
Compositional Changes of the High-Fat Diet-Induced Gut Microbiota upon Consumption of Common Pulses

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. 16S rRNA Gene Library Preparation and Sequencing
2.3. Sequence Processing
2.4. Statistical and Bioinformatics Analyses
3. Results
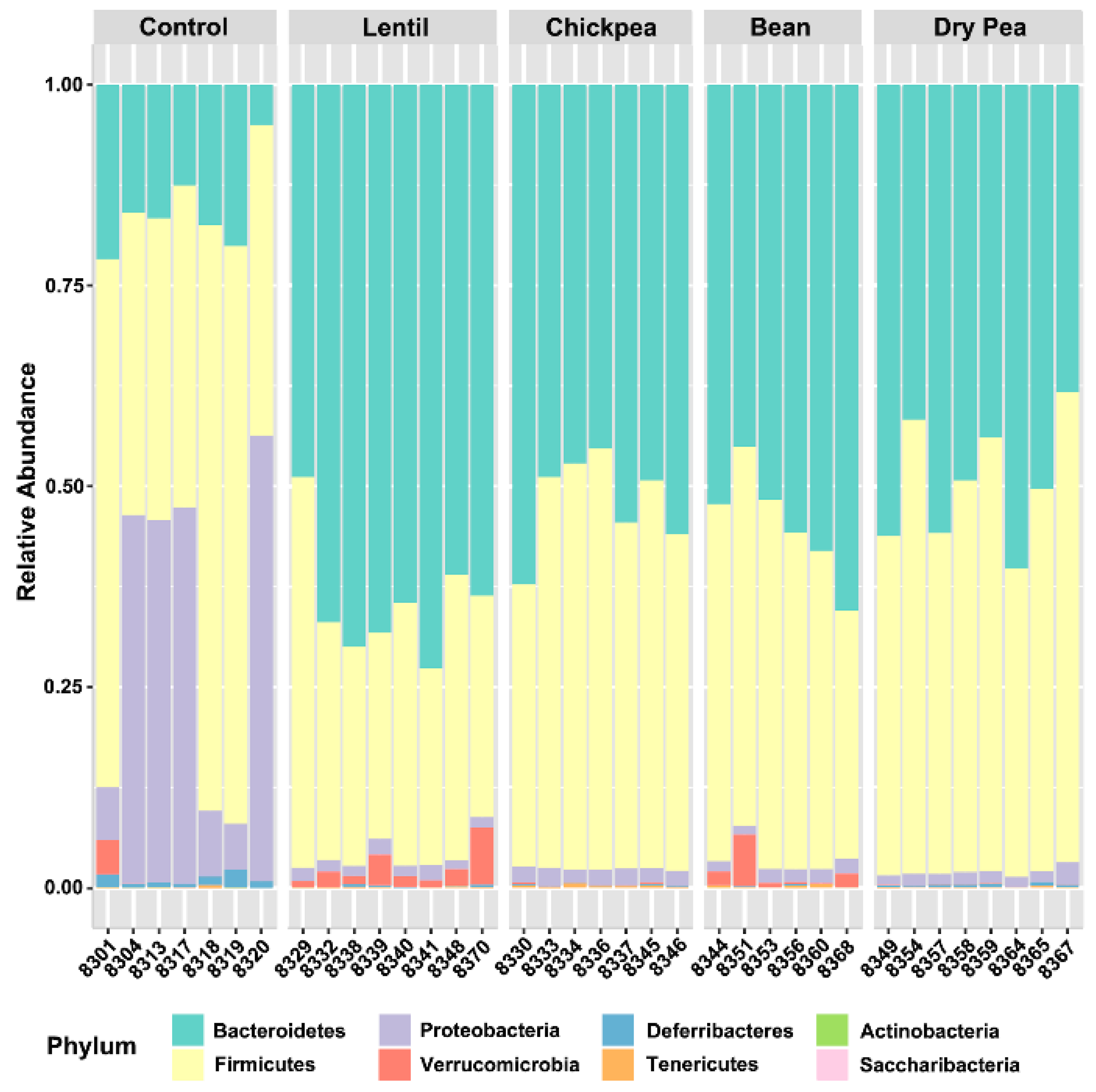
3.1. Overall Response to Pulse Consumption
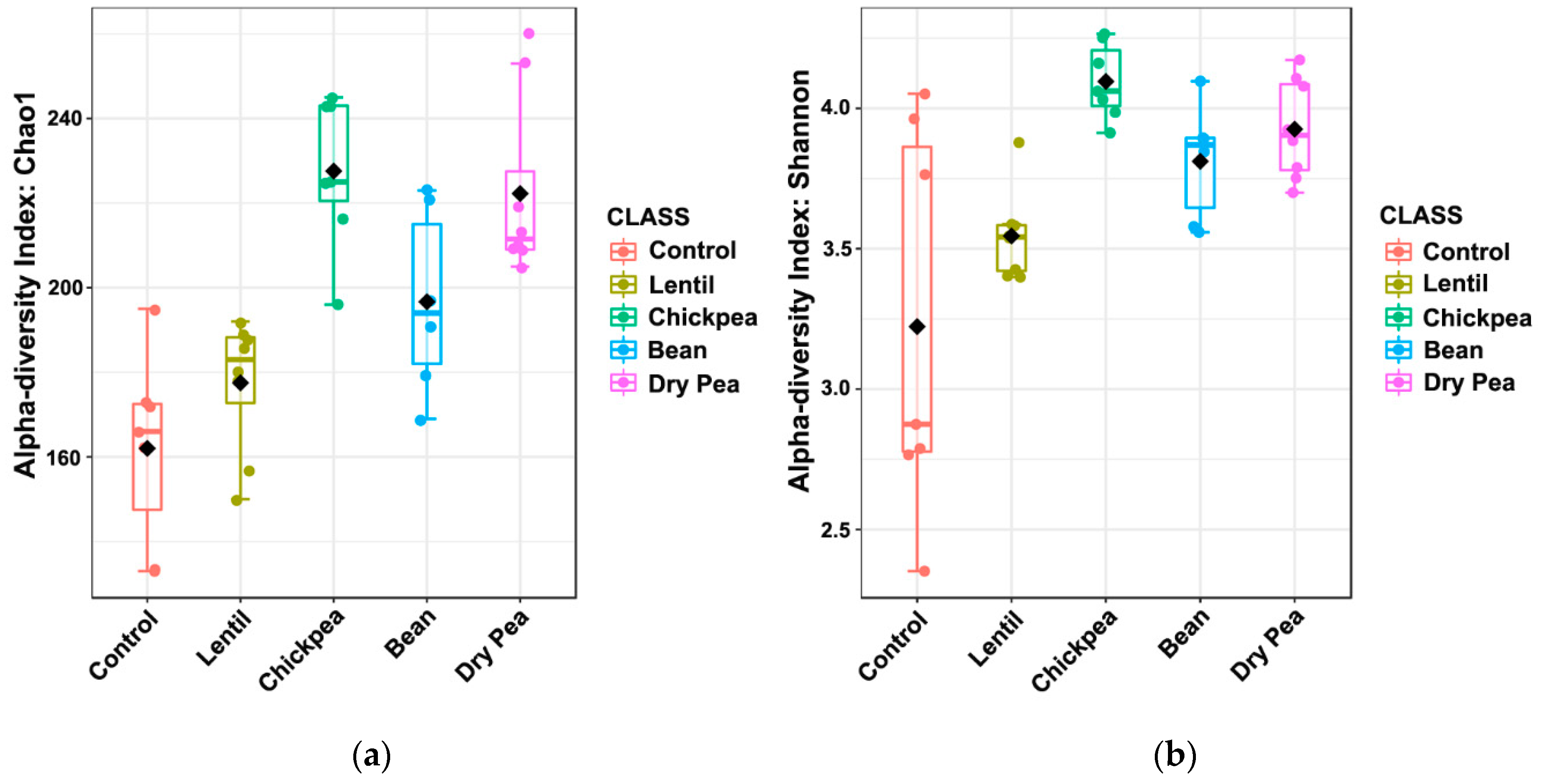
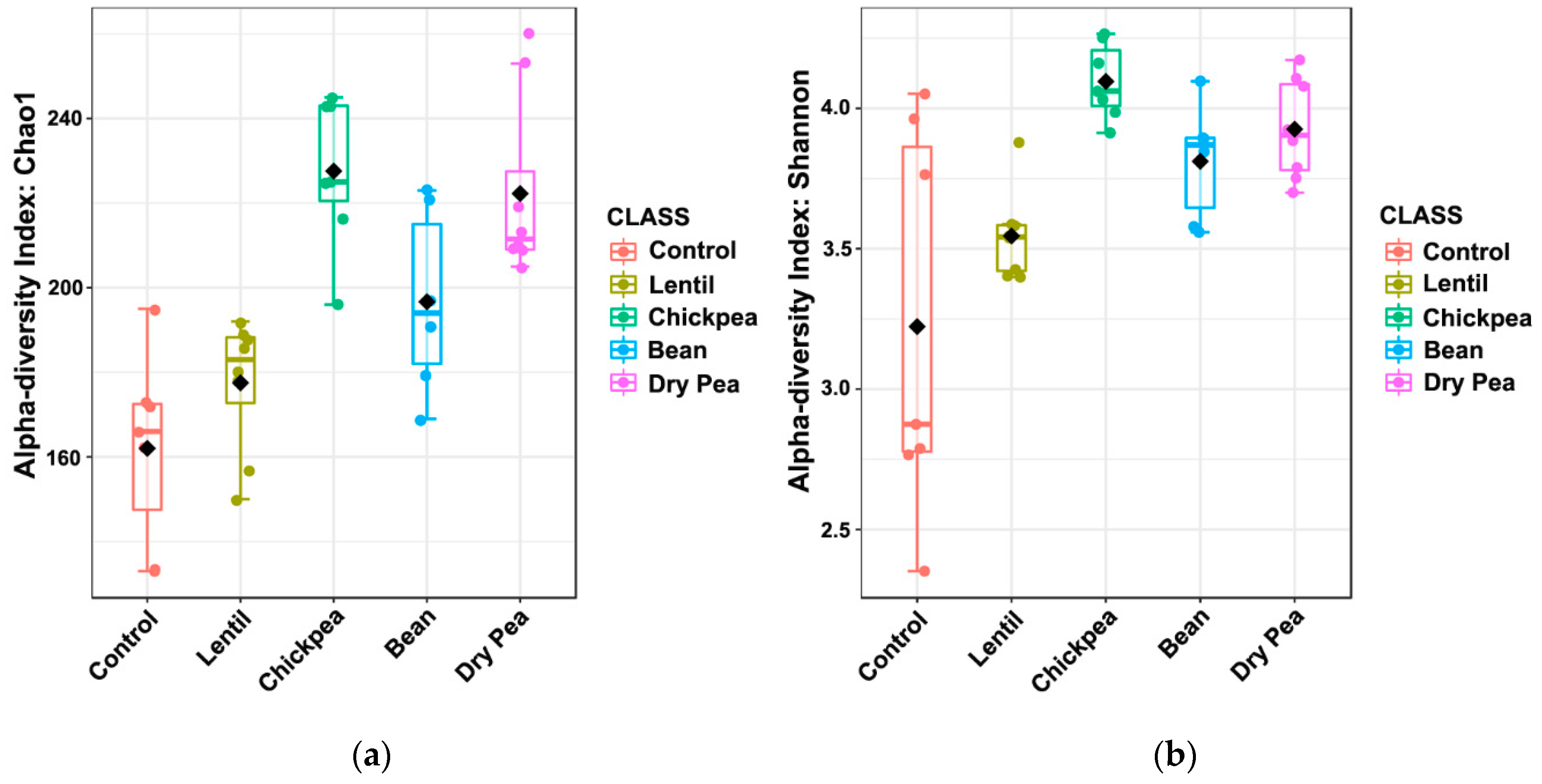
3.2. Effects on α-Diversity
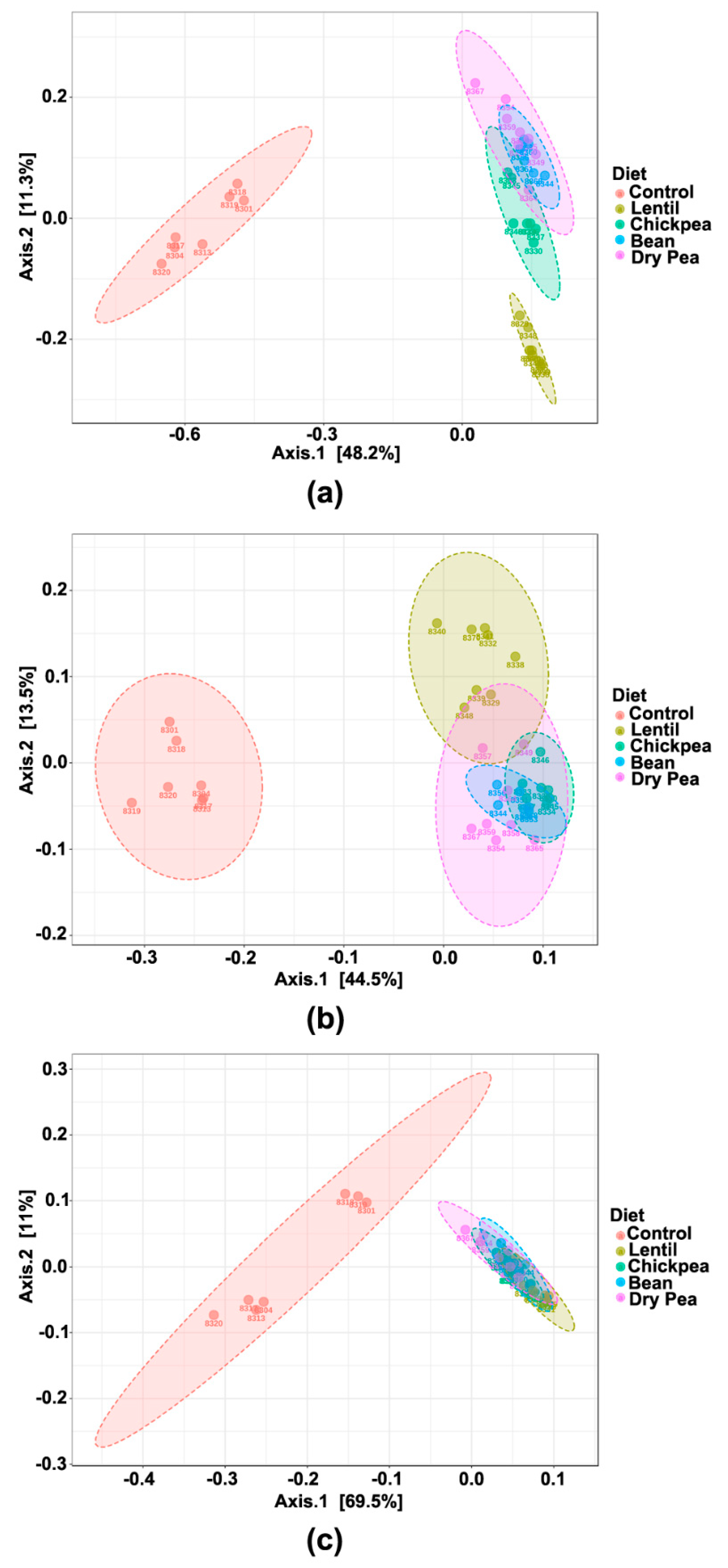
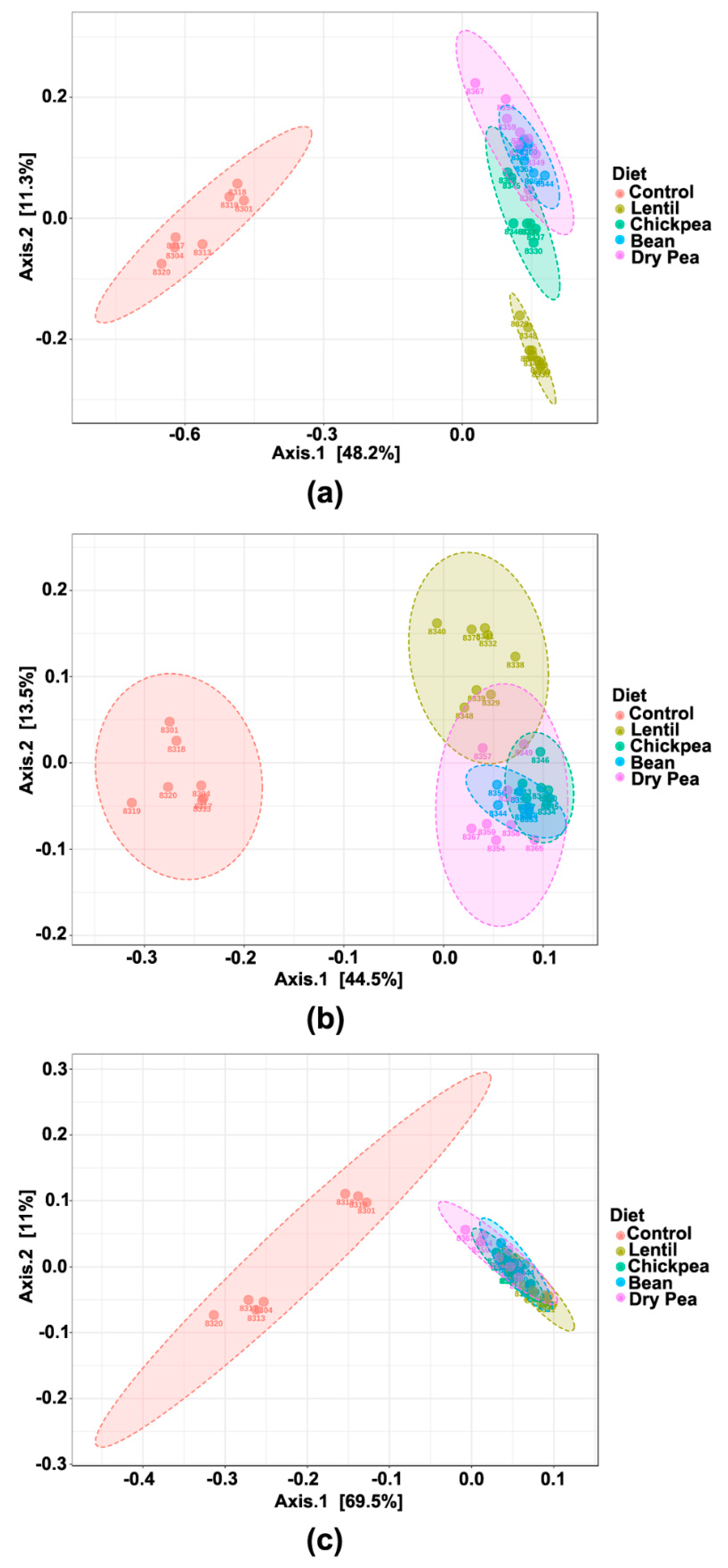
3.3. Effect on β-Diversity
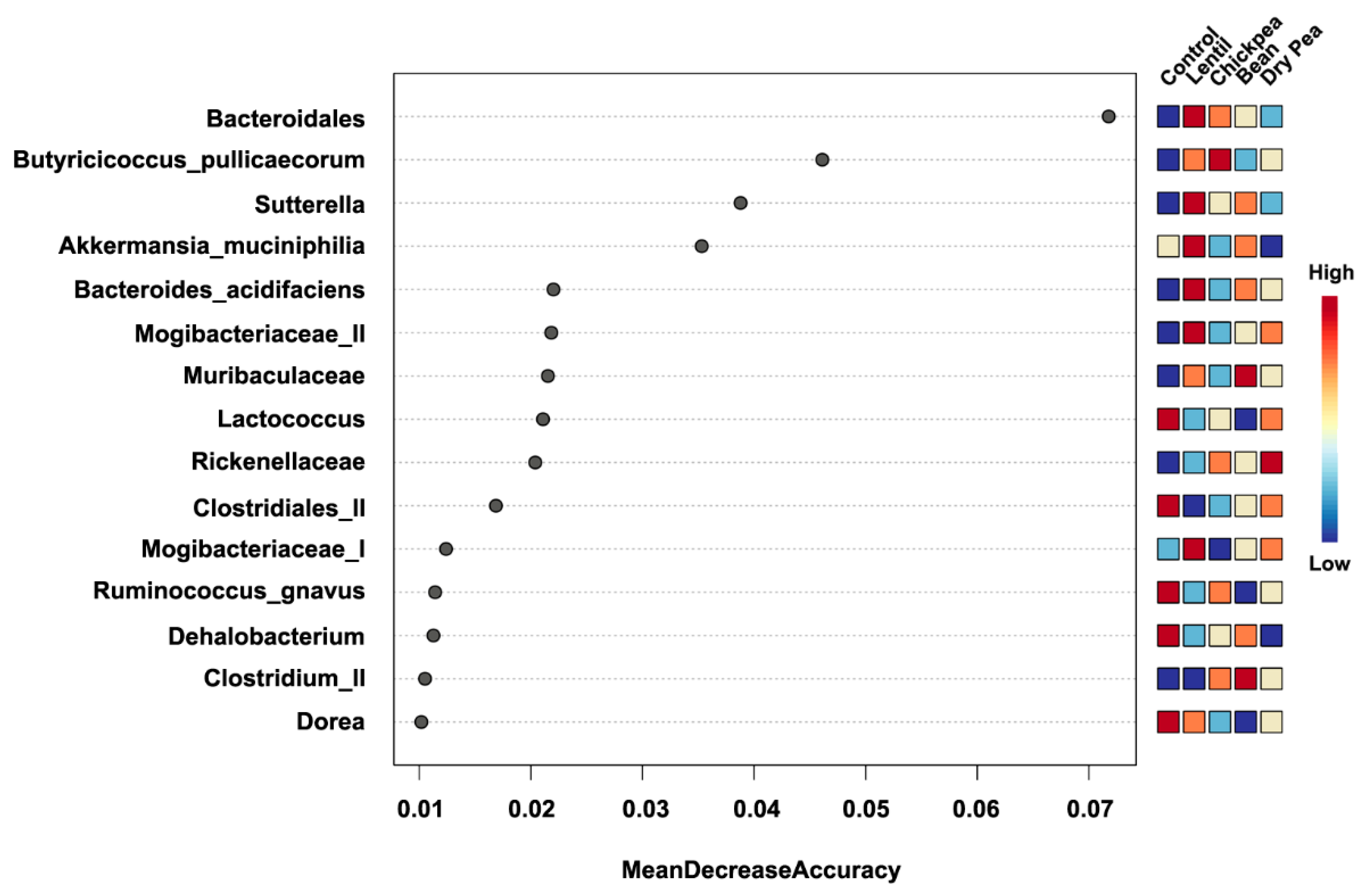
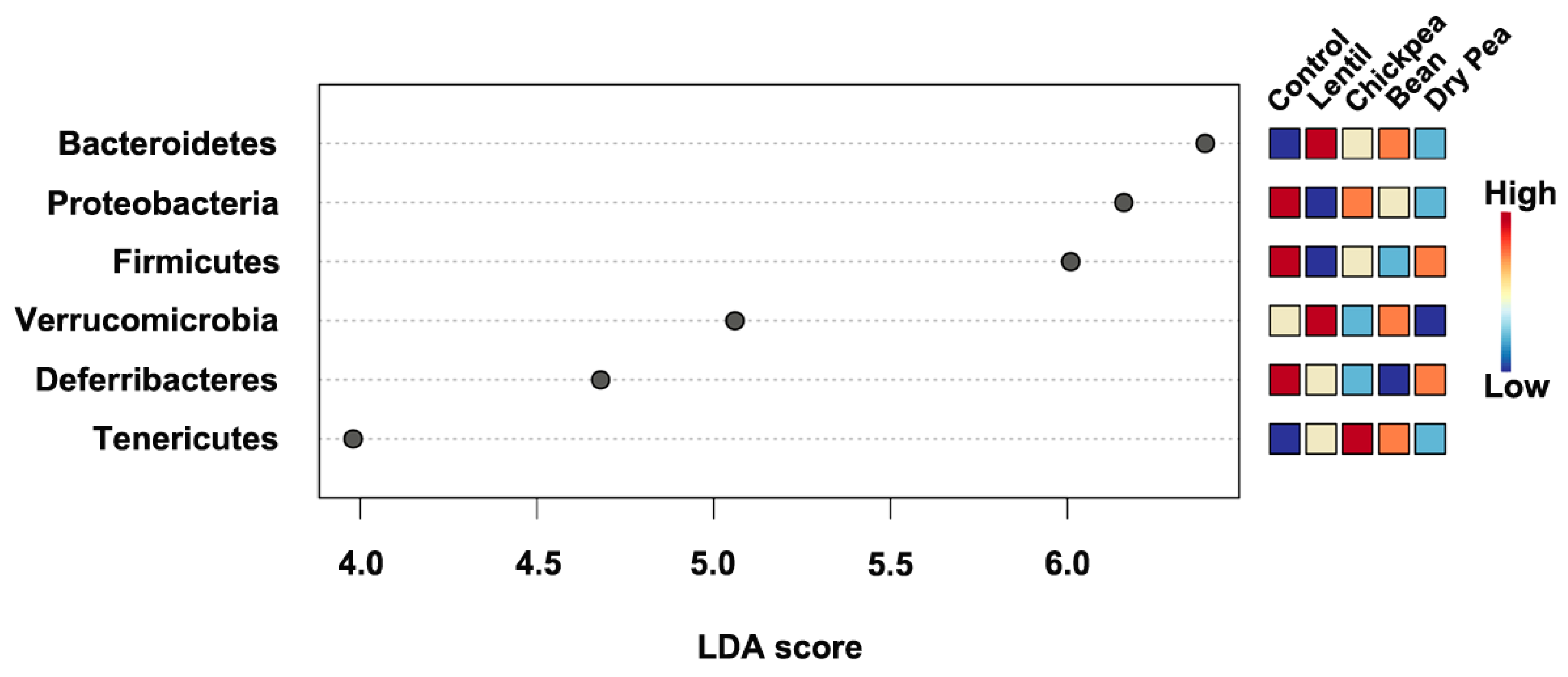
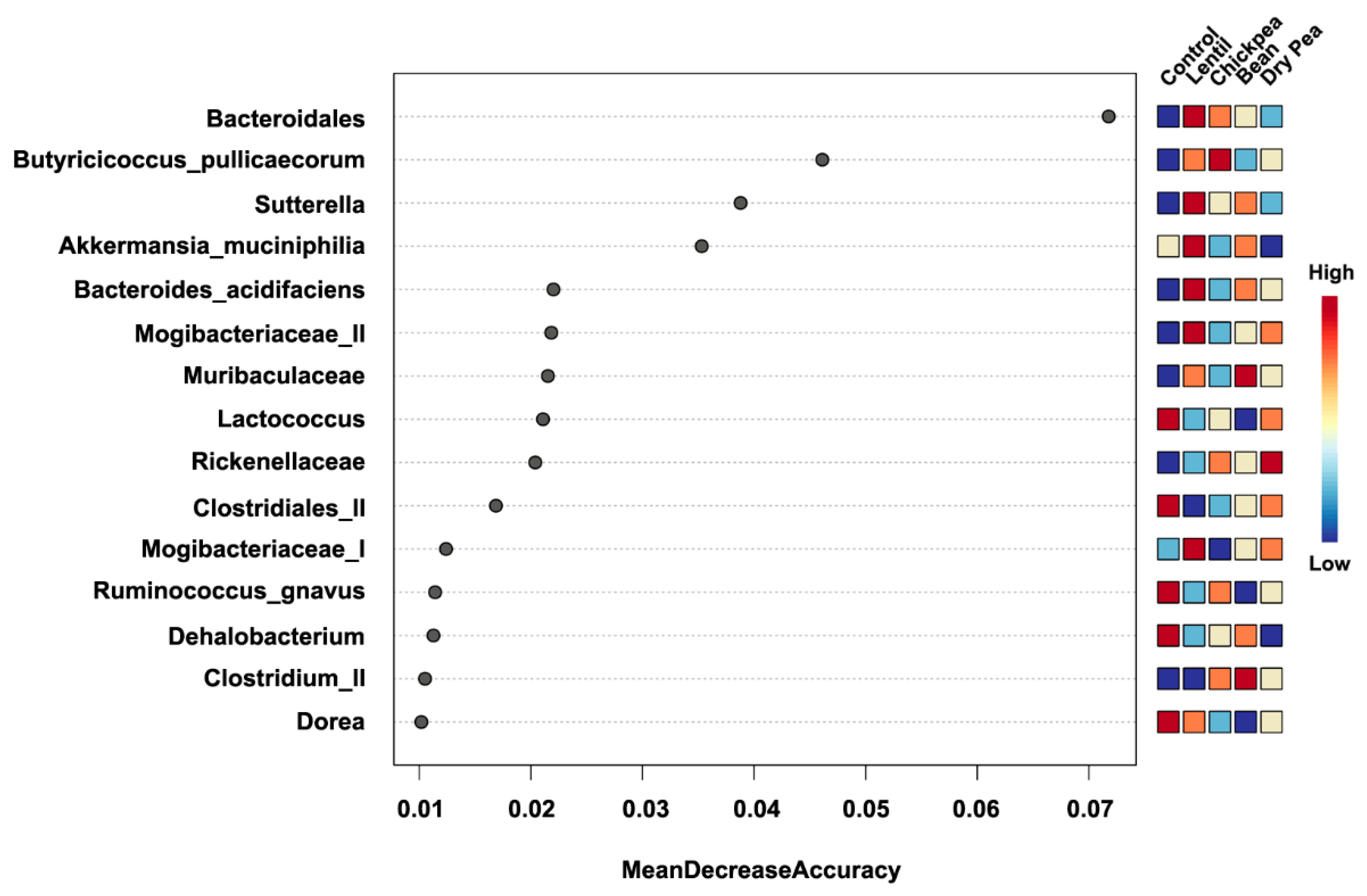
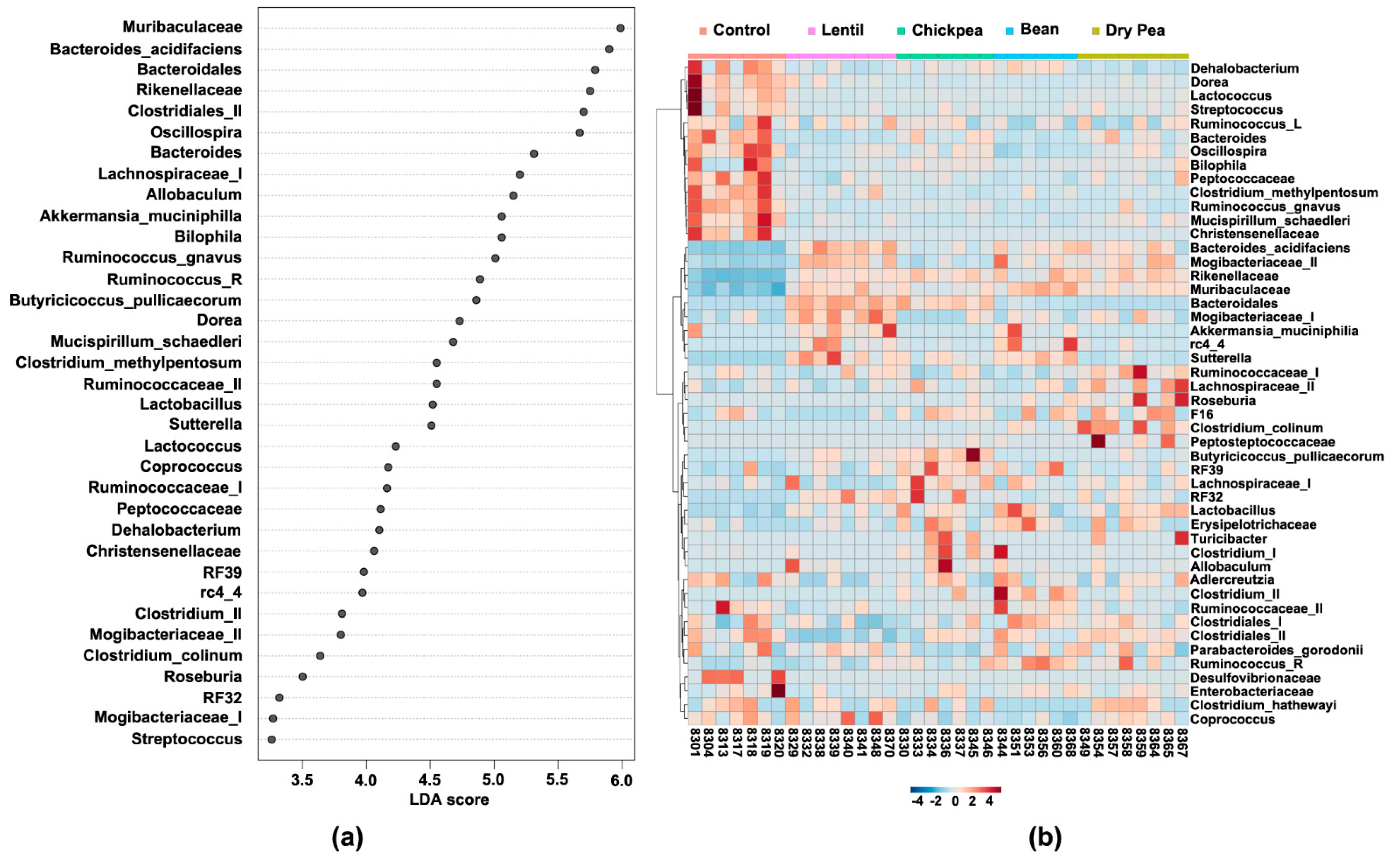
3.4. Which Bacteria Are the Major Players in Accounting for Differences Due to Pulse Consumption?
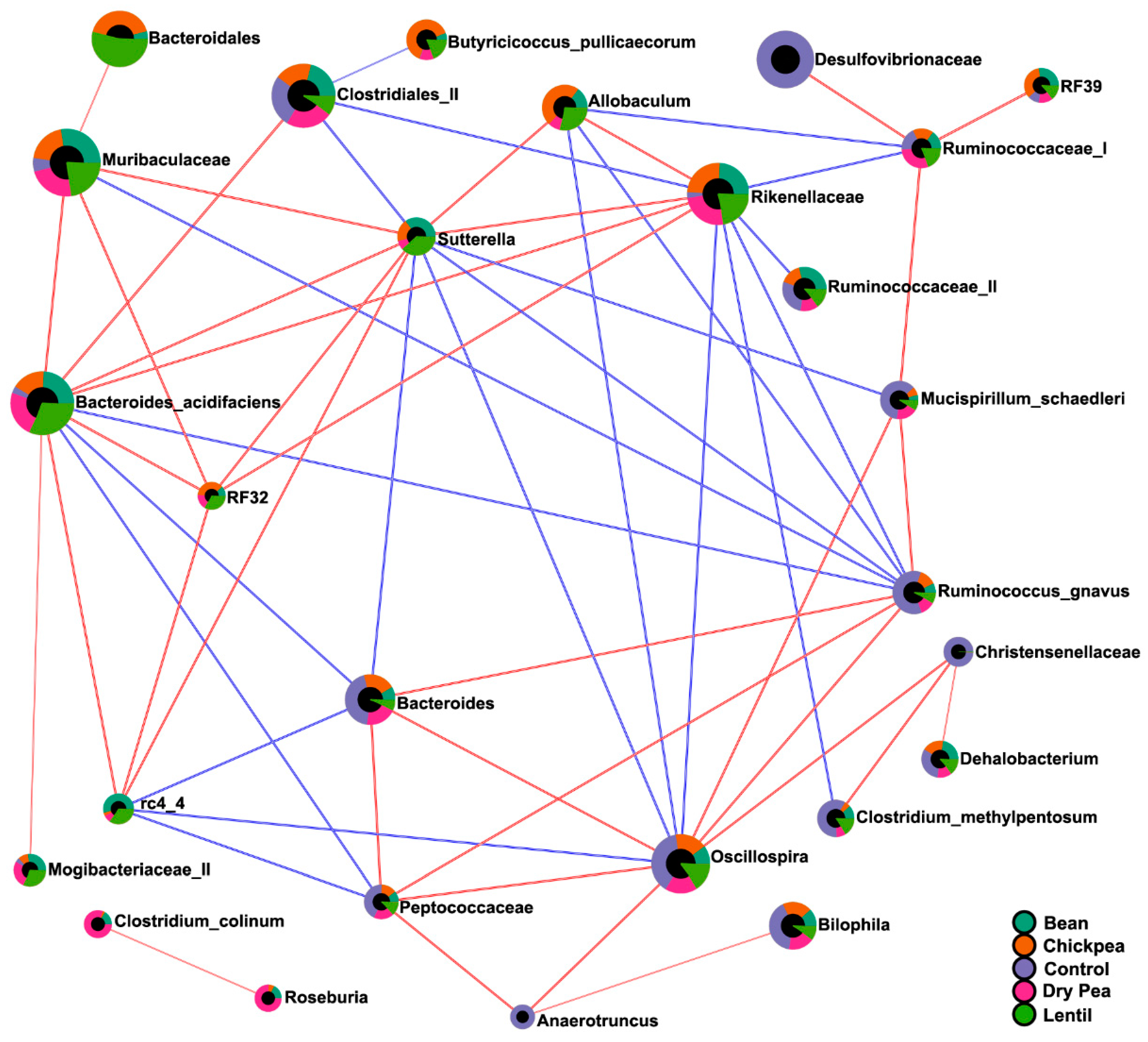
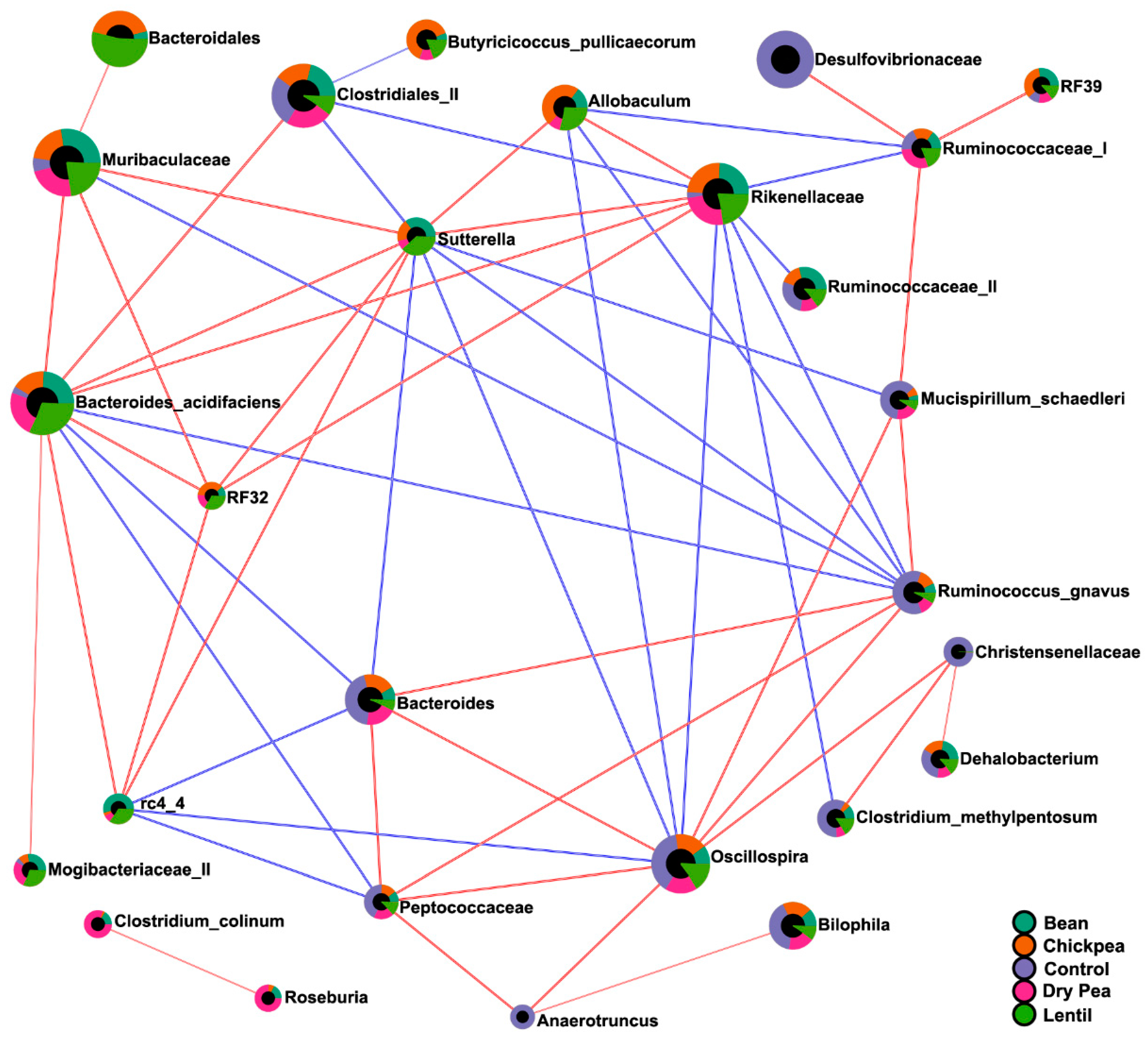
3.5. Diet-Specific Microbial Ecosystems
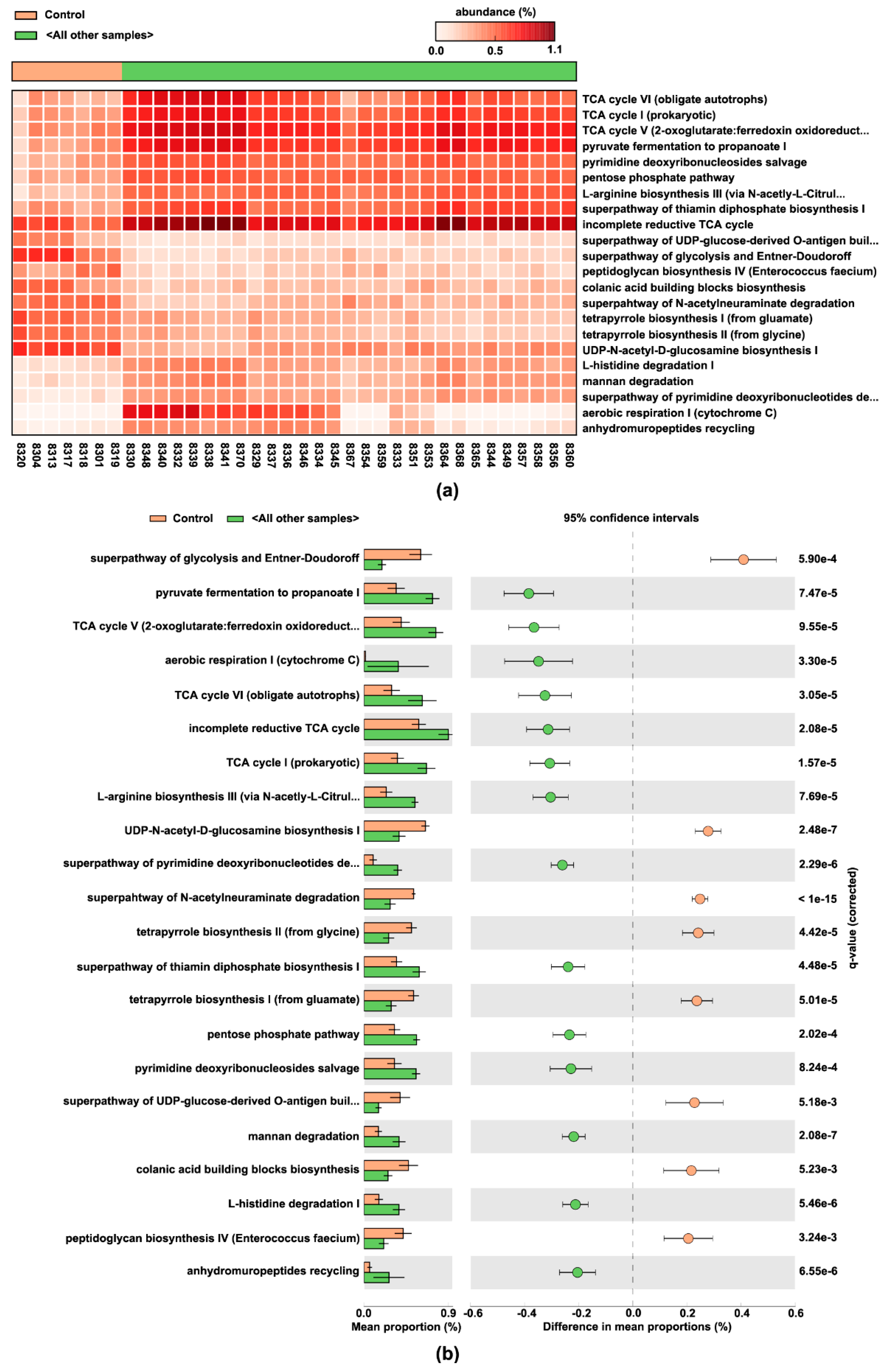
3.6. Pulse-Predicted Microbial Function
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- US Department of Agriculture and US National Institutes of Health. Dietary Guidelines for Americans 2020–2025; USDA: Washington, DC, USA, 2020.
- NIH. The Science behind Healthy Eating Patterns; NIH: Bethesda, MD, USA, 2020.
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2021, 70, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association. Get Smart about Superfoods. 2020. Available online: https://www.heart.org/en/healthy-living/healthy-eating/eat-smart/nutrition-basics/get-smart-about-superfoods-infographic (accessed on 20 August 2021).
- Didinger, C.; Thompson, H. Motivating Pulse-Centric Eating Patterns to Benefit Human and Environmental Well-Being. Nutrients 2020, 12, 3500. [Google Scholar] [CrossRef] [PubMed]
- Didinger, C.; Thompson, H.J. Defining Nutritional and Functional Niches of Legumes: A Call for Clarity to Distinguish a Future Role for Pulses in the Dietary Guidelines for Americans. Nutrients 2021, 13, 1100. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.; Vasconcelos, M.; Gil, A.M.; Pinto, E. Benefits of pulse consumption on metabolism and health: A systematic review of randomized controlled trials. Crit. Rev. Food Sci. Nutr. 2021, 61, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pandey, G. Biofortification of pulses and legumes to enhance nutrition. Heliyon 2020, 6, e03682. [Google Scholar] [CrossRef] [PubMed]
- Didinger, C.F.; Bunning, M.; Thomppson, H.J. Nutrition and Human Health Benefits of Dry Beans and Other Pulses. In Dry Beans and Pulses Book, 2nd ed.; Siddiq, M.U.M., Ed.; Wiley: Hoboken, NJ, USA, 2021. [Google Scholar]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Barratt, M.J.; Lebrilla, C.; Shapiro, H.-Y.; Gordon, J.I. The Gut Microbiota, Food Science, and Human Nutrition: A Timely Marriage. Cell Host Microbe 2017, 22, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 866–877. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yi, B.; Zhong, R.; Wang, M.; Zhang, S.; Ma, J.; Yin, Y.; Yin, J.; Chen, L.; Zhang, H. From gut microbiota to host appetite: Gut microbiota-derived metabolites as key regulators. Microbiome 2021, 9, 162. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.; Huey, S.L.; McDonald, D.; Knight, R.; Finkelstein, J.L. Nutritional Interventions and the Gut Microbiome in Children. Annu. Rev. Nutr. 2021, 41, 479–510. [Google Scholar] [CrossRef] [PubMed]
- Raman, A.S.; Gehrig, J.L.; Venkatesh, S.; Chang, H.-W.; Hibberd, M.C.; Subramanian, S.; Kang, G.; Bessong, P.O.; Lima, A.A.M.; Kosek, M.N.; et al. A sparse covarying unit that describes healthy and impaired human gut microbiota development. Science 2019, 365, eaau4735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinley, J.N.; Fitzgerald, V.K.; Neil, E.S.; Omerigic, H.M.; Heuberger, A.L.; Weir, T.L.; McGee, R.; Vandemark, G.; Thompson, H.J. Pulse Crop Effects on Gut Microbial Populations, Intestinal Function, and Adiposity in a Mouse Model of Diet-Induced Obesity. Nutrients 2020, 12, 593. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.M.; Battson, M.L.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Weir, T.L.; Gentile, C.L. SGLT2 inhibition via dapagliflozin improves generalized vascular dysfunction and alters the gut microbiota in type 2 diabetic mice. Cardiovasc. Diabetol. 2018, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Robeson, M.S.; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020, 326504. [Google Scholar] [CrossRef]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–W188. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Costello, E.K.; Knight, R. Supervised classification of human microbiota. FEMS Microbiol. Rev. 2011, 35, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively Parallel Evolutionary Placement of Genetic Sequences. Syst. Biol. 2018, 68, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Czech, L.; Barbera, P.; Stamatakis, A. Genesis and Gappa: Processing, analyzing and visualizing phylogenetic (placement) data. Bioinformatics 2020, 36, 3263–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louca, S.; Doebeli, M. Efficient comparative phylogenetics on large trees. Bioinformatics 2017, 34, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Doak, T.G. A Parsimony Approach to Biological Pathway Reconstruction/Inference for Genomes and Metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Fulcher, C.A.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; et al. The MetaCyc database of metabolic pathways and enzymes. Nucleic Acids Res. 2018, 46, D633–D639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Gehrig, J.L.; Venkatesh, S.; Chang, H.-W.; Hibberd, M.C.; Kung, V.L.; Cheng, J.; Chen, R.Y.; Subramanian, S.; Cowardin, C.A.; Meier, M.F.; et al. Effects of microbiota-directed foods in gnotobiotic animals and undernourished children. Science 2019, 365, eaau4732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagkouvardos, I.; Lesker, T.R.; Hitch, T.C.A.; Gálvez, E.J.C.; Smit, N.; Neuhaus, K.; Wang, J.; Baines, J.F.; Abt, B.; Stecher, B.; et al. Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 2019, 7, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormerod, K.L.; Wood, D.L.A.; Lachner, N.; Gellatly, S.L.; Daly, J.N.; Parsons, J.D.; Dal’Molin, C.G.O.; Palfreyman, R.W.; Nielsen, L.K.; Cooper, M.A.; et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome 2016, 4, 36. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Song, Z.; Li, H.; Chang, L.; Pan, T.; Gu, X.; He, X.; Fan, Z. Honokiol Ameliorates High-Fat-Diet-Induced Obesity of Different Sexes of Mice by Modulating the Composition of the Gut Microbiota. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Then, C.K.; Paillas, S.; Wang, X.; Hampson, A.; Kiltie, A.E. Association of Bacteroides acidifaciens relative abundance with high-fibre diet-associated radiosensitisation. BMC Biol. 2020, 18, 102. [Google Scholar] [CrossRef]
- Ottosson, F.; Brunkwall, L.; Ericson, U.; Nilsson, P.M.; Almgren, P.; Fernandez, C.; Melander, O.; Orho-Melander, M. Connection Between BMI-Related Plasma Metabolite Profile and Gut Microbiota. J. Clin. Endocrinol. Metab. 2018, 103, 1491–1501. [Google Scholar] [CrossRef] [Green Version]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farup, P.G.; Lydersen, S.; Valeur, J. Are Nonnutritive Sweeteners Obesogenic? Associations between Diet, Faecal Microbiota, and Short-Chain Fatty Acids in Morbidly Obese Subjects. J. Obes. 2019, 2019, 4608315. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Devkumar, P.; Chattopadhyay, I. Could dysbiosis of inflammatory and anti-inflammatory gut bacteria have an implications in the development of type 2 diabetes? A pilot investigation. BMC Res. Notes 2021, 14, 52. [Google Scholar] [CrossRef] [PubMed]
- Herp, S.; Durai Raj, A.C.; Salvado Silva, M.; Woelfel, S.; Stecher, B. The human symbiont Mucispirillum schaedleri: Causality in health and disease. Med. Microbiol. Immunol. 2021, 210, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jang, J.-Y.; Kwon, M.-S.; Lim, S.K.; Kim, N.; Lee, J.; Park, H.K.; Yun, M.; Shin, M.-Y.; Jo, H.E.; et al. Mixture of Two Lactobacillus plantarum Strains Modulates the Gut Microbiota Structure and Regulatory T Cell Response in Diet-Induced Obese Mice. Mol. Nutr. Food Res. 2018, 62, 1800329. [Google Scholar] [CrossRef]
- Qian, M.; Lyu, Q.; Liu, Y.; Hu, H.; Wang, S.; Pan, C.; Duan, X.; Gao, Y.; Qi, L.-W.; Liu, W.; et al. Chitosan Oligosaccharide Ameliorates Nonalcoholic Fatty Liver Disease (NAFLD) in Diet-Induced Obese Mice. Mar. Drugs 2019, 17, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Kong, Q.; Li, X.; Zhao, J.; Zhang, H.; Chen, W.; Wang, G. A High-Fat Diet Increases Gut Microbiota Biodiversity and Energy Expenditure Due to Nutrient Difference. Nutrients 2020, 12, 3197. [Google Scholar] [CrossRef]
- Henke, M.T.; Kenny, D.J.; Cassilly, C.D.; Vlamakis, H.; Xavier, R.J.; Clardy, J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. USA 2019, 116, 12672–12677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toya, T.; Corban, M.T.; Marrietta, E.; Horwath, I.E.; Lerman, L.O.; Murray, J.A.; Lerman, A. Coronary artery disease is associated with an altered gut microbiome composition. PLoS ONE 2020, 15, e0227147. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Wang, S.; Li, H.; Lu, Z.; Shi, J.; Xu, Z. Mannan-oligosaccharide modulates the obesity and gut microbiota in high-fat diet-fed mice. Food Funct. 2018, 9, 3916–3929. [Google Scholar] [CrossRef] [PubMed]
- Petriz, B.A.; Castro, A.P.; Almeida, J.A.; Gomes, C.P.; Fernandes, G.R.; Kruger, R.H.; Pereira, R.W.; Franco, O.L. Exercise induction of gut microbiota modifications in obese, non-obese and hypertensive rats. BMC Genom. 2014, 15, 511. [Google Scholar] [CrossRef] [Green Version]
- Chua, H.-H.; Chou, H.-C.; Tung, Y.-L.; Chiang, B.-L.; Liao, C.-C.; Liu, H.-H.; Ni, Y.-H. Intestinal Dysbiosis Featuring Abundance of Ruminococcus gnavus Associates with Allergic Diseases in Infants. Gastroenterology 2018, 154, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Yassour, M.; Sauk, J.; Garner, A.; Jiang, X.; Arthur, T.; Lagoudas, G.K.; Vatanen, T.; Fornelos, N.; Wilson, R.; et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428. [Google Scholar] [CrossRef] [PubMed]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P.J. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe 2019, 26, 265–272.e264. [Google Scholar] [CrossRef]
- Fan, S.; Raychaudhuri, S.; Page, R.; Shahinozzaman, M.; Obanda, D.N. Metagenomic insights into the effects of Urtica dioica vegetable on the gut microbiota of C57BL/6J obese mice, particularly the composition of Clostridia. J. Nutr. Biochem. 2021, 91, 108594. [Google Scholar] [CrossRef]
- Zhao, F.; Song, S.; Ma, Y.; Xu, X.; Zhou, G.; Li, C. A Short-Term Feeding of Dietary Casein Increases Abundance of Lactococcus lactis and Upregulates Gene Expression Involving Obesity Prevention in Cecum of Young Rats Compared with Dietary Chicken Protein. Front. Microbiol. 2019, 10, 2411. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van de Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Hellman, J.; Horswill, A.R.; Crosby, H.A.; Francis, K.P.; Prakash, A. Elevated Gut Microbiome-Derived Propionate Levels Are Associated with Reduced Sterile Lung Inflammation and Bacterial Immunity in Mice. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- El Hage, R.; Hernandez-Sanabria, E.; Calatayud Arroyo, M.; Props, R.; Van de Wiele, T. Propionate-Producing Consortium Restores Antibiotic-Induced Dysbiosis in a Dynamic in vitro Model of the Human Intestinal Microbial Ecosystem. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Parca, F.; Koca, Y.O.; Ünay, A.n. Nutritional and Antinutritional Factors of Some Pulses Seed and Their Effects on Human Health. Int. J. Second. Metab. 2018, 5, 331–342. [Google Scholar] [CrossRef]
- Gao, R.; Duff, W.; Chizen, D.; Zello, G.A.; Chilibeck, P.D. The Effect of a Low Glycemic Index Pulse-Based Diet on Insulin Sensitivity, Insulin Resistance, Bone Resorption and Cardiovascular Risk Factors during Bed Rest. Nutrients 2019, 11, 2012. [Google Scholar] [CrossRef] [Green Version]
- Tang, Q.; Jin, G.; Wang, G.; Liu, T.; Liu, X.; Wang, B.; Cao, H. Current Sampling Methods for Gut Microbiota: A Call for More Precise Devices. Front. Cell. Infect. Microbiol. 2020, 10, 51. [Google Scholar] [CrossRef]
- Kim, D.; Hofstaedter, C.E.; Zhao, C.; Mattei, L.; Tanes, C.; Clarke, E.; Lauder, A.; Sherrill-Mix, S.; Chehoud, C.; Kelsen, J.; et al. Optimizing methods and dodging pitfalls in microbiome research. Microbiome 2017, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Laukens, D.; Brinkman, B.M.; Raes, J.; De Vos, M.; Vandenabeele, P. Heterogeneity of the gut microbiome in mice: Guidelines for optimizing experimental design. FEMS Microbiol. Rev. 2016, 40, 117–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witjes, V.M.; Boleij, A.; Halffman, W. Reducing versus Embracing Variation as Strategies for Reproducibility: The Microbiome of Laboratory Mice. Animals 2020, 10, 2415. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | High-Fat Pulse-Free Control 1 | High-Fat Pulse-Based Diet 1,5 |
|---|---|---|
| g/100 g | g/100 g | |
| Solka-Floc | 6.46 | 0 |
| Pulse Crop 5 | 0 | 40 |
| Casein | 25.85 | 17.05 |
| Cerelose | 16.15 | 0 |
| Sucrose | 8.89 | 0.3 |
| Vitamin mix 2 | 1.29 | 1.29 |
| DL-Methionine | 0.39 | 0.39 |
| L-Tryptophan 3 | 0 | 0.01 |
| Choline bitartrate (41% choline) | 0.26 | 0.26 |
| Mineral mix 4 | 5.82 | 5.82 |
| Soybean oil | 3.23 | 3.23 |
| Lard | 31.66 | 31.66 |
| Phyla | Control, % | Lentil, % | Chickpea, % | Bean, % | Dry Pea, % |
|---|---|---|---|---|---|
| Actinobacteria | 0.020 | 0.008 | 0.016 | 0.023 | 0.017 |
| Bacteroidetes | 15.533 | 64.941 1,*** | 52.212 1,* | 54.500 | 49.689 1,* |
| Deferribacteres | 0.966 | 0.166 1,** | 0.109 1,** | 0.071 1,*** | 0.294 |
| Firmicutes | 51.189 | 30.838 1,* | 45.475 | 41.992 | 48.367 2,* |
| Proteobacteria | 31.654 | 1.496 1,** | 1.898 | 1.545 1,*** | 1.484 1,** |
| Saccharibacteria | 0.000 | 0.001 | 0.013 1,*; 2,* | 0.012 | 0.015 1,*; 2,* |
| Tenericutes | 0.089 | 0.100 | 0.213 1,* | 0.239 | 0.106 |
| Verrucomicrobia | 0.549 | 2.450 | 0.064 1,*; 2,* | 1.619 | 0.026 1,*; 2,* |
| Cecal Bacteria | Lentil | Chickpea | Bean | Dry Pea |
|---|---|---|---|---|
| Adlercreutzia | ≈ | ≈ | ≈ | ≈ |
| Akkermansia muciniphila * | ↑ | ≈ | ↑ | ≈ |
| Allobaculum | ↑ | ↑ | ↑ | ↑ |
| Anaerotruncus | ↓ | ↓ | ≈ | ↓ |
| Bacteroidales * | ↑ | ↑ | ↑ | — |
| Bacteroides acidifaciens * | ↑ | ↑ | ↑ | ↑ |
| Bacteroides * | ↓ | ≈ | ↓ | ≈ |
| Bilophila * | ≈ | ≈ | ≈ | ≈ |
| Butyricicoccus pullicaecorum * | ↑ | ↑ | ↑ | ↑ |
| Christensenellaceae | ↓ | ↓ | ↓ | ↓ |
| Clostridiales (I) | ≈ | ≈ | ≈ | ≈ |
| Clostridiales (II) * | ↓ | ≈ | ≈ | ≈ |
| Clostridium colinum * | — | ≈ | — | ↑ |
| Clostridium hathewayi | ≈ | ≈ | ≈ | ≈ |
| Clostridium (I) | ≈ | ≈ | — | ≈ |
| Clostridium (II) * | — | ↑ | ↑ | ≈ |
| Clostridium methylpentosum | ↓ | ↓ | ↓ | ↓ |
| Coprococcus | ≈ | ≈ | ≈ | ≈ |
| Dehalobacterium * | ↓ | ≈ | ≈ | ↓ |
| Desulfovibrionaceae | ≈ | ≈ | ≈ | ≈ |
| Dorea * | ↓ | ↓ | ↓ | ↓ |
| Enterobacteriaceae | ≈ | ≈ | ≈ | ≈ |
| Erysipelotrichaceae | ≈ | ≈ | ≈ | ≈ |
| F16 | ≈ | ≈ | ≈ | ≈ |
| Lachnospiraceae (I) | ≈ | ↑ | ≈ | ≈ |
| Lachnospiraceae (II) | ≈ | ≈ | ≈ | ↑ |
| Lactobacillus (I) | ↑ | ↑ | ≈ | ↑ |
| Lactobacillus (II) | — | — | ≈ | — |
| Lactococcus | ↓ | ↓ | ↓ | ↓ |
| Mogibacteriaceae (I) * | ↑ | ≈ | ≈ | ≈ |
| Mogibacteriaceae (II) * | ↑ | ↑ | ↑ | ↑ |
| Mucispirillum schaedleri * | ↓ | ↓ | ↓ | ↓ |
| Muribaculaceae | ↑ | ↑ | ↑ | ↑ |
| Oscillospira | ↓ | ↓ | ↓ | ↓ |
| Parabacteroides gordonii | ≈ | ≈ | ≈ | ≈ |
| Peptococcaceae | ↓ | ↓ | ↓ | ↓ |
| Peptostreptococcaceae | — | — | — | ≈ |
| rc4 4 * | ↑ | ↑ | ↑ | ↑ |
| RF32 | ↑ | ↑ | ↑ | ↑ |
| RF39 | ≈ | ↑ | ≈ | ≈ |
| Rikenellaceae | ↑ | ↑ | ↑ | ↑ |
| Roseburia * | — | — | — | ↑ |
| Ruminococcaceae (I) | ≈ | ≈ | ≈ | ≈ |
| Ruminococcaceae (II) * | ≈ | ≈ | ≈ | ↓ |
| Ruminococcus gnavus | ↓ | ↓ | ↓ | ↓ |
| Ruminococcus (Lachnospiraceae) | ≈ | ≈ | ≈ | ≈ |
| Ruminococcus (Ruminococcaceae) | ≈ | ≈ | ↑ | ≈ |
| Streptococcus | ↓ | ↓ | ↓ | ↓ |
| Sutterella * | ↑ | ↑ | ↑ | ↑ |
| Eco-Groups | Microbial Composition |
|---|---|
| Pulse-enhanced | Allobaculum Bacteroides acidifaciens Butyricicoccus pullicaecorum Mogibacteriaceae (II) Muribaculaceae rc4 4 (Peptococcaceae) RF32 (Alphaproteobacteria) Rikenellaceae Sutterella |
| Pulse-suppressed | Christensenellaceae Clostridium methylpentosum Dorea Lactococcus Mucispirillum schaedleri Oscillospira Peptococcaceae Ruminococcus gnavus Streptococcus |
| Pulse-indifferent | Adlercreutzia Bilophila Clostridiales (I) Clostridium hathewayi Coprococcus Desulfovibrionaceae Enterobacteriaceae Erysipelotrichaceae F16 Parabacteroides gordonii Ruminococcaceae (I) Ruminococcus (Lachnospiraceae). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutsiv, T.; Weir, T.L.; McGinley, J.N.; Neil, E.S.; Wei, Y.; Thompson, H.J. Compositional Changes of the High-Fat Diet-Induced Gut Microbiota upon Consumption of Common Pulses. Nutrients 2021, 13, 3992. https://doi.org/10.3390/nu13113992
Lutsiv T, Weir TL, McGinley JN, Neil ES, Wei Y, Thompson HJ. Compositional Changes of the High-Fat Diet-Induced Gut Microbiota upon Consumption of Common Pulses. Nutrients. 2021; 13(11):3992. https://doi.org/10.3390/nu13113992
Chicago/Turabian StyleLutsiv, Tymofiy, Tiffany L. Weir, John N. McGinley, Elizabeth S. Neil, Yuren Wei, and Henry J. Thompson. 2021. "Compositional Changes of the High-Fat Diet-Induced Gut Microbiota upon Consumption of Common Pulses" Nutrients 13, no. 11: 3992. https://doi.org/10.3390/nu13113992
APA StyleLutsiv, T., Weir, T. L., McGinley, J. N., Neil, E. S., Wei, Y., & Thompson, H. J. (2021). Compositional Changes of the High-Fat Diet-Induced Gut Microbiota upon Consumption of Common Pulses. Nutrients, 13(11), 3992. https://doi.org/10.3390/nu13113992