Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sources of Oxidative Stress in the Kidney
3. Evidence That Oxidative Stress Is Operant in CKD
4. NRF2 in Kidney Disease
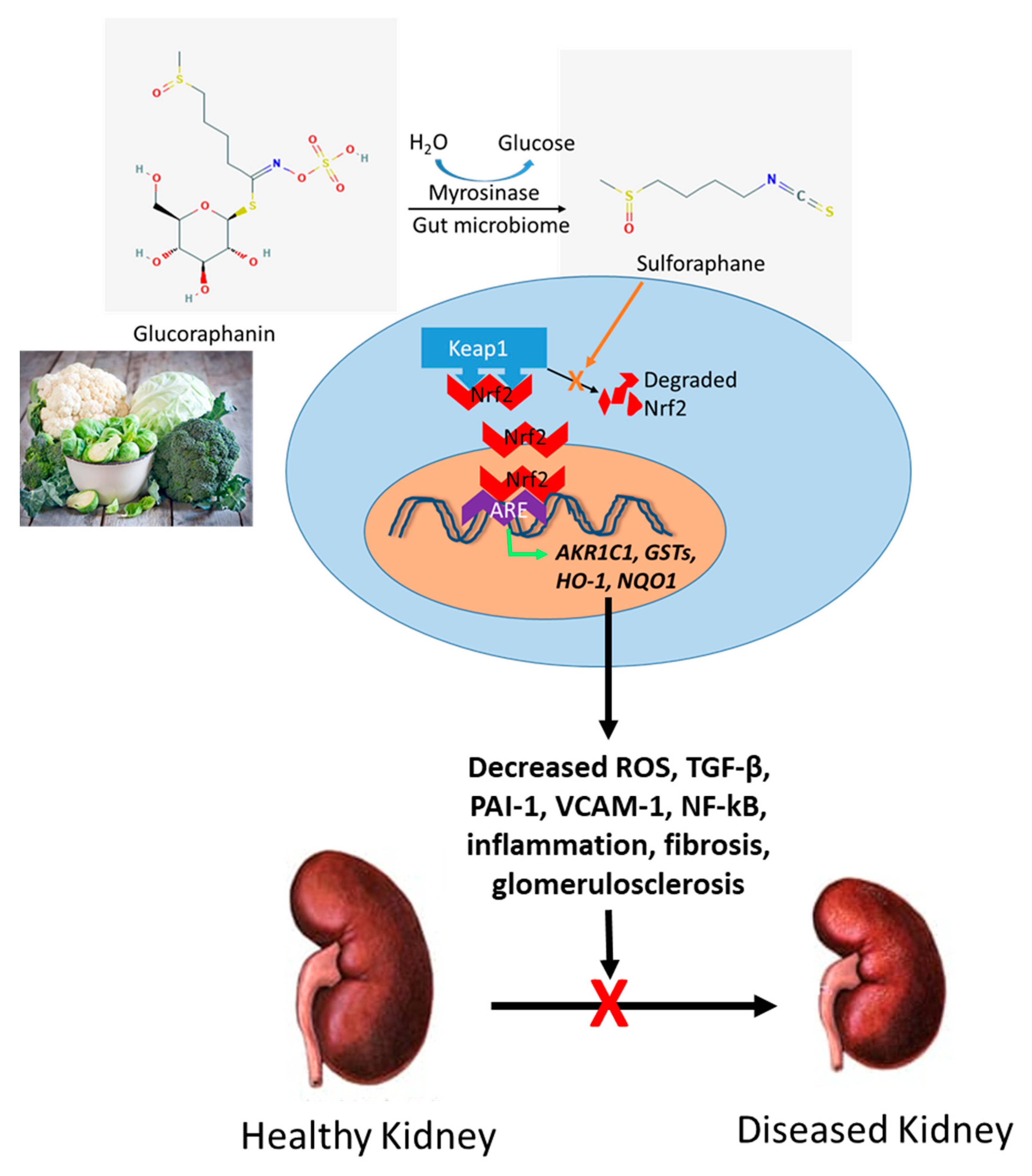
5. Potential Role for Sulforaphane (SFN) in Kidney Disease
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Health Information Center. Kidney Disease Statistics for the United States. The National Institute of Diabetes and Digestive and Kidney Diseases. Available online: https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease (accessed on 21 May 2020).
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.-y. Chronic Kidney Disease and the Risks of Death, Cardiovascular Events, and Hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- United States Renal Data System. 2018 USRDS Annual Data Report: Epidemiology of Kidney Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2018. [Google Scholar]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney Inter. Suppl. 2013, 3, 1–150. [Google Scholar]
- Rapa, S.F.; Di Iorio, B.R.; Campiglia, P.; Heidland, A.; Marzocco, S. Inflammation and Oxidative Stress in Chronic Kidney Disease-Potential Therapeutic Role of Minerals, Vitamins and Plant-Derived Metabolites. Int. J. Mol. Sci. 2019, 21, 263. [Google Scholar] [CrossRef] [PubMed]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Halliwell, B. Biochemistry of oxidative stress. Biochem. Soc. Trans. 2007, 35, 1147–1150. [Google Scholar] [CrossRef]
- Flemming, N.B.; Gallo, L.A.; Forbes, J.M. Mitochondrial Dysfunction and Signaling in Diabetic Kidney Disease: Oxidative Stress and Beyond. Semin Nephrol. 2018, 38, 101–110. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Irazabal, M.V.; Torres, V.E. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells 2020, 9, 1342. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol 2017, 13, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, D.; La Russa, D.; Marrone, A. Oxidative Imbalance and Kidney Damage: New Study Perspectives from Animal Models to Hospitalized Patients. Antioxidants 2019, 8, 594. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.S.; Wilcox, C.S. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006, 8, 1597–1607. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Griendling, K.K. Redox signaling, vascular function, and hypertension. Antioxid Redox Signal. 2008, 10, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Ettinger, L.; Liebman, S.E. Plant-Based Diets and Hypertension. Am. J. Lifestyle Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Conti, V.; Damato, A.; Ambrosio, M.; Puca, A.A.; Sciarretta, S.; Frati, G.; Vecchione, C.; Carrizzo, A. Targeting Nitric Oxide with Natural Derived Compounds as a Therapeutic Strategy in Vascular Diseases. Oxidative Med. Cell. Longev. 2016, 2016, 20. [Google Scholar] [CrossRef]
- Forbes, M.S.; Thornhill, B.A.; Park, M.H.; Chevalier, R.L. Lack of endothelial nitric-oxide synthase leads to progressive focal renal injury. Am. J. Pathol. 2007, 170, 87–99. [Google Scholar] [CrossRef]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef]
- Lehners, A.; Lange, S.; Niemann, G.; Rosendahl, A.; Meyer-Schwesinger, C.; Oh, J.; Stahl, R.; Ehmke, H.; Benndorf, R.; Klinke, A.; et al. Myeloperoxidase deficiency ameliorates progression of chronic kidney disease in mice. Am. J. Physiol. Ren. Physiol. 2014, 307, F407–F417. [Google Scholar] [CrossRef]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine dehydrogenase/xanthine oxidase and oxidative stress. Age (Omaha) 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Watanabe, S.; Kang, D.H.; Feng, L.; Nakagawa, T.; Kanellis, J.; Lan, H.; Mazzali, M.; Johnson, R.J. Uric acid, hominoid evolution, and the pathogenesis of salt-sensitivity. Hypertension 2002, 40, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Ota, T.; Tamura, Y.; Chang, W.X.; Shibata, S.; Uchida, S. Time to target uric acid to retard CKD progression. Clin. Exp. Nephrol. 2017, 21, 182–192. [Google Scholar] [CrossRef]
- Johnson, R.J.; Nakagawa, T.; Jalal, D.; Sánchez-Lozada, L.G.; Kang, D.H.; Ritz, E. Uric acid and chronic kidney disease: Which is chasing which? Nephrol. Dial. Transpl. 2013, 28, 2221–2228. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-h.; Kang, K.-S.; Kwak, M.-K. Effect of redox modulating NRF2 activators on chronic kidney disease. Molecules 2014, 19, 12727–12759. [Google Scholar] [CrossRef] [PubMed]
- Gulcin, İ. Antioxidants and antioxidant methods: An updated overview. Arch. Toxicol. 2020, 94, 651–715. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hai, C. Novel insights into redox system and the mechanism of redox regulation. Mol. Biol. Rep. 2016, 43, 607–628. [Google Scholar] [CrossRef]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis. 2006, 48, 752–760. [Google Scholar] [CrossRef]
- Kuchta, A.; Pacanis, A.; Kortas-Stempak, B.; Cwiklińska, A.; Ziętkiewicz, M.; Renke, M.; Rutkowski, B. Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press Res. 2011, 34, 12–19. [Google Scholar] [CrossRef]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef]
- Tbahriti, H.F.; Kaddous, A.; Bouchenak, M.; Mekki, K. Effect of different stages of chronic kidney disease and renal replacement therapies on oxidant-antioxidant balance in uremic patients. Biochem. Res. Int. 2013, 2013, 358985. [Google Scholar] [CrossRef] [PubMed]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, D.; Capili, A.; Choi, M.E. Mitochondrial dysfunction in kidney injury, inflammation, and disease: Potential therapeutic approaches. Kidney Res. Clin. Pract. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in Protecting the Kidney from Oxidative Damage. Int. J. Mol. Sci. 2020, 21, 2951. [Google Scholar] [CrossRef]
- Villeneuve, N.F.; Lau, A.; Zhang, D.D. Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin proteasome system: An insight into cullin-ring ubiquitin ligases. Antioxid Redox Signal. 2010, 13, 1699–1712. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Jiang, T.; Huang, Z.; Lin, Y.; Zhang, Z.; Fang, D.; Zhang, D.D. The protective role of Nrf2 in streptozotocin-induced diabetic nephropathy. Diabetes 2010, 59, 850–860. [Google Scholar] [CrossRef]
- Aleksunes, L.M.; Goedken, M.J.; Rockwell, C.E.; Thomale, J.; Manautou, J.E.; Klaassen, C.D. Transcriptional regulation of renal cytoprotective genes by Nrf2 and its potential use as a therapeutic target to mitigate cisplatin-induced nephrotoxicity. J. Pharm. Exp. 2010, 335, 2–12. [Google Scholar] [CrossRef]
- Liu, M.; Grigoryev, D.N.; Crow, M.T.; Haas, M.; Yamamoto, M.; Reddy, S.P.; Rabb, H. Transcription factor Nrf2 is protective during ischemic and nephrotoxic acute kidney injury in mice. Kidney Int. 2009, 76, 277–285. [Google Scholar] [CrossRef]
- Liu, M.; Reddy, N.M.; Higbee, E.M.; Potteti, H.R.; Noel, S.; Racusen, L.; Kensler, T.W.; Sporn, M.B.; Reddy, S.P.; Rabb, H. The Nrf2 triterpenoid activator, CDDO-imidazolide, protects kidneys from ischemia-reperfusion injury in mice. Kidney Int. 2014, 85, 134–141. [Google Scholar] [CrossRef]
- Ma, F.; Wu, J.; Jiang, Z.; Huang, W.; Jia, Y.; Sun, W.; Wu, H. P53/NRF2 mediates SIRT1’s protective effect on diabetic nephropathy. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1272–1281. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Shimizu, A.; Pastan, I.; Taguchi, K.; Naganuma, E.; Suzuki, T.; Hosoya, T.; Yokoo, T.; Saito, A.; Miyata, T.; et al. Keap1 inhibition attenuates glomerulosclerosis. Nephrol. Dial. Transpl. 2014, 29, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Souma, T.; Yu, L.; Suzuki, T.; Saigusa, D.; Ito, S.; Suzuki, N.; Yamamoto, M. Transcription factor Nrf2 hyperactivation in early-phase renal ischemia-reperfusion injury prevents tubular damage progression. Kidney Int. 2017, 91, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.; Martina, M.N.; Bandapalle, S.; Racusen, L.C.; Potteti, H.R.; Hamad, A.R.; Reddy, S.P.; Rabb, H. T Lymphocyte-Specific Activation of Nrf2 Protects from AKI. J. Am. Soc. Nephrol. 2015, 26, 2989–3000. [Google Scholar] [CrossRef]
- Tan, R.J.; Chartoumpekis, D.V.; Rush, B.M.; Zhou, D.; Fu, H.; Kensler, T.W.; Liu, Y. Keap1 hypomorphism protects against ischemic and obstructive kidney disease. Sci. Rep. 2016, 6, 36185. [Google Scholar] [CrossRef]
- Tanaka, Y.; Aleksunes, L.M.; Goedken, M.J.; Chen, C.; Reisman, S.A.; Manautou, J.E.; Klaassen, C.D. Coordinated induction of Nrf2 target genes protects against iron nitrilotriacetate (FeNTA)-induced nephrotoxicity. Toxicol. Appl. Pharm. 2008, 231, 364–373. [Google Scholar] [CrossRef]
- Yoh, K.; Hirayama, A.; Ishizaki, K.; Yamada, A.; Takeuchi, M.; Yamagishi, S.; Morito, N.; Nakano, T.; Ojima, M.; Shimohata, H.; et al. Hyperglycemia induces oxidative and nitrosative stress and increases renal functional impairment in Nrf2-deficient mice. Genes Cells 2008, 13, 1159–1170. [Google Scholar] [CrossRef]
- Zheng, H.; Whitman, S.A.; Wu, W.; Wondrak, G.T.; Wong, P.K.; Fang, D.; Zhang, D.D. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 2011, 60, 3055–3066. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Hirayama, A.; Itoh, K.; Nose, M.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 deficiency improves autoimmune nephritis caused by the fas mutation lpr. Kidney Int. 2004, 65, 1703–1713. [Google Scholar] [CrossRef]
- Zhao, S.; Ghosh, A.; Lo, C.S.; Chenier, I.; Scholey, J.W.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Nrf2 Deficiency Upregulates Intrarenal Angiotensin-Converting Enzyme-2 and Angiotensin 1-7 Receptor Expression and Attenuates Hypertension and Nephropathy in Diabetic Mice. Endocrinology 2018, 159, 836–852. [Google Scholar] [CrossRef]
- Kanda, H.; Yamawaki, K. Bardoxolone methyl: Drug development for diabetic kidney disease. Clin. Exp. Nephrol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Kurzrock, R.; Supko, J.G.; He, X.; Naing, A.; Wheler, J.; Lawrence, D.; Eder, J.P.; Meyer, C.J.; Ferguson, D.A.; et al. A phase I first-in-human trial of bardoxolone methyl in patients with advanced solid tumors and lymphomas. Clin. Cancer Res. 2012, 18, 3396–3406. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and Stage 3b-4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- de Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.P.; Wrolstad, D.; Bakris, G.L.; Chertow, G.M.; de Zeeuw, D.; Goldsberry, A.; Linde, P.G.; McCullough, P.A.; McMurray, J.J.; Wittes, J.; et al. Risk factors for heart failure in patients with type 2 diabetes mellitus and stage 4 chronic kidney disease treated with bardoxolone methyl. J. Card Fail. 2014, 20, 953–958. [Google Scholar] [CrossRef]
- Nangaku, M.; Kanda, H.; Takama, H.; Ichikawa, T.; Hase, H.; Akizawa, T. Randomized Clinical Trial on the Effect of Bardoxolone Methyl on GFR in Diabetic Kidney Disease Patients (TSUBAKI Study). Kidney Int. Rep. 2020, 5, 879–890. [Google Scholar] [CrossRef]
- Reata Announces Positive Topline Year One Results From Pivotal Phase 3 Cardinal Study of Bardoxolone Methyl in Patients With Alport Syndrome: Reata Pharmaceuticals 2019. Available online: https://www.reatapharma.com/press-releases/reata-announces-positive-topline-year-one-results-from-pivotal-phase-3-cardinal-study-of-bardoxolone-methyl-in-patients-with-alport-syndrome/ (accessed on 19 October 2020).
- Baigent, C.; Lennon, R. Should We Increase GFR with Bardoxolone in Alport Syndrome? J. Am. Soc. Nephrol. 2018, 29, 357–359. [Google Scholar] [CrossRef]
- Toto, R.D. Bardoxolone-the Phoenix? J. Am. Soc. Nephrol. 2018, 29, 360–361. [Google Scholar] [CrossRef]
- Tan, S.M.; Sharma, A.; Stefanovic, N.; Yuen, D.Y.; Karagiannis, T.C.; Meyer, C.; Ward, K.W.; Cooper, M.E.; de Haan, J.B. Derivative of bardoxolone methyl, dh404, in an inverse dose-dependent manner lessens diabetes-associated atherosclerosis and improves diabetic kidney disease. Diabetes 2014, 63, 3091–3103. [Google Scholar] [CrossRef]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharm. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, C.; Torres, E. Biochemical pharmacology of functional foods and prevention of chronic diseases of aging. Biomed. Pharmacother. 2003, 57, 251–260. [Google Scholar] [CrossRef]
- Eggler, A.L.; Savinov, S.N. Chemical and biological mechanisms of phytochemical activation of Nrf2 and importance in disease prevention. Recent Adv. Phytochem. 2013, 43, 121–155. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Shao, S.; Zhou, T.; Tsao, R. 4.53—Antimicrobials from Plants—Food Preservation and Shelf-Life Extension. In Comprehensive Biotechnology, 2nd ed.; Moo-Young, M., Ed.; Academic Press: Burlington, VT, USA, 2011; pp. 645–658. [Google Scholar] [CrossRef]
- McWalter, G.K.; Higgins, L.G.; McLellan, L.I.; Henderson, C.J.; Song, L.; Thornalley, P.J.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J. Nutr. 2004, 134, 3499s–3506s. [Google Scholar] [CrossRef]
- Kwok, C.S.; Gulati, M.; Michos, E.D.; Potts, J.; Wu, P.; Watson, L.; Loke, Y.K.; Mallen, C.; Mamas, M.A. Dietary components and risk of cardiovascular disease and all-cause mortality: A review of evidence from meta-analyses. Eur. J. Prev. Cardiol. 2019, 26, 1415–1429. [Google Scholar] [CrossRef]
- Mori, N.; Shimazu, T.; Charvat, H.; Mutoh, M.; Sawada, N.; Iwasaki, M.; Yamaji, T.; Inoue, M.; Goto, A.; Takachi, R.; et al. Cruciferous vegetable intake and mortality in middle-aged adults: A prospective cohort study. Clin. Nutr. 2019, 38, 631–643. [Google Scholar] [CrossRef]
- Zhang, X.; Shu, X.O.; Xiang, Y.B.; Yang, G.; Li, H.; Gao, J.; Cai, H.; Gao, Y.T.; Zheng, W. Cruciferous vegetable consumption is associated with a reduced risk of total and cardiovascular disease mortality. Am. J. Clin. Nutr. 2011, 94, 240–246. [Google Scholar] [CrossRef]
- Genkinger, J.M.; Platz, E.A.; Hoffman, S.C.; Comstock, G.W.; Helzlsouer, K.J. Fruit, vegetable, and antioxidant intake and all-cause, cancer, and cardiovascular disease mortality in a community-dwelling population in Washington County, Maryland. Am. J. Epidemiol. 2004, 160, 1223–1233. [Google Scholar] [CrossRef]
- Blekkenhorst, L.C.; Bondonno, C.P.; Lewis, J.R.; Devine, A.; Zhu, K.; Lim, W.H.; Woodman, R.J.; Beilin, L.J.; Prince, R.L.; Hodgson, J.M. Cruciferous and Allium Vegetable Intakes are Inversely Associated With 15-Year Atherosclerotic Vascular Disease Deaths in Older Adult Women. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Zurbau, A.; Au-Yeung, F.; Blanco Mejia, S.; Khan, T.A.; Vuksan, V.; Jovanovski, E.; Leiter, L.A.; Kendall, C.W.C.; Jenkins, D.J.A.; Sievenpiper, J.L. Relation of Different Fruit and Vegetable Sources With Incident Cardiovascular Outcomes: A Systematic Review and Meta-Analysis of Prospective Cohort Studies. J. Am. Heart Assoc. 2020, 9, e017728. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Fang, J.C.; Gao, Z.H.; Zhang, C.; Xie, S.Y. Higher intake of fruits, vegetables or their fiber reduces the risk of type 2 diabetes: A meta-analysis. J. Diabetes Investig. 2016, 7, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhong, L.; Song, Y.; Hu, Y.; Wang, G.; Sun, S. Consumption of citrus and cruciferous vegetables with incident type 2 diabetes mellitus based on a meta-analysis of prospective study. Prim. Care Diabetes 2016, 10, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, L. Cruciferous vegetables intake is associated with lower risk of renal cell carcinoma: Evidence from a meta-analysis of observational studies. PLoS ONE 2013, 8, e75732. [Google Scholar] [CrossRef] [PubMed]
- Farvid, M.S.; Holmes, M.D.; Chen, W.Y.; Rosner, B.A.; Tamimi, R.M.; Willett, W.C.; Eliassen, A.H. Postdiagnostic Fruit and Vegetable Consumption and Breast Cancer Survival: Prospective Analyses in the Nurses’ Health Studies. Cancer Res. 2020, 80, 5134–5143. [Google Scholar] [CrossRef]
- Wu, Q.J.; Yang, G.; Zheng, W.; Li, H.L.; Gao, J.; Wang, J.; Gao, Y.T.; Shu, X.O.; Xiang, Y.B. Pre-diagnostic cruciferous vegetables intake and lung cancer survival among Chinese women. Sci. Rep. 2015, 5, 10306. [Google Scholar] [CrossRef]
- Kim, H.; Caulfield, L.E.; Garcia-Larsen, V.; Steffen, L.M.; Grams, M.E.; Coresh, J.; Rebholz, C.M. Plant-Based Diets and Incident CKD and Kidney Function. Clin. J. Am. Soc. Nephrol. 2019, 14, 682. [Google Scholar] [CrossRef]
- Khatri, M.; Moon, Y.P.; Scarmeas, N.; Gu, Y.; Gardener, H.; Cheung, K.; Wright, C.B.; Sacco, R.L.; Nickolas, T.L.; Elkind, M.S.V. The Association between a Mediterranean-Style Diet and Kidney Function in the Northern Manhattan Study Cohort. Clin. J. Am. Soc. Nephrol. 2014, 9, 1868–1875. [Google Scholar] [CrossRef]
- Bernier-Jean, A.; Prince, R.L.; Lewis, J.R.; Craig, J.C.; Hodgson, J.M.; Lim, W.H.; Teixeira-Pinto, A.; Wong, G. Dietary plant and animal protein intake and decline in estimated glomerular filtration rate among elderly women: A 10-year longitudinal cohort study. Nephrol. Dial. Transpl. 2020. [Google Scholar] [CrossRef]
- Hu, E.A.; Coresh, J.; Anderson, C.A.M.; Appel, L.J.; Grams, M.E.; Crews, D.C.; Mills, K.T.; He, J.; Scialla, J.; Rahman, M.; et al. Adherence to Healthy Dietary Patterns and Risk of CKD Progression and All-Cause Mortality: Findings From the CRIC (Chronic Renal Insufficiency Cohort) Study. Am. J. Kidney Dis. 2020. [Google Scholar] [CrossRef]
- Houghton, C.A. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxidative Med. Cell. Longev. 2019, 2019, 2716870. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Fahey, J.W.; Dinkova-Kostova, A.T.; Kensler, T.W. Broccoli or Sulforaphane: Is It the Source or Dose That Matters? Molecules 2019, 24, 3593. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, N.; Coldham, N.; Gielbert, A.; Kuhnert, N.; Sauer, M.J.; King, L.J.; Ioannides, C. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br. J. Nutr. 2008, 99, 559–564. [Google Scholar] [CrossRef] [PubMed]
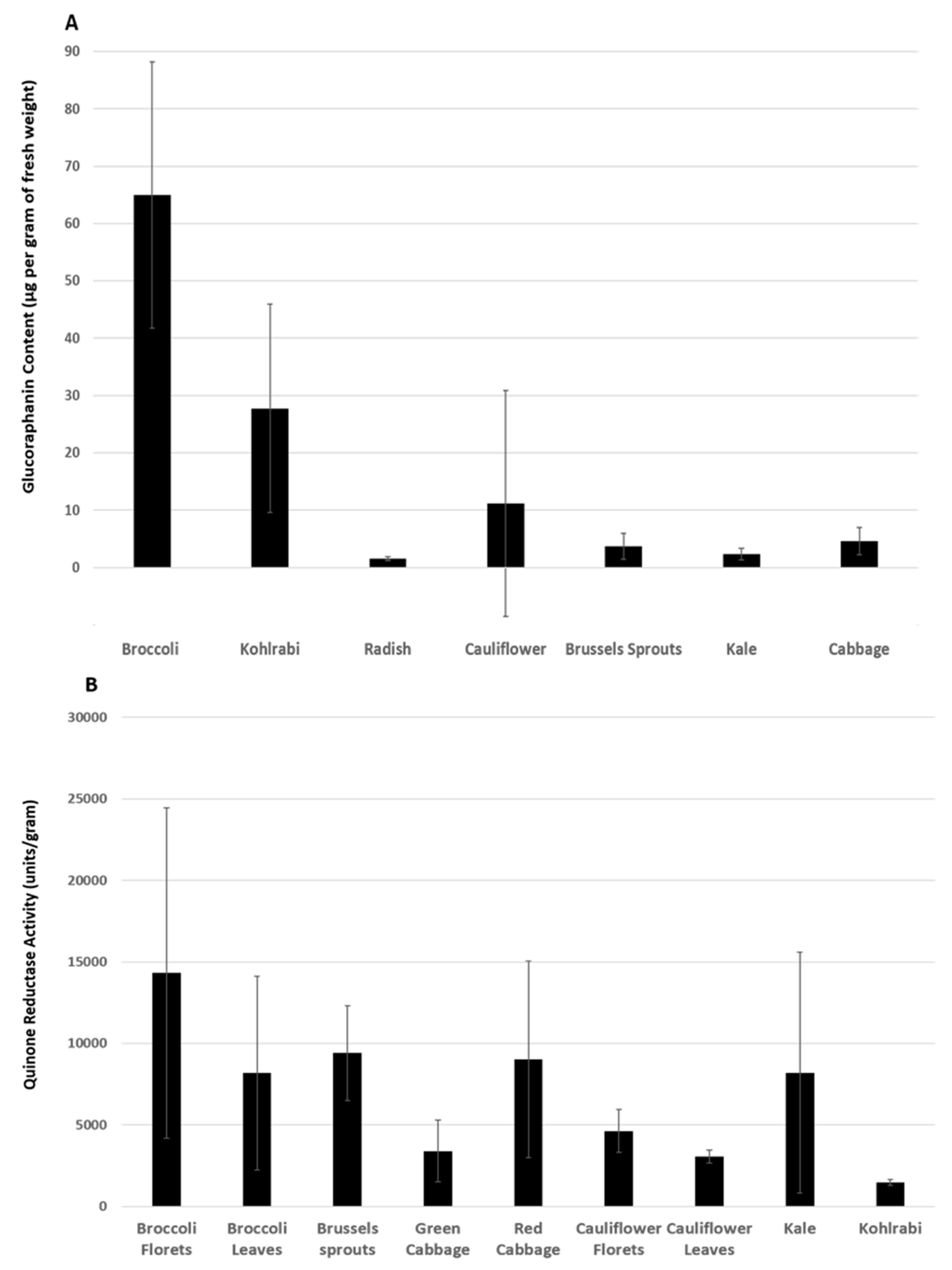
- West, L.G.; Meyer, K.A.; Balch, B.A.; Rossi, F.J.; Schultz, M.R.; Haas, G.W. Glucoraphanin and 4-hydroxyglucobrassicin contents in seeds of 59 cultivars of broccoli, raab, kohlrabi, radish, cauliflower, brussels sprouts, kale, and cabbage. J. Agric. Food Chem. 2004, 52, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Prochaska, H.J.; Santamaria, A.B.; Talalay, P. Rapid detection of inducers of enzymes that protect against carcinogens. Proc. Natl. Acad. Sci. USA 1992, 89, 2394–2398. [Google Scholar] [CrossRef] [PubMed]
- Sivapalan, T.; Melchini, A.; Saha, S.; Needs, P.W.; Traka, M.H.; Tapp, H.; Dainty, J.R.; Mithen, R.F. Bioavailability of Glucoraphanin and Sulforaphane from High-Glucoraphanin Broccoli. Mol. Nutr. Food Res. 2018, 62, e1700911. [Google Scholar] [CrossRef]
- Conaway, C.C.; Getahun, S.M.; Liebes, L.L.; Pusateri, D.J.; Topham, D.K.; Botero-Omary, M.; Chung, F.L. Disposition of glucosinolates and sulforaphane in humans after ingestion of steamed and fresh broccoli. Nutr. Cancer 2000, 38, 168–178. [Google Scholar] [CrossRef]
- Baenas, N.; Marhuenda, J.; García-Viguera, C.; Zafrilla, P.; Moreno, D.A. Influence of Cooking Methods on Glucosinolates and Isothiocyanates Content in Novel Cruciferous Foods. Foods 2019, 8, 257. [Google Scholar] [CrossRef]
- Wang, G.C.; Farnham, M.; Jeffery, E.H. Impact of thermal processing on sulforaphane yield from broccoli (Brassica oleracea L. ssp. italica). J. Agric. Food Chem. 2012, 60, 6743–6748. [Google Scholar] [CrossRef]
- Jones, R.B.; Frisina, C.L.; Winkler, S.; Imsic, M.; Tomkins, R.B. Cooking method significantly effects glucosinolate content and sulforaphane production in broccoli florets. Food Chem. 2010, 123, 237–242. [Google Scholar] [CrossRef]
- Tabart, J.; Pincemail, J.; Kevers, C.; Defraigne, J.-O.; Dommes, J. Processing effects on antioxidant, glucosinolate, and sulforaphane contents in broccoli and red cabbage. Eur. Food Res. Technol. 2018, 244, 2085–2094. [Google Scholar] [CrossRef]
- Lu, Y.; Pang, X.; Yang, T. Microwave cooking increases sulforaphane level in broccoli. Food Sci. Nutr. 2020, 8, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Galgano, F.; Favati, F.; Caruso, M.; Pietrafesa, A.; Natella, S. The influence of processing and preservation on the retention of health-promoting compounds in broccoli. J. Food Sci. 2007, 72, S130–S135. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Hollands, W.; Teucher, B.; Needs, P.W.; Narbad, A.; Ortori, C.A.; Barrett, D.A.; Rossiter, J.T.; Mithen, R.F.; Kroon, P.A. Isothiocyanate concentrations and interconversion of sulforaphane to erucin in human subjects after consumption of commercial frozen broccoli compared to fresh broccoli. Mol. Nutr. Food Res. 2012, 56, 1906–1916. [Google Scholar] [CrossRef]
- Okunade, O.; Niranjan, K.; Ghawi, S.K.; Kuhnle, G.; Methven, L. Supplementation of the Diet by Exogenous Myrosinase via Mustard Seeds to Increase the Bioavailability of Sulforaphane in Healthy Human Subjects after the Consumption of Cooked Broccoli. Mol. Nutr. Food Res. 2018, 62, e1700980. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Fahey, J.W.; Kostov, R.V.; Kensler, T.W. KEAP1 and Done? Targeting the NRF2 Pathway with Sulforaphane. Trends Food Sci. Technol. 2017, 69, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxid Med. Cell Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [PubMed]
- Shang, G.; Tang, X.; Gao, P.; Guo, F.; Liu, H.; Zhao, Z.; Chen, Q.; Jiang, T.; Zhang, N.; Li, H. Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway. J. Nutr. Biochem. 2015, 26, 596–606. [Google Scholar] [CrossRef]
- Khaleel, S.A.; Raslan, N.A.; Alzokaky, A.A.; Ewees, M.G.; Ashour, A.A.; Abdel-Hamied, H.E.; Abd-Allah, A.R. Contrast media (meglumine diatrizoate) aggravates renal inflammation, oxidative DNA damage and apoptosis in diabetic rats which is restored by sulforaphane through Nrf2/HO-1 reactivation. Chem. Biol. Interact. 2019, 309, 108689. [Google Scholar] [CrossRef]
- Wu, H.; Kong, L.; Cheng, Y.; Zhang, Z.; Wang, Y.; Luo, M.; Tan, Y.; Chen, X.; Miao, L.; Cai, L. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic Biol. Med. 2015, 89, 431–442. [Google Scholar] [CrossRef]
- Cui, W.; Bai, Y.; Miao, X.; Luo, P.; Chen, Q.; Tan, Y.; Rane, M.J.; Miao, L.; Cai, L. Prevention of diabetic nephropathy by sulforaphane: Possible role of Nrf2 upregulation and activation. Oxid Med. Cell Longev. 2012, 2012, 821936. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Yang, X.; Tang, K.; Ye, T.; Duan, C.; Lv, P.; Yan, L.; Wu, X.; Chen, Z.; Liu, J.; et al. Sulforaphane elicts dual therapeutic effects on Renal Inflammatory Injury and crystal deposition in Calcium Oxalate Nephrocalcinosis. Theranostics 2020, 10, 7319–7334. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hu, Z.; Xu, X.; Li, Z.; Chen, Y.; Dong, J. The associations of plant-based protein intake with all-cause and cardiovascular mortality in patients on peritoneal dialysis. Nutr. Metab. Cardiovasc. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.D.; Lai, T.Y.; Chien, C.T.; Yu, H.J. Activating Nrf-2 signaling depresses unilateral ureteral obstruction-evoked mitochondrial stress-related autophagy, apoptosis and pyroptosis in kidney. PLoS ONE 2012, 7, e47299. [Google Scholar] [CrossRef]
- Noorafshan, A.; Karbalay-Doust, S.; Poorshahid, M. Stereological survey of the ameliorative effects of sulforaphane and quercetin on renal tissue in unilateral ureteral obstruction in rats. Acta Clin. Croat. 2012, 51, 555–562. [Google Scholar]
- Lu, Y.; Zhang, Y.; Lou, Y.; Cui, W.; Miao, L. Sulforaphane suppresses obesity-related glomerulopathy-induced damage by enhancing autophagy via Nrf2. Life Sci. 2020, 258, 118153. [Google Scholar] [CrossRef]
- Briones-Herrera, A.; Avila-Rojas, S.H.; Aparicio-Trejo, O.E.; Cristóbal, M.; León-Contreras, J.C.; Hernández-Pando, R.; Pinzón, E.; Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Tapia, E. Sulforaphane prevents maleic acid-induced nephropathy by modulating renal hemodynamics, mitochondrial bioenergetics and oxidative stress. Food Chem. Toxicol. 2018, 115, 185–197. [Google Scholar] [CrossRef]
- Briones-Herrera, A.; Ramírez-Camacho, I.; Zazueta, C.; Tapia, E.; Pedraza-Chaverri, J. Altered proximal tubule fatty acid utilization, mitophagy, fission and supercomplexes arrangement in experimental Fanconi syndrome are ameliorated by sulforaphane-induced mitochondrial biogenesis. Free Radic Biol. Med. 2020, 153, 54–70. [Google Scholar] [CrossRef]
- Lv, D.; Zhou, Q.; Xia, Y.; You, X.; Zhao, Z.; Li, Y.; Zou, H. The Association Between Oxidative Stress Alleviation via Sulforaphane-Induced Nrf2-HO-1/NQO-1 Signaling Pathway Activation and Chronic Renal Allograft Dysfunction Improvement. Kidney Blood Press Res. 2018, 43, 191–205. [Google Scholar] [CrossRef]
- Cekauskas, A.; Bruns, H.; Manikas, M.; Herr, I.; Gross, M.L.; Zorn, M.; Jankevicius, F.; Strupas, K.; Schemmer, P. Sulforaphane decreases kidney injury after transplantation in rats: Role of mitochondrial damage. Ann. Transpl. 2013, 18, 488–496. [Google Scholar] [CrossRef]
- Senanayake, G.V.; Banigesh, A.; Wu, L.; Lee, P.; Juurlink, B.H. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. Am. J. Hypertens 2012, 25, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T. The role of methylation in gene expression. Nat. Educ. 2008, 1, 116. [Google Scholar]
- Shokeir, A.A.; Barakat, N.; Hussein, A.M.; Awadalla, A.; Harraz, A.M.; Khater, S.; Hemmaid, K.; Kamal, A.I. Activation of Nrf2 by ischemic preconditioning and sulforaphane in renal ischemia/reperfusion injury: A comparative experimental study. Physiol. Res. 2015, 64, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.Y.; Kang, N.I.; Lee, H.K.; Jang, K.Y.; Park, J.W.; Park, B.H. Sulforaphane protects kidneys against ischemia-reperfusion injury through induction of the Nrf2-dependent phase 2 enzyme. Biochem. Pharm. 2008, 75, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Thangapandiyan, S.; Ramesh, M.; Miltonprabu, S.; Hema, T.; Jothi, G.B.; Nandhini, V. Sulforaphane potentially attenuates arsenic-induced nephrotoxicity via the PI3K/Akt/Nrf2 pathway in albino Wistar rats. Env. Sci. Pollut. Res. Int. 2019, 26, 12247–12263. [Google Scholar] [CrossRef]
- Rubio-Navarro, A.; Vázquez-Carballo, C.; Guerrero-Hue, M.; García-Caballero, C.; Herencia, C.; Gutiérrez, E.; Yuste, C.; Sevillano, Á.; Praga, M.; Egea, J.; et al. Nrf2 Plays a Protective Role Against Intravascular Hemolysis-Mediated Acute Kidney Injury. Front. Pharm. 2019, 10, 740. [Google Scholar] [CrossRef]
- Negrette-Guzmán, M.; Huerta-Yepez, S.; Medina-Campos, O.N.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Torres, I.; Tapia, E.; Pedraza-Chaverri, J. Sulforaphane Attenuates Gentamicin-Induced Nephrotoxicity: Role of Mitochondrial Protection. Evid. -Based Complementary Altern. Med. 2013, 2013, 135314. [Google Scholar] [CrossRef]
- Loboda, A.; Stachurska, A.; Sobczak, M.; Podkalicka, P.; Mucha, O.; Jozkowicz, A.; Dulak, J. Nrf2 deficiency exacerbates ochratoxin A-induced toxicity in vitro and in vivo. Toxicology 2017, 389, 42–52. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Liu, X.; Wang, L.; Ren, F.; Wang, X.; Leng, X. Hippuric Acid Promotes Renal Fibrosis by Disrupting Redox Homeostasis via Facilitation of NRF2-KEAP1-CUL3 Interactions in Chronic Kidney Disease. Antioxidants 2020, 9, 783. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, K.M.; Jeong, J.U.; Shin, J.M.; Kang, J.H.; Bang, K.; Kim, J.H. Nrf2-Heme Oxygenase-1 Attenuates High-Glucose-Induced Epithelial-to-Mesenchymal Transition of Renal Tubule Cells by Inhibiting ROS-Mediated PI3K/Akt/GSK-3β Signaling. J. Diabetes Res. 2019, 2019, 2510105. [Google Scholar] [CrossRef]
- Atilano-Roque, A.; Wen, X.; Aleksunes, L.M.; Joy, M.S. Nrf2 activators as potential modulators of injury in human kidney cells. Toxicol. Rep. 2016, 3, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Beltrán, C.E.; Calderón-Oliver, M.; Martínez-Abundis, E.; Tapia, E.; Zarco-Márquez, G.; Zazueta, C.; Pedraza-Chaverri, J. Protective effect of sulforaphane against cisplatin-induced mitochondrial alterations and impairment in the activity of NAD(P)H: Quinone oxidoreductase 1 and γ glutamyl cysteine ligase: Studies in mitochondria isolated from rat kidney and in LLC-PK1 cells. Toxicol. Lett. 2010, 199, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zheng, J.; Zhao, T.; Tang, X.; Hu, N. Uranium-induced rat kidney cell cytotoxicity is mediated by decreased endogenous hydrogen sulfide (H2S) generation involved in reduced Nrf2 levels. Toxicol. Res. 2016, 5, 660–673. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef]
- Cross, J.V.; Rady, J.M.; Foss, F.W.; Lyons, C.E.; Macdonald, T.L.; Templeton, D.J. Nutrient isothiocyanates covalently modify and inhibit the inflammatory cytokine macrophage migration inhibitory factor (MIF). Biochem. J. 2009, 423, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, J.C.; Tin, A.; Pourafshar, S.; Cechova, S.; Wang, Y.T.; Sung, S.J.; Bodonyi-Kovacs, G.; Cross, J.V.; Yang, G.; Nguyen, N.; et al. GSTM1 Deletion Exaggerates Kidney Injury in Experimental Mouse Models and Confers the Protective Effect of Cruciferous Vegetables in Mice and Humans. J. Am. Soc. Nephrol. 2020, 31, 102–116. [Google Scholar] [CrossRef]
- Langston-Cox, A.; Anderson, D.; Creek, D.J.; Palmer, K.; Wallace, E.M.; Marshall, S.A. Measuring Sulforaphane and Its Metabolites in Human Plasma: A High Throughput Method. Molecules 2020, 25, 829. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, G.; Tian, H.; Hu, Y.; Wu, S.; Geng, Y.; Lin, K.; Wu, W. Sulforaphane metabolites cause apoptosis via microtubule disruption in cancer. Endocr. Relat Cancer 2018, 25, 255–268. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Y.; Zheng, Z.; Li, J.; Yan, Y.; Wu, W. Sulforaphane metabolites reduce resistance to paclitaxel via microtubule disruption. Cell Death Dis. 2018, 9, 1134. [Google Scholar] [CrossRef]
- Berhane, K.; Widersten, M.; Engström, A.; Kozarich, J.W.; Mannervik, B. Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. USA 1994, 91, 1480–1484. [Google Scholar] [CrossRef]
- Hubatsch, I.; Ridderström, M.; Mannervik, B. Human glutathione transferase A4-4: An alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem. J. 1998, 330 Pt 1, 175–179. [Google Scholar] [CrossRef]
- Paumi, C.M.; Smitherman, P.K.; Townsend, A.J.; Morrow, C.S. Glutathione S-transferases (GSTs) inhibit transcriptional activation by the peroxisomal proliferator-activated receptor gamma (PPAR gamma) ligand, 15-deoxy-delta 12,14prostaglandin J2 (15-d-PGJ2). Biochemistry 2004, 43, 2345–2352. [Google Scholar] [CrossRef] [PubMed]
- Bernardini, S.; Hirvonen, A.; Järventaus, H.; Norppa, H. Trans-stilbene oxide-induced sister chromatid exchange in cultured human lymphocytes: Influence of GSTM1 and GSTT1 genotypes. Mutagenesis 2001, 16, 277–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chang, J.; Ma, J.Z.; Zeng, Q.; Cechova, S.; Gantz, A.; Nievergelt, C.; O’Connor, D.; Lipkowitz, M.; Le, T.H. Loss of GSTM1, a NRF2 target, is associated with accelerated progression of hypertensive kidney disease in the African American Study of Kidney Disease (AASK). Am. J. Physiol. Ren. Physiol. 2013, 304, F348–F355. [Google Scholar] [CrossRef] [PubMed]
- Tin, A.; Scharpf, R.; Estrella, M.M.; Yu, B.; Grove, M.L.; Chang, P.P.; Matsushita, K.; Köttgen, A.; Arking, D.E.; Boerwinkle, E.; et al. The Loss of GSTM1 Associates with Kidney Failure and Heart Failure. J. Am. Soc. Nephrol. 2017, 28, 3345–3352. [Google Scholar] [CrossRef] [PubMed]
- Gasper, A.V.; Al-Janobi, A.; Smith, J.A.; Bacon, J.R.; Fortun, P.; Atherton, C.; Taylor, M.A.; Hawkey, C.J.; Barrett, D.A.; Mithen, R.F. Glutathione S-transferase M1 polymorphism and metabolism of sulforaphane from standard and high-glucosinolate broccoli. Am. J. Clin. Nutr. 2005, 82, 1283–1291. [Google Scholar] [CrossRef]
- Szczesny-Malysiak, E.; Stojak, M.; Campagna, R.; Grosicki, M.; Jamrozik, M.; Kaczara, P.; Chlopicki, S. Bardoxolone Methyl Displays Detrimental Effects on Endothelial Bioenergetics, Suppresses Endothelial ET-1 Release, and Increases Endothelial Permeability in Human Microvascular Endothelium. Oxid. Med. Cell Longev. 2020, 2020, 4678252. [Google Scholar] [CrossRef]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker Exploration in Human Peripheral Blood Mononuclear Cells for Monitoring Sulforaphane Treatment Responses in Autism Spectrum Disorder. Sci. Rep. 2020, 10, 5822. [Google Scholar] [CrossRef]
- Atwell, L.L.; Hsu, A.; Wong, C.P.; Stevens, J.F.; Bella, D.; Yu, T.-W.; Pereira, C.B.; Löhr, C.V.; Christensen, J.M.; Dashwood, R.H.; et al. Absorption and chemopreventive targets of sulforaphane in humans following consumption of broccoli sprouts or a myrosinase-treated broccoli sprout extract. Mol. Nutr. Food Res. 2015, 59, 424–433. [Google Scholar] [CrossRef]
- Ye, L.; Dinkova-Kostova, A.T.; Wade, K.L.; Zhang, Y.; Shapiro, T.A.; Talalay, P. Quantitative determination of dithiocarbamates in human plasma, serum, erythrocytes and urine: Pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin. Chim. Acta 2002, 316, 43–53. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebman, S.E.; Le, T.H. Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients 2021, 13, 266. https://doi.org/10.3390/nu13010266
Liebman SE, Le TH. Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients. 2021; 13(1):266. https://doi.org/10.3390/nu13010266
Chicago/Turabian StyleLiebman, Scott E., and Thu H. Le. 2021. "Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease" Nutrients 13, no. 1: 266. https://doi.org/10.3390/nu13010266
APA StyleLiebman, S. E., & Le, T. H. (2021). Eat Your Broccoli: Oxidative Stress, NRF2, and Sulforaphane in Chronic Kidney Disease. Nutrients, 13(1), 266. https://doi.org/10.3390/nu13010266