
Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response

 ,
,  , ,
, ,  ,
,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
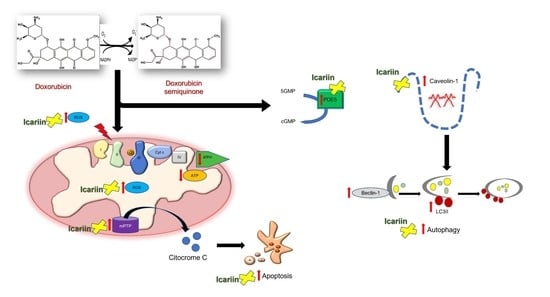
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
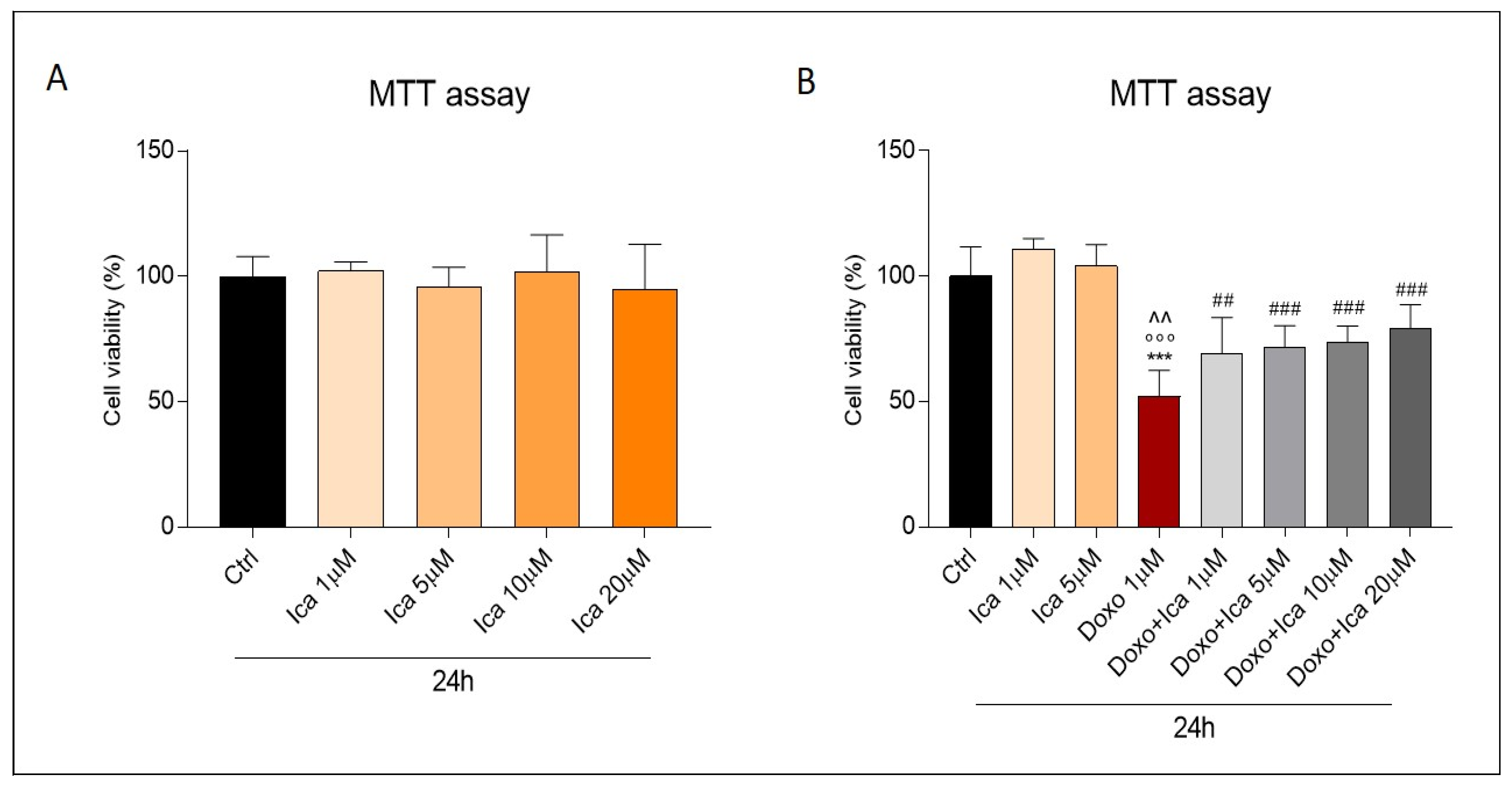
- The cells were treated with different concentrations (1 µM, 5 µM, 10 µM, 20 µM) of Ica (Sigma Aldrich # I1286) and incubated for 24 h to evaluate the related viability.
- Increasing concentrations of Ica (1 µM, 5 µM, 10 µM, 20 µM) were used to pretreat H9c2 for 3 h. Subsequently, they were exposed to 1 µM Doxo (Sigma Aldrich # D1515) for an additional 24 h to assess relative viability.
- Ica concentrations of 1 µM and 5 µM were used to pretreat H9c2 for 3 h, which were subsequently exposed to 1 µM Doxo for an additional 24 h to evaluate ROS production. The same experimental design was used to evaluate cell viability, immunofluorescence staining and protein expression levels.
2.2. MTT Assay
2.3. Intracellular ROS Detection
2.4. Apoptosis
2.5. Mitochondrial Permeability Transition Pore
2.6. MitoSOX
2.7. Protein Extraction and SDS-PAGE Western Blot Analysis
2.8. PDE5a Activity Assay
2.9. Statistical Analysis
3. Results
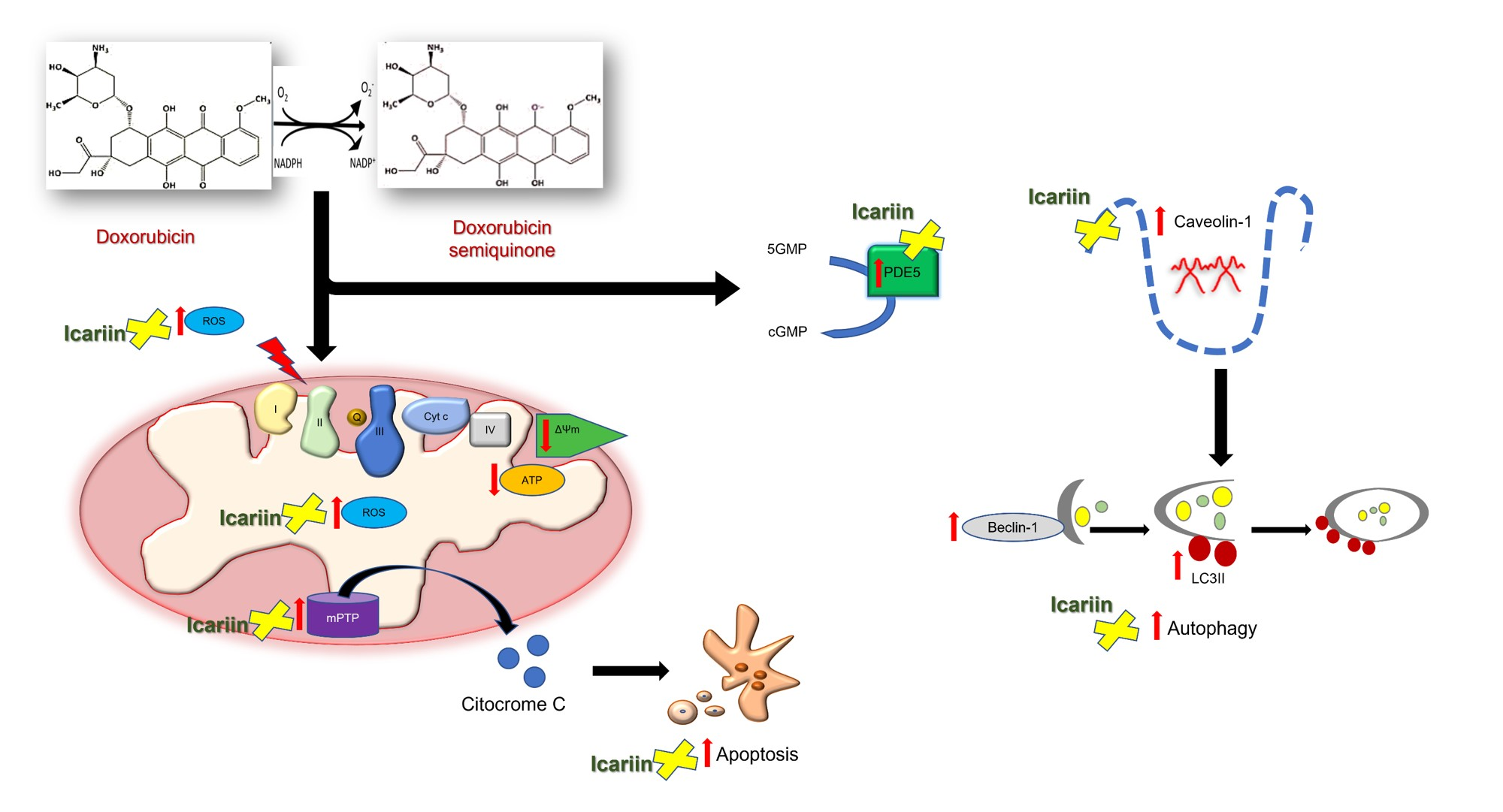
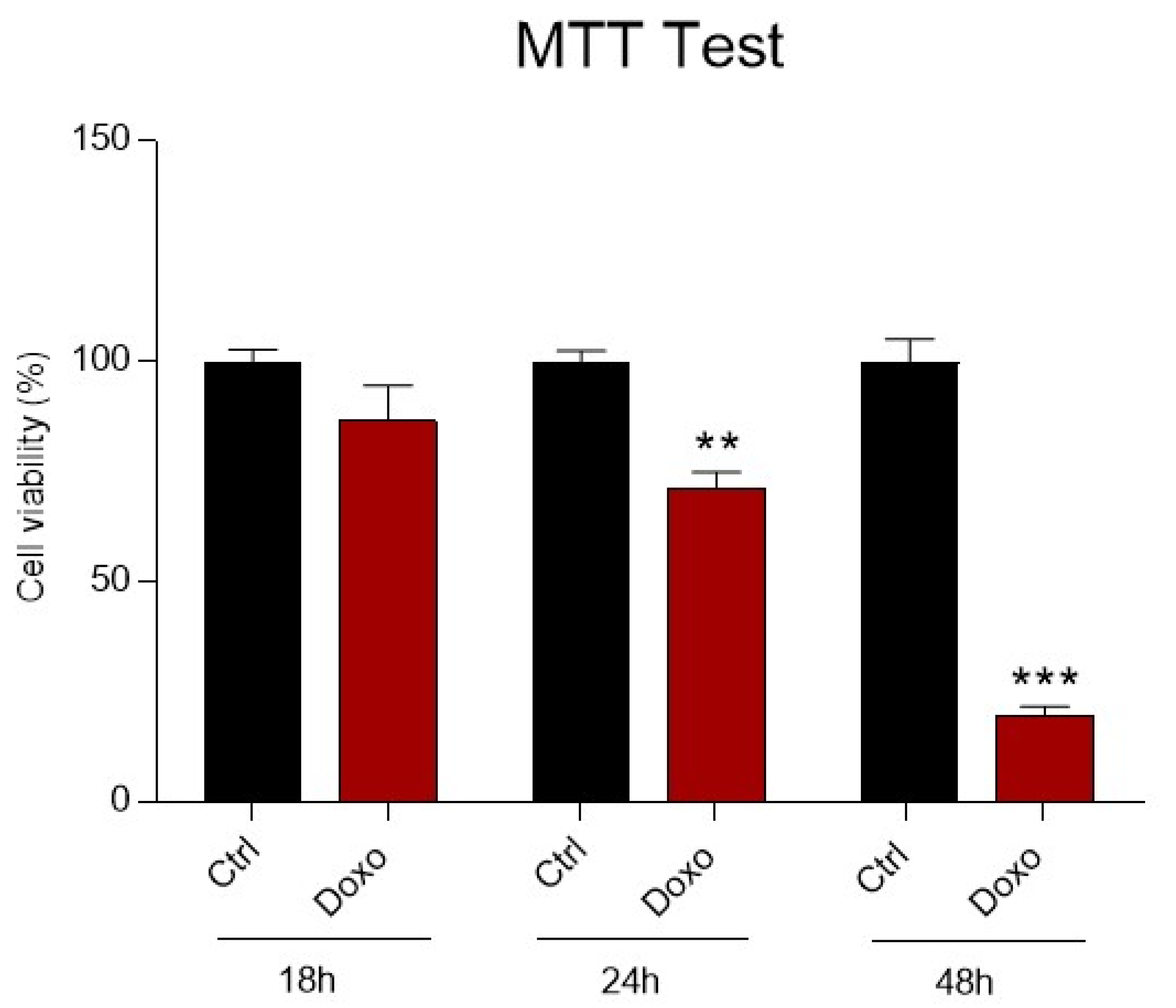
3.1. The Detrimental Effects of Doxo on Mitochondrial Metabolism and Cell Viability in H9c2 Cardiomyoblasts
3.2. The Protective Effects of Ica on Cell Viability in Doxo Treated H9c2 Cells
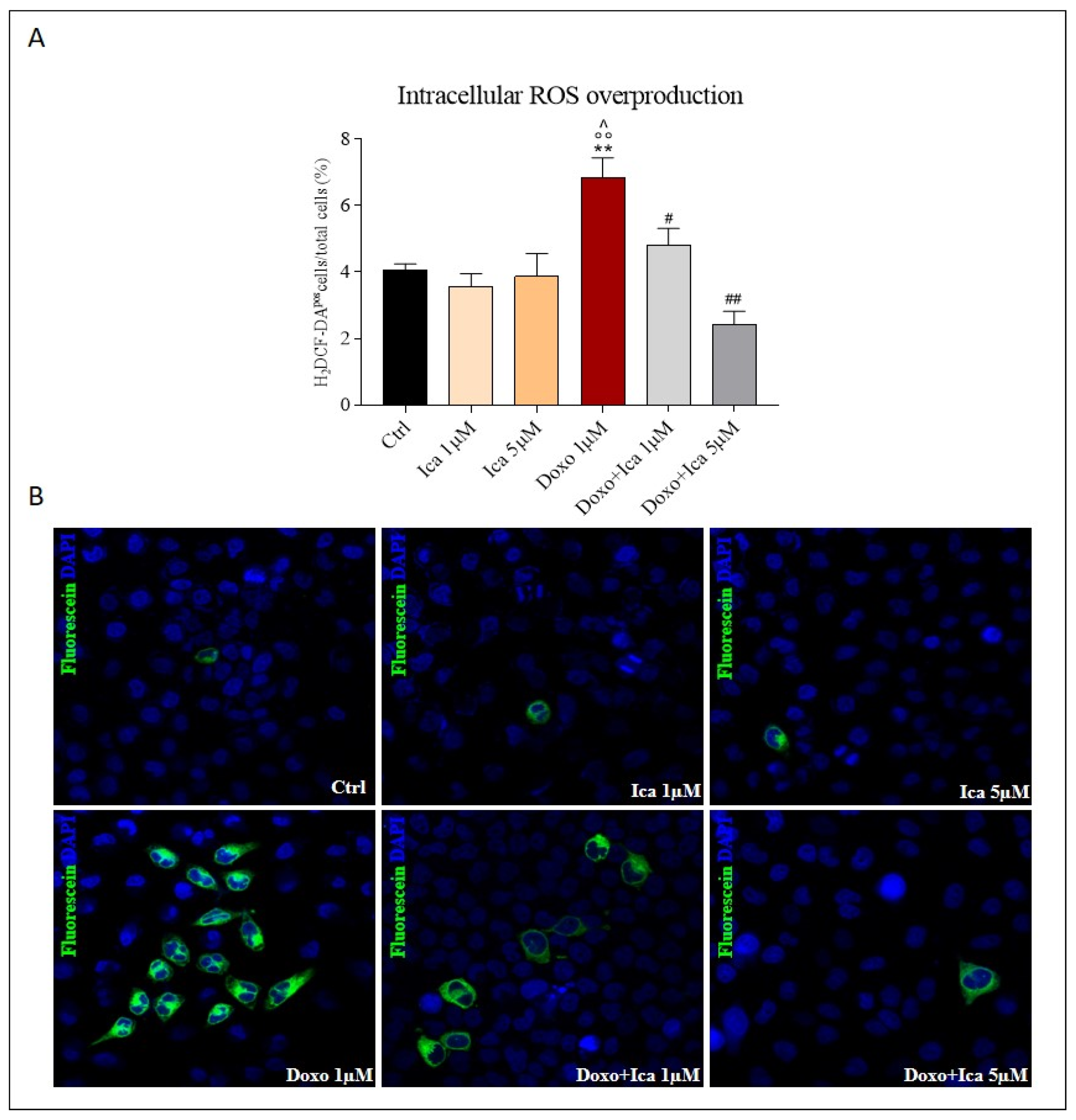
3.3. Ica Reduces Doxo-Induced ROS Overproduction
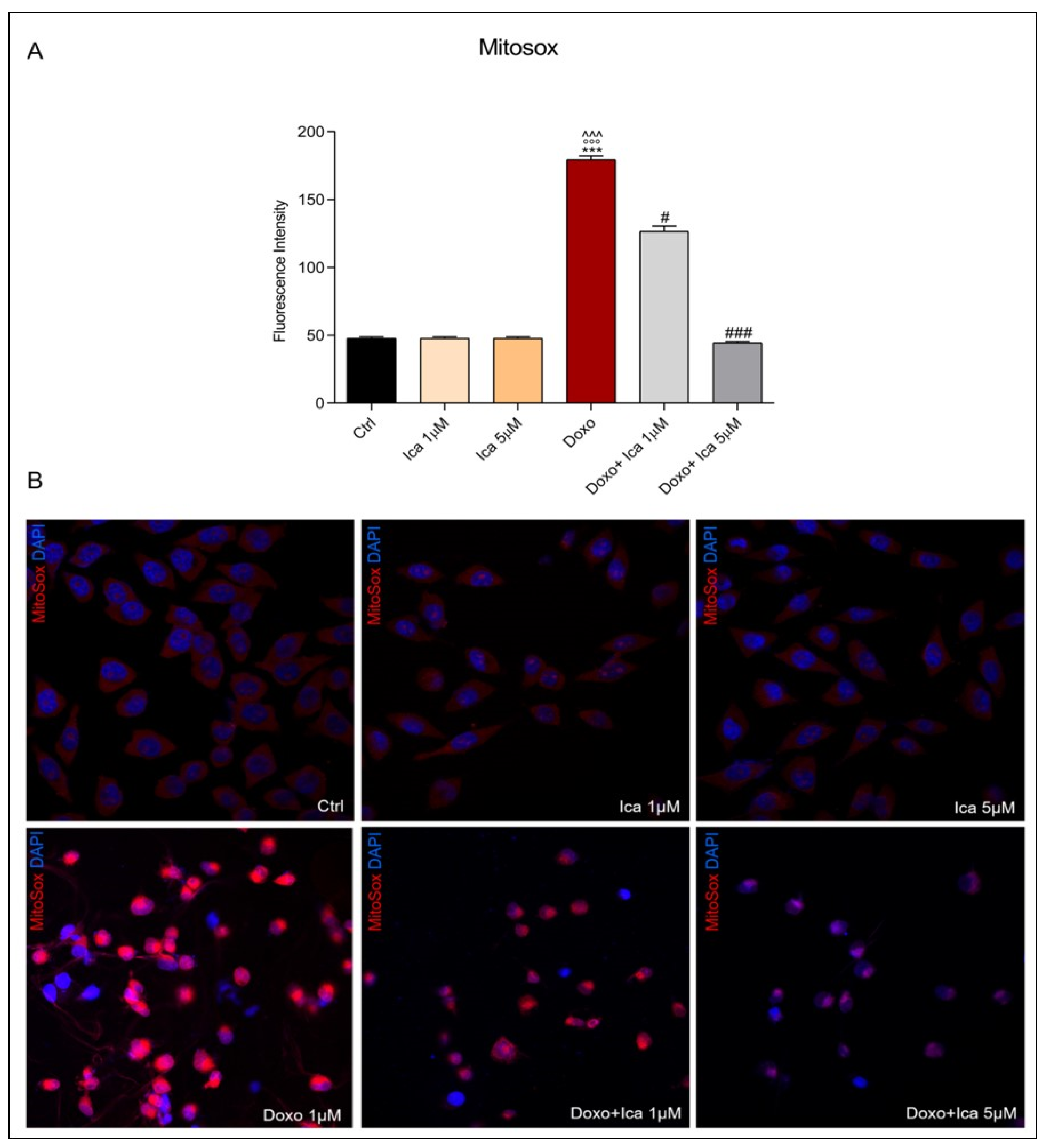
3.4. Ica Inhibits Doxo-Induced O2− Overproduction
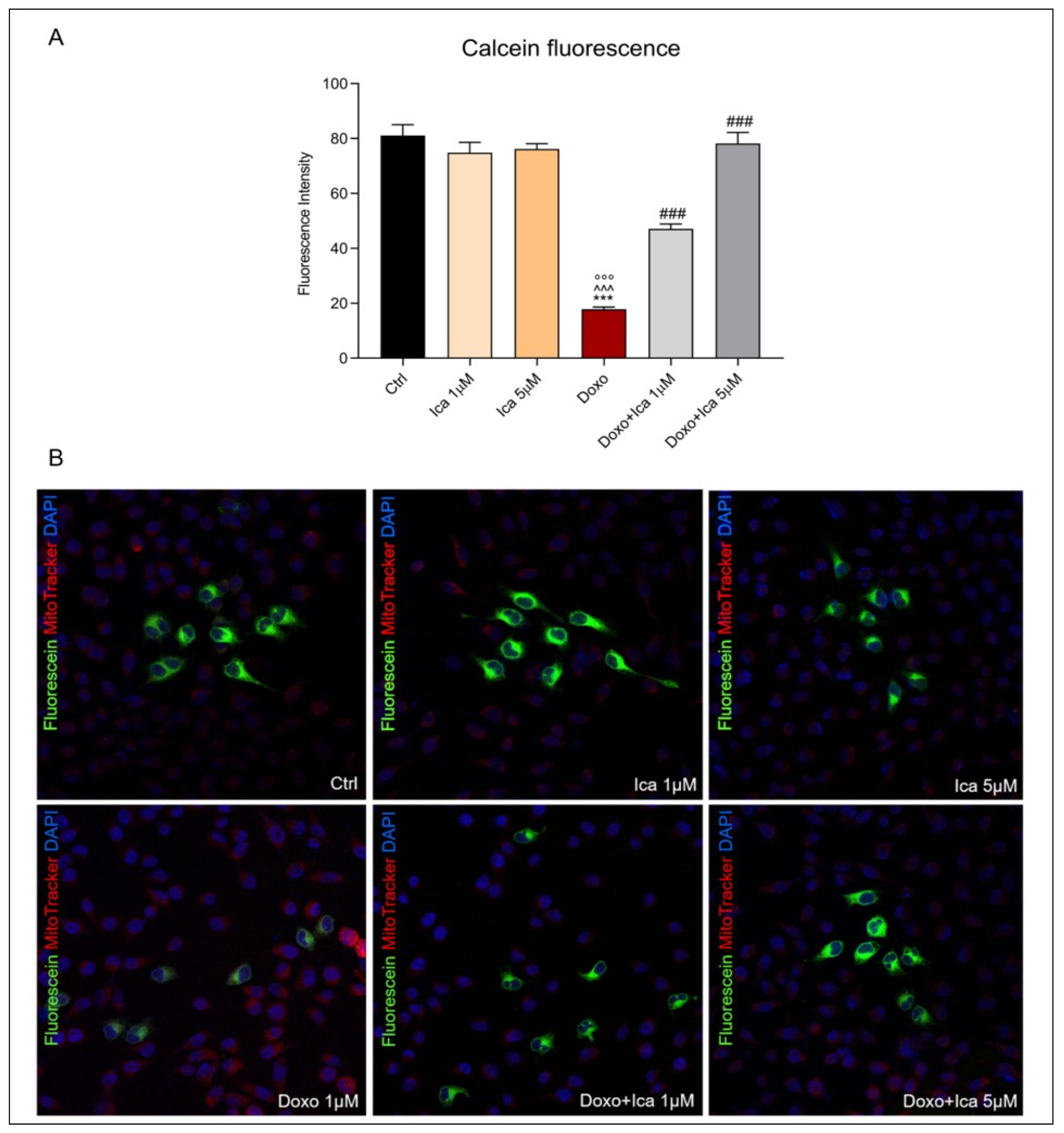
3.5. Modulatory Effect of Ica on mPTP Opening in Doxo-Treated H9c2 Cells
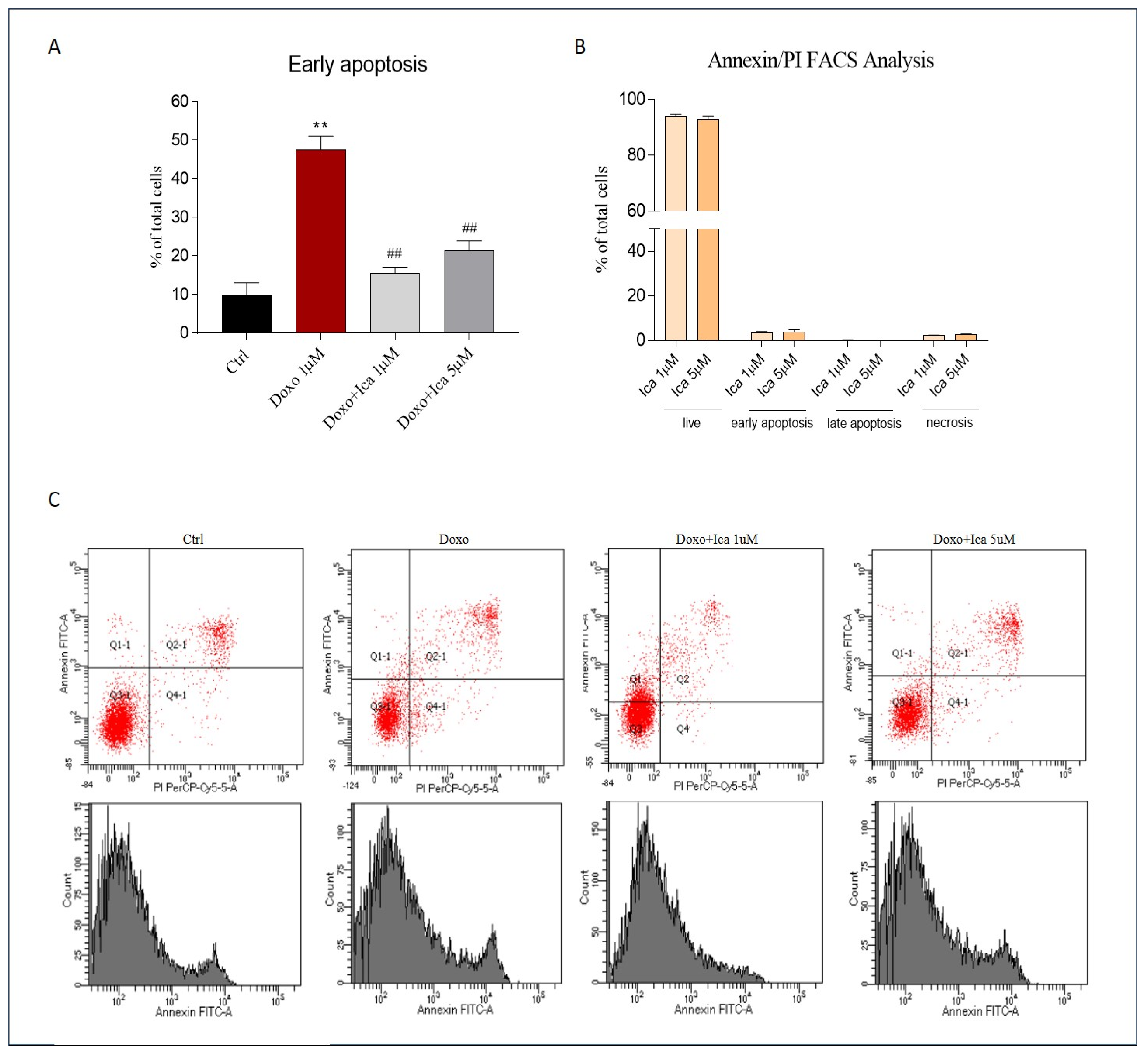
3.6. The Protective Effect of Ica against Doxo-Induced Apoptotic Cell Death
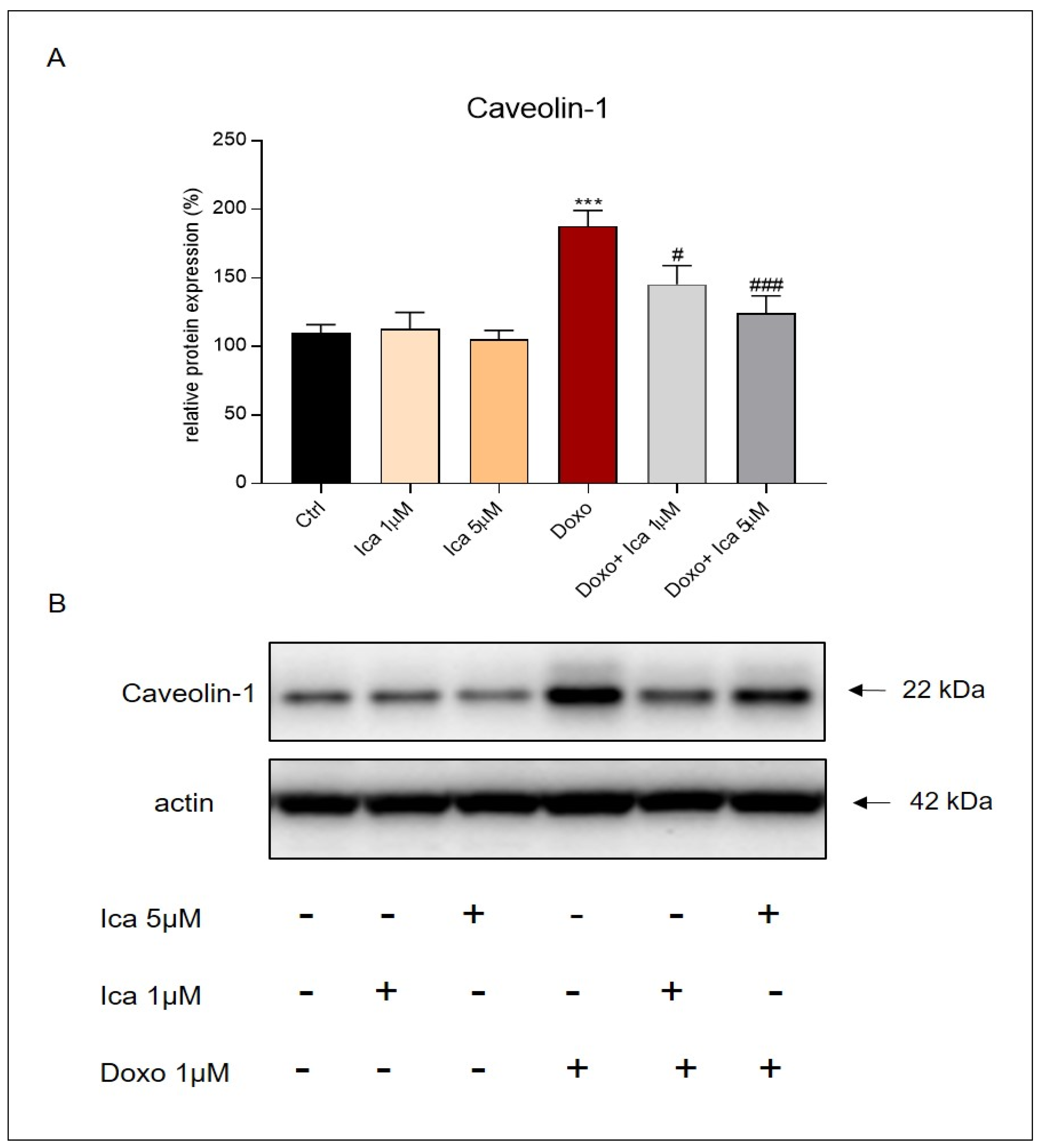
3.7. The Role of Ica on Cav-1 Expression Levels in Doxo-Induced Cardiotoxicity
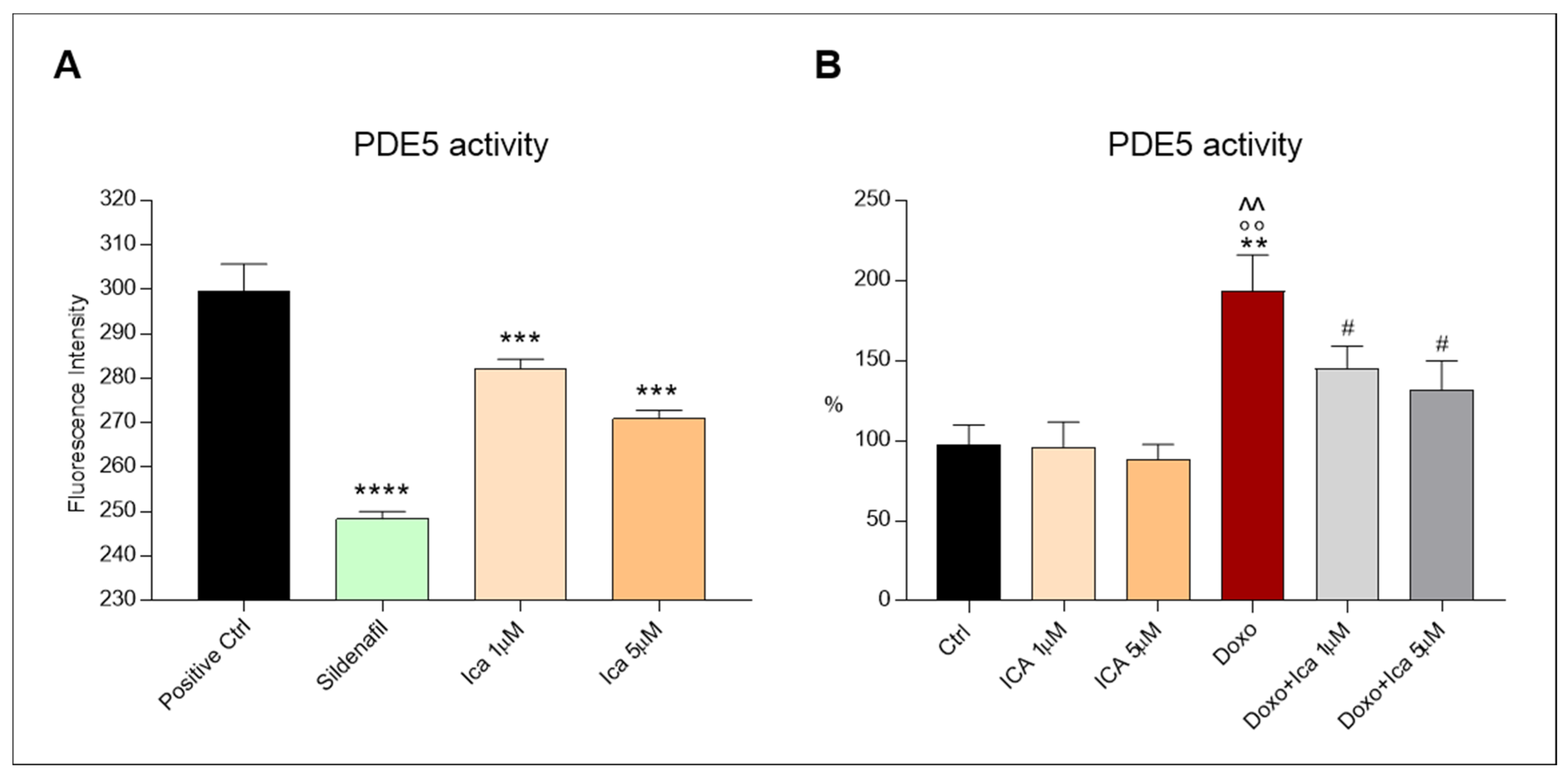
3.8. Ica Inhibits PDE5a Activity in Doxo-Treated H9c2 Cells
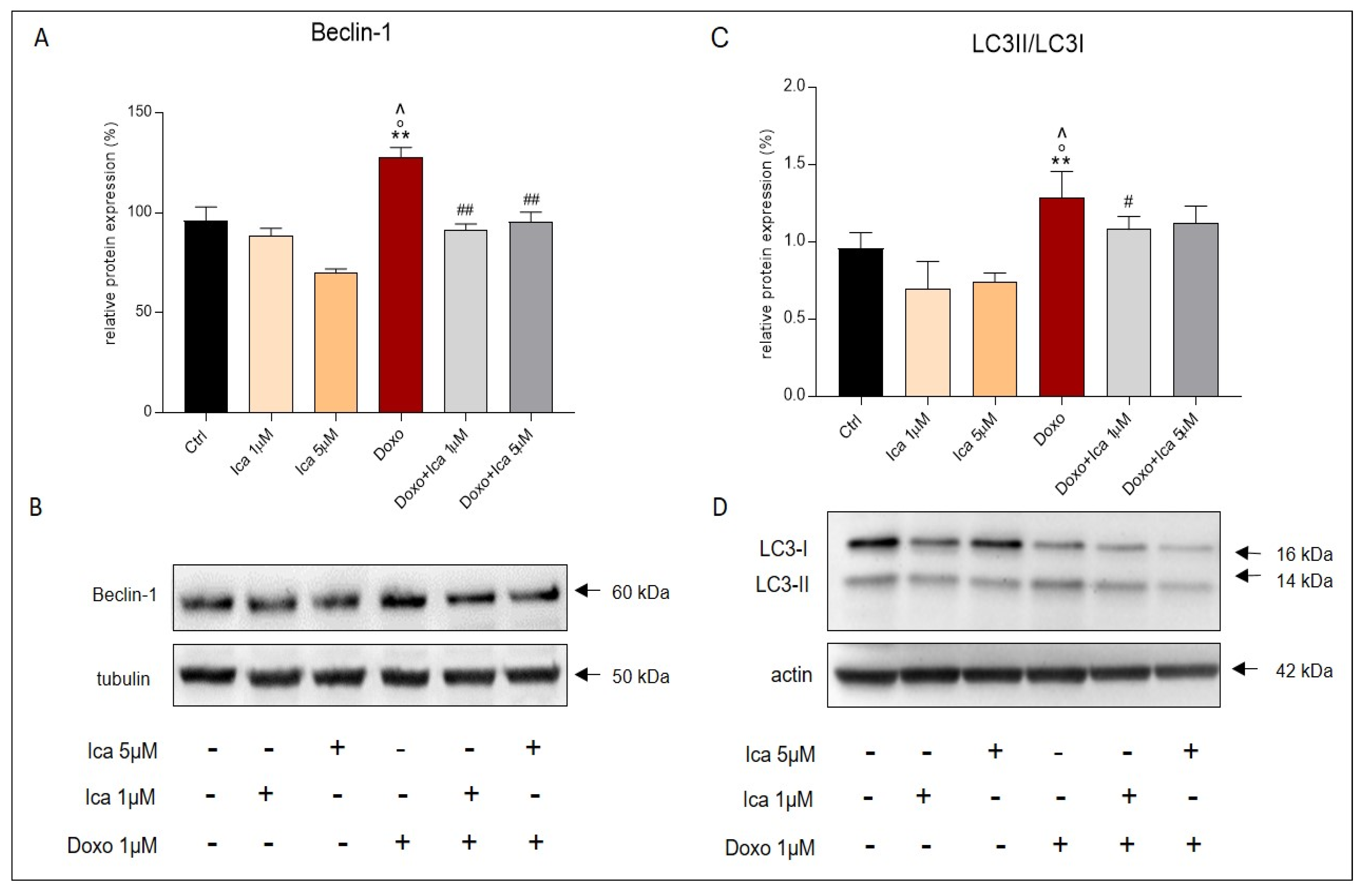
3.9. The Protective Role of Ica in the Autophagic Pathway in Doxo-Treated H9c2 Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Doxo | doxorubicin |
| ROS | reactive oxygen species |
| Ica | icariin |
| CVDs | cardiovascular diseases |
| HF | heart failure |
| mPTP | mitochondrial permeability transition pore |
| ETC | electron transport chain |
| CypD | cyclophilin d |
| Ca2+ | calcium |
| RNS | reactive nitrogen species |
| O2− | anion superoxide |
| NO | nitric oxide |
| I/R | ischemia-reperfusion |
| sGC | soluble guanylyl cyclase |
| cGMP | cyclic gmp |
| PDE5a | phosphodiesterase 5a |
References
- Aleman, B.M.; Moser, E.C.; Nuver, J.; Suter, T.M.; Maraldo, M.V.; Specht, L.; Vrieling, C.; Darby, S.C. Cardiovascular disease after cancer therapy. Eur. J. Cancer Suppl. 2014, 12, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Sturgeon, K.M.; Deng, L.; Bluethmann, S.M.; Zhou, S.; Trifiletti, D.M.; Jiang, C. A population-based study of cardio-vascular disease mortality risk in US cancer patients. Eur. Heart J. 2019, 40, 3889–3897. [Google Scholar] [CrossRef]
- Blaes, A.H.; Shenoy, C. Is it time to include cancer in cardiovascular risk prediction tools? Lancet 2019, 394, 986–988. [Google Scholar] [CrossRef]
- McGowan, J.V.; Chung, R.; Maulik, A.; Piotrowska, I.; Walker, J.M.; Yellon, D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017, 31, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-Hydroxydaunomycin, a new antitumor antibiotic fromS. peucetius var.caesius. Biotechnol. Bioeng. 2000, 67, 704–713. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altamana, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharm. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Rahman, A.M.; Yusuf, S.W.; Ewer, M.S. Anthracycline-induced cardiotoxicity and the cardiac-sparing effect of liposomal formulation. Int. J. Nanomed. 2007, 2, 567–583. [Google Scholar]
- dos Santos, D.B.; dos Santos Goldenberg, R.C. Doxorubicin-Induced Cardiotoxicity: From Mechanisms to Development of Efficient Therapy. In Wenyong Tan Cardiotoxicity; IntechOpen Limited: London, UK, 2018. [Google Scholar]
- Volkova, M.; Russell, R., III. Anthracycline Cardiotoxicity: Prevalence, Pathogenesis and Treatment. Curr. Cardiol. Rev. 2011, 7, 214–220. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular Advances and Pharmacologic Developments in Antitumor Activity and Cardiotoxicity. Pharm. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Granados-Principal, S.; Ramirez-Tortosa, C.L.; Sanchez-Rovira, P.; Ramirez-Tortosa, M.C. New advances in molecular mechanisms and the prevention of adriamycin toxicity by antioxidant nutrients. Food Chem. Toxicol. 2010, 48, 1425–1438. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant Polyphenols as Dietary Antioxidants in Human Health and Disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Brenner, C.; Moulin, M. Physiological Roles of the Permeability Transition Pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Wacquier, B.; Combettes, L.; Dupont, G. Dual dynamics of mitochondrial permeability transition pore opening. Sci. Rep. 2020, 10, 10. [Google Scholar] [CrossRef]
- Hurst, S.; Hoek, J.; Sheu, S.-S. Mitochondrial Ca2+ and regulation of the permeability transition pore. J. Bioenergy Biomembr. 2017, 49, 27–47. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Yang, J.J.; Zhang, H.S. Carvedilol (CAR) combined with carnosic acid (CAA) attenuates doxorubi-cin-induced cardiotoxicity by suppressing excessive oxidative stress, inflammation, apoptosis and autophagy. Biomed. Pharm. 2019, 109, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Liu, J.; Nguyen, T.; Liu, C.; Sun, J.; Teng, Y.; Fergusson, M.M.; Rovira, I.I.; Allen, M.; Springer, D.A.; et al. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat. Cell Biol. 2013, 15, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26. [Google Scholar] [CrossRef]
- Gharanei, M.; Hussain, A.; Janneh, O.; Maddock, H.L. Doxorubicin induced myocardial injury is exacerbated fol-lowing ischaemic stress via opening of the mitochondrial permeability transition pore. Toxicol. Appl. Pharm. 2013, 268, 149–156. [Google Scholar] [CrossRef]
- Ghimire, K.; Altmann, H.M.; Straub, A.C.; Isenberg, J.S. Nitric oxide: What’s new to NO? Am. J. Physiol. Cell Physiol. 2017, 312, C254–C262. [Google Scholar] [CrossRef]
- Galbiati, F.; Volonte’, D.; Liu, J.; Capozza, F.; Frank, P.; Zhu, L.; Pestell, R.G.; Lisanti, M.P. Caveolin-1 Expression Negatively Regulates Cell Cycle Progression by Inducing G0/G1 Arrest via a p53/p21WAF1/Cip1-dependent Mechanism. Mol. Biol. Cell 2001, 12, 2229–2244. [Google Scholar] [CrossRef]
- Volonte, D.; McTiernan, C.F.; Drab, M.; Kasper, M.; Galbiati, F. Caveolin-1 and caveolin-3 form heterooligomeric complexes in atrial cardiac myocytes that are required for doxorubicin-induced apoptosis. Am. J. Physiol. Circ. Physiol. 2008, 294, H392–H401. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017, 7, 11. [Google Scholar] [CrossRef]
- Yamaoka, M.; Yamaguchi, S.; Suzuki, T.; Okuyama, M.; Nitobe, J.; Nakamura, N.; Mitsui, Y.; Tomoike, H. Apoptosis in Rat Cardiac Myocytes Induced by Fas Ligand: Priming for Fas-mediated Apoptosis with Doxorubicin. J. Mol. Cell Cardiol. 2000, 32, 881–889. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, C.; Zhang, X.M.; Yuan, C.H.; Wen, M.D.; Chen, Z.; Dong, D.-C.; Gao, Y.-H.; Liu, C.; Zhang, Z. Ophiopogonin D attenuates doxorubi-cin-induced autophagic cell death by relieving mitochondrial damage in vitro and in vivo. J. Pharm. Exp. Ther. 2015, 352, 166–174. [Google Scholar] [CrossRef]
- Dimitrakis, P.; Romay-Ogando, M.-I.; Timolati, F.; Suter, T.M.; Zuppinger, C. Effects of doxorubicin cancer therapy on autophagy and the ubiquitin-proteasome system in long-term cultured adult rat cardiomyocytes. Cell Tissue Res. 2012, 350, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Vinson, J.A.; Liang, X.; Proch, J.; Hontz, B.A.; Dancel, J.; Sandone, N. Polyphenol Antioxidants in Citrus Juices: In vitro and in vivo Studies Relevant to Heart Disease. Adv. Exp. Med. Biol. 2002, 505, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Abushouk, A.I.; Ismail, A.; Salem, A.M.A.; Afifi, A.M.; Abdel-Daim, M.M. Cardioprotective mechanisms of phyto-chemicals against doxorubicin-induced cardiotoxicity. Biomed. Pharm. 2017, 90, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.T.; Ibrahim, Y.F.; Espey, M.G.; Suzuki, Y.J. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharm. 2013, 72, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Carresi, C.; Musolino, V.; Gliozzi, M.; Maiuolo, J.; Mollace, R.; Nucera, S.; Marettaa, A.; Sergib, D.; Muscolib, S.; Gratteri, S. Anti-oxidant effect of bergamot polyphenolic fraction counteracts doxorubicin-induced cardiomyopathy: Role of autophagy and ckit-posCD45negCD31neg cardiac stem cell activation. J. Mol. Cell. Cardiol. 2018, 119, 10–18. [Google Scholar] [CrossRef]
- Sergazy, S.; Shulgau, Z.; Fedotovskikh, G.; Chulenbayeva, L.; Nurgozhina, A.; Nurgaziyev, M.; Krivyh, E.; Kamyshanskiy, Y.; Kushugulova, A.; Gulyayev, A.; et al. Cardioprotective effect of grape polyphenol extract against doxorubicin induced cardiotoxicity. Sci. Rep. 2020, 10, 12. [Google Scholar] [CrossRef]
- Abe, J.; Yamada, Y.; Takeda, A.; Harashima, H. Cardiac progenitor cells activated by mitochondrial delivery of resveratrol enhance the survival of a doxorubicin-induced cardiomyopathy mouse model via the mitochondrial activation of a damaged myocardium. J. Control. Release 2018, 269, 177–188. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, L.; Li, H.; Wang, J.; Li, J.; Yang, Y.; Wang, Y.; Cai, H.; Li, R.; Zhao, Y. Salsolinol Attenuates Doxoru-bicin-Induced Chronic Heart Failure in Rats and Improves Mitochondrial Function in H9c2 Cardiomyocytes. Front. Pharm. 2019, 10, 1135. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, T.Y.; Wu, Y.-T.; Lin, L.-C.; Chiu, A.W.; Lin, C.-H.; Tsai, T.-H. Herb-Drug Interaction of Epimedium sagittatum (Sieb. et Zucc.) Maxim Extract on the Pharmacokinetics of Sildenafil in Rats. Molecules 2013, 18, 7323–7335. [Google Scholar] [CrossRef]
- Wu, H.; Lien, E.J.; Len, L.L. Chemical and pharmacological investigations of Epimedium species: A survey. Progr. Drug Res. 2003, 60, 1–57. [Google Scholar]
- Ma, H.; He, X.; Yang, Y.; Li, M.; Hao, D.; Jia, Z. The genus Epimedium: An ethnopharmacological and phytochemical review. J. Ethnopharmacol. 2011, 134, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.-L.; Chan, K.-G.; Pusparajah, P.; Saokaew, S.; Duangjai, A.; Lee, L.-H.; Goh, B.-H. Anti-Cancer Properties of the Naturally Occurring Aphrodisiacs: Icariin and Its Derivatives. Front. Pharm. 2016, 7, 191. [Google Scholar] [CrossRef]
- Fang, J.; Zhang, Y. Icariin, an Anti-atherosclerotic Drug from Chinese Medicinal Herb Horny Goat Weed. Front. Pharm. 2017, 8, 734. [Google Scholar] [CrossRef] [PubMed]
- Shen, R.; Wang, J.-H. The effect of icariin on immunity and its potential application. Am. J. Clin. Exp. Immunol. 2018, 7, 50–56. [Google Scholar]
- Wang, F.-Y.; Jia, J.; Song, H.-H.; Jia, C.-M.; Chen, C.-B.; Ma, J. Icariin protects vascular endothelial cells from oxidative stress through inhibiting endoplasmic reticulum stress. J. Integr. Med. 2019, 17, 205–212. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Wang, Z.; Shi, J. Pharmacological effects of icariin. Stud. Surf. Sci. Catal. 2020, 87, 179–203. [Google Scholar] [CrossRef]
- Song, Y.H.; Cai, C.H.; Zhao, Z.M.; Chang, W.J.; Gu, N.; Cao, S.P.; Wu, M.L. Icariin attenuated oxidative stress in-duced-cardiac apoptosis by mitochondria protection and ERK activation. Biomed. Pharm. 2016, 83, 1089–1094. [Google Scholar] [CrossRef]
- Song, Y.-H.; Cai, H.; Gu, N.; Qian, C.-F.; Cao, S.-P.; Zhao, Z.-M. Icariin attenuates cardiac remodelling through down-regulating myocardial apoptosis and matrix metalloproteinase activity in rats with congestive heart failure. J. Pharm. Pharm. 2011, 63, 541–549. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, H.; Wang, S.; Liu, M.; Feng, Y.; Wang, X. Icariin Protects Rat Cardiac H9c2 Cells from Apoptosis by Inhibiting Endoplasmic Reticulum Stress. Int. J. Mol. Sci. 2013, 14, 17845–17860. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zeng, M.; Cao, Z.; Liu, Z.; Hao, J.; Zhang, P.; Tian, Y.; Zhang, P.; Ma, J. Icariin, a Novel Blocker of Sodium and Calcium Channels, Eliminates Early and Delayed Afterdepolarizations, As Well As Triggered Activity, in Rabbit Cardiomyocytes. Front. Physiol. 2017, 8, 342. [Google Scholar] [CrossRef]
- Meng, X.; Pei, H.; Lan, C. Icariin Exerts Protective Effect Against Myocardial Ischemia/Reperfusion Injury in Rats. Cell Biophys. 2015, 73, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Ke, Z.; Liu, J.; Xu, P.; Gao, A.; Wang, L.; Ji, L. The Cardioprotective Effect of Icariin on Ischemia-Reperfusion Injury in Isolated Rat Heart: Potential Involvement of the PI3K-Akt Signaling Pathway. Cardiovasc. Ther. 2015, 33, 134–140. [Google Scholar] [CrossRef]
- Qi, C.; Shao, Y.; Liu, X.; Wang, D.; Li, X. The cardioprotective effects of icariin on the isoprenaline-induced tako-tsubo-like rat model: Involvement of reactive oxygen species and the TLR4/NF-κB signaling pathway. Int. Immunopharmacol. 2019, 74, 105733. [Google Scholar] [CrossRef] [PubMed]
- Sandner, P.; Hütter, J.; Tinel, H.; Ziegelbauer, K.; Bischoff, E. PDE5 inhibitors beyond erectile dysfunction. Int. J. Impot. Res. 2007, 19, 533–543. [Google Scholar] [CrossRef]
- Ockaili, R.; Salloum, F.; Hawkins, J.; Kukreja, R.C. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1263–H1269. [Google Scholar] [CrossRef]
- Das, A.; Salloum, F.; Xi, L.; Rao, Y.J.; Kukreja, R.C. ERK phosphorylation mediates sildenafil-induced myocardial protection against ischemia-reperfusion injury in mice. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1236–H1243. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.W.; Salloum, F.; Das, A.; Hyder, H.; Kukreja, R.C. Phosphodiesterase-5 Inhibition With Sildenafil Attenuates Cardiomyocyte Apoptosis and Left Ventricular Dysfunction in a Chronic Model of Doxorubicin Cardiotoxicity. Circulation 2005, 111, 1601–1610. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.N.; Daset, A. Role of cGMP Signaling and Phosphodiesterase-5 inhibitors in Cardioprotection. J. Am. Coll. Cardiol. 2012, 59, 1921–1927. [Google Scholar] [CrossRef]
- Musolino, V.; Gliozzi, M.; Scarano, F.; Bosco, F.; Scicchitano, M.; Nucera, S.; Carresi, C.; Ruga, S.; Zito, M.C.; Maiuolo, J.; et al. Bergamot Polyphenols Improve Dyslipidemia and Pathophysiological Features in a Mouse Model of Non-Alcoholic Fatty Liver Disease. Sci. Rep. 2020, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Carresi, C.; Gliozzi, M.; Giancotta, C.; Scarcella, A.; Scarano, F.; Bosco, F.; Mollace, R.; Tavernese, A.; Vitale, C.; Musolino, V. Studies on the protective role of Bergamot polyphenols in doxorubicin-induced cardiotoxicity. PharmaNutrition 2016, 4, S19–S26. [Google Scholar] [CrossRef]
- Štěrba, M.; Popelová, O.; Vávrova, A.; Jirkovský, E.; Kovaříková, P.; Geršl, V.; Simůnek, T. Oxidative Stress, Redox Signaling, and Metal Chelation in Anthracycline Cardiotoxicity and Pharmacological Cardioprotection. Antioxid. Redox Signal. 2013, 18, 899–929. [Google Scholar] [CrossRef]
- Green, P.S.; Leeuwenburgh, C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2002, 1588, 94–101. [Google Scholar] [CrossRef]
- Hanf, A.; Oelze, M.; Manea, A.; Li, H.; Münzel, T.; Daiber, A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chem. Interact. 2019, 313, 108834. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Q.; Qi, H.; Wang, C.; Wang, C.; Zhang, J.; Dong, L. Doxorubicin-Induced Systemic Inflammation Is Driven by Upregulation of Toll-Like Receptor TLR4 and Endotoxin Leakage. Cancer Res. 2016, 76, 6631–6642. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Abdullah, C.S.; Alam, S.; Aishwarya, R.; Miriyala, S.; Bhuiyan, M.A.N.; Panchatcharam, M.; Pattillo, C.B.; Orr, A.W.; Sadoshima, J.; Hill, J.A.; et al. Doxorubicin-induced cardiomyopathy associated with inhibition of autophagic degradation process and defects in mitochondrial respi-ration. Sci. Rep. 2019, 9, 2002. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomy-opathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.M.; Simic, V.D. Doxorubicin-Induced Oxidative Injury of Cardiomyocytes—Do We Have Right Strategies for Prevention? In Cardiotoxicity of Oncology Treatment; Manuela Fiuza, Ed.; IntechOpen Limited: London, UK, 2012. [Google Scholar]
- Siasos, G.; Tsigkou, V.; Kosmopoulos, M.; Theodosiadis, D.; Simantiris, S.; Tagkou, N.M. Mitochondria and cardio-vascular diseases-from pathophysiology to treatment. Ann. Transl. Med. 2018, 6, 256. [Google Scholar] [CrossRef]
- de Tassigny, A.D.; Assaly, R.; Schaller, S.; Pruss, R.M.; Berdeaux, A.; Morin, D. Mitochondrial translocator protein (TSPO) ligands prevent doxorubicin-induced mechanical dysfunction and cell death in isolated cardiomyocytes. Mitochondrion 2013, 13, 688–697. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Smaili, S.S.; Russell, J.T.; Fiskum, G. Elevation of resting mitochondrial membrane potential of neural cells by cyclosporin A, BAPTA-AM, and bcl-2. Am. J. Physiol. Physiol. 2000, 279, C852–C859. [Google Scholar] [CrossRef]
- Mitry, M.A.; Edwards, J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. IJC Heart Vasc. 2016, 10, 17–24. [Google Scholar] [CrossRef]
- Osataphan, N.; Phrommintikul, A.; Chattipakorn, S.C.; Chattipakorn, N. Effects of doxorubicin-induced cardiotoxi-city on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. J. Cell Mol. Med. 2020, 22, 6534–6557. [Google Scholar] [CrossRef]
- Nijholt, K.T.; Westenbrink, B.D.; de Boer, R.A. Mitochondrial therapy for doxorubicin cardiomyopathy: Nuclear factor-κB to the rescue? Cardiovasc. Res. 2020, 116, 1092–1094. [Google Scholar] [CrossRef]
- Tacar, O.; Dass, C.R. Doxorubicin-induced death in tumour cells and cardiomyocytes: Is autophagy the key to improving future clinical outcomes? J. Pharm. Pharm. 2013, 65, 1577–1589. [Google Scholar] [CrossRef]
- Cappetta, D.; De Angelis, A.; Sapio, L.; Prezioso, L.; Illiano, M.; Quaini, F.; Rossi, F.; Berrino, L.; Naviglio, S.; Urbanek, K. Oxidative Stress and Cellular Response to Doxorubicin: A Common Factor in the Complex Milieu of Anthracycline Cardiotoxicity. Oxidative Med. Cell. Longev. 2017, 2017, 1521020. [Google Scholar] [CrossRef]
- Dell’Agli, M.; Galli, G.V.; Cero, E.D.; Belluti, F.; Matera, R.; Zironi, E.; Pagliuca, G.; Bosisio, E. Potent inhibition of human phos-phodiesterase-5 by icariin derivatives. J. Nat. Prod. 2008, 71, 1513–1517. [Google Scholar] [CrossRef]
- Ning, H.; Xin, Z.-C.; Lin, G.; Banie, L.; Lue, T.F.; Lin, C.-S. Effects of icariin on phosphodiesterase-5 activity in vitro and cyclic guanosine monophosphate level in cavernous smooth muscle cells. Urology 2006, 68, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.; Li, F.-S.; Levsh, O.; Weng, J.-K. Exploration of icariin analog structure space reveals key features driving potent inhibition of human phosphodiesterase-5. PLoS ONE 2019, 14, e0222803. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Lee, S.; Traverse, J.H.; Zhu, G.; Fassett, J.; Tao, Y.; Zhang, P.; dos Remedios, C.; et al. Oxidative Stress Regulates Left Ventricular PDE5 Expression in the Failing Heart. Circulation 2010, 121, 1474–1483. [Google Scholar] [CrossRef]
- Verit, A.; Savas, M.; Ciftci, H.; Aksoy, N.; Taskin, A.; Topal, U. Assessment of the acute effects of tadalafil on the cardiovascular system based on examination of serum oxidative status and paraoxonase activity in men with erectile dysfunction: A preliminary study. Int. J. Impot. Res. 2010, 22, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Mullershausen, F.; Russwurm, M.; Friebe, A.; Koesling, D. Inhibition of Phosphodiesterase Type 5 by the Activator of Nitric Oxide–Sensitive Guanylyl Cyclase BAY 41-2272. Circulation 2004, 109, 1711–1713. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Ockaili, R.; Salloum, F.; Yin, C.; Hawkins, J.; Das, A.; Xi, L. Cardioprotection with phosphodiesterase-5 inhibition-a novel preconditioning strateg. J. Mol. Cell Cardiol. 2004, 36, 165–173. [Google Scholar] [CrossRef]
- Kukreja, R.C.; Salloum, F.; Das, A.; Ockaili, R.; Yin, C.; Bremer, Y.A.; Fisher, P.W.; Wittkamp, M.; Hawkins, J.; Chou, E.; et al. Pharmacological preconditioning with sildenafil: Basic mechanisms and clinical implications. Vasc. Pharm. 2005, 42, 219–232. [Google Scholar] [CrossRef]
- Wehinger, S.; Ortiz, R.; Díaz, M.I.; Aguirre, A.; Valenzuela, M.; Llanos, P.; Mc Master, C.; Leyton, L.; Quest, A.F. Phosphorylation of caveolin-1 on tyrosine-14 induced by ROS enhances palmitate-induced death of beta-pancreatic cells. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 693–708. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quest, A.F.; Lobos-Gonzalez, L.; Nunez, S.; Sanhueza, C.; Fernandez, J.G.; Aguirre, A.; Rodriguez, D.; Leyton, L.; Torres, V. The caveolin-1 connection to cell death and survival. Curr. Mol. Med. 2013, 13, 266–281. [Google Scholar] [CrossRef]
- Bosch, M.; Marí, M.; Herms, A.; Fernández, A.; Fajardo, A.; Kassan, A.; Giralt, A.; Colell, A.; Balgoma, D.; Barbero, E.; et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr. Biol. 2011, 21, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Sagua, R.; Parra, V.; Ortiz-Sandoval, C.; Navarro-Márquez, M.; Rodríguez, A.E.; Valdivia, N.D.; Sanhueza, C.; Lopez-Crisosto, C.; Tahbaz, N.; Rothermel, B.A.; et al. Caveolin-1 impairs PKA-DRP1-mediated remodelling of ER–mitochondria communication during the early phase of ER stress. Cell Death Differ. 2019, 26, 1195–1212. [Google Scholar] [CrossRef]
- Takaguri, A.; Kamato, M.; Satoh, Y.; Ohtsuki, K.; Satoh, K. Effect of alteration of caveolin-1 expression on doxoru-bicin-induced apoptosis in H9c2 cardiac cells. Cell Biol. Int. 2015, 39, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Song, Z.; Wu, S.; Wei, Z.; Xu, Y.; Shen, X. Toll-like receptor 4 contributes to a myofibroblast phenotype in cardiac fibroblasts and is associated with autophagy after myocardial infarction in a mouse model. Atherosclerosis 2018, 279, 23–31. [Google Scholar] [CrossRef]
- Ding, Z.; Liu, S.; Wang, X.; Dai, Y.; Khaidakov, M.; Romeo, F.; Mehta, J.L. LOX-1, oxidant stress, mtDNA damage, autophagy, and immune response in atherosclerosis. Can. J. Physiol. Pharm. 2014, 92, 524–530. [Google Scholar] [CrossRef]
- Shihata, W.A.; Michell, D.L.; Andrews, K.L.; Chin-Dusting, J.P.F. Caveolae: A Role in Endothelial Inflammation and Mechanotransduction? Front. Physiol. 2016, 7, 628. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Li, B.; Zhen, Y.; Chen, J.; Wang, P.; Fei, Y. NF-κB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem. Biophys. Re Commun. 2013, 436, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2011, 441, 523–540. [Google Scholar] [CrossRef]
- Sishi, B.J.; Loos, B.; van Rooyen, J.; Engelbrecht, A.-M. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem. Pharm. 2013, 85, 124–134. [Google Scholar] [CrossRef]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.-S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scicchitano, M.; Carresi, C.; Nucera, S.; Ruga, S.; Maiuolo, J.; Macrì, R.; Scarano, F.; Bosco, F.; Mollace, R.; Cardamone, A.; et al. Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response. Nutrients 2021, 13, 4070. https://doi.org/10.3390/nu13114070
Scicchitano M, Carresi C, Nucera S, Ruga S, Maiuolo J, Macrì R, Scarano F, Bosco F, Mollace R, Cardamone A, et al. Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response. Nutrients. 2021; 13(11):4070. https://doi.org/10.3390/nu13114070
Chicago/Turabian StyleScicchitano, Miriam, Cristina Carresi, Saverio Nucera, Stefano Ruga, Jessica Maiuolo, Roberta Macrì, Federica Scarano, Francesca Bosco, Rocco Mollace, Antonio Cardamone, and et al. 2021. "Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response" Nutrients 13, no. 11: 4070. https://doi.org/10.3390/nu13114070
APA StyleScicchitano, M., Carresi, C., Nucera, S., Ruga, S., Maiuolo, J., Macrì, R., Scarano, F., Bosco, F., Mollace, R., Cardamone, A., Coppoletta, A. R., Guarnieri, L., Zito, M. C., Bava, I., Cariati, L., Greco, M., Foti, D. P., Palma, E., Gliozzi, M., ... Mollace, V. (2021). Icariin Protects H9c2 Rat Cardiomyoblasts from Doxorubicin-Induced Cardiotoxicity: Role of Caveolin-1 Upregulation and Enhanced Autophagic Response. Nutrients, 13(11), 4070. https://doi.org/10.3390/nu13114070