mothers to Infants by Strain-Level Metagenomic Profiling. mSystems 2017, 2, e00164-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duranti, S.; Lugli, G.A.; Mancabelli, L.; Armanini, F.; Turroni, F.; James, K.; Ferretti, P.; Gorfer, V.; Ferrario, C.; Milani, C.; et al. Maternal inheritance of bifidobacterial communities and bifidophages in infants through vertical transmission. Microbiome 2017, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Dzidic, M.; Mira, A.; Artacho, A.; Abrahamsson, T.R.; Jenmalm, M.C.; Collado, M.C. Allergy development is associated with consumption of breastmilk with a reduced microbial richness in the first month of life. Pediatr. Allergy Immunol. 2020, 31, 250–257. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stinson, L.F.; Trevenen, M.L.; Geddes, D.T. The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile. Nutrients 2021, 13, 4445. https://doi.org/10.3390/nu13124445
Stinson LF, Trevenen ML, Geddes DT. The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile. Nutrients. 2021; 13(12):4445. https://doi.org/10.3390/nu13124445
Chicago/Turabian StyleStinson, Lisa F., Michelle L. Trevenen, and Donna T. Geddes. 2021. "The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile" Nutrients 13, no. 12: 4445. https://doi.org/10.3390/nu13124445
APA StyleStinson, L. F., Trevenen, M. L., & Geddes, D. T. (2021). The Viable Microbiome of Human Milk Differs from the Metataxonomic Profile. Nutrients, 13(12), 4445. https://doi.org/10.3390/nu13124445