An Examination of Serum Acylcarnitine and Amino Acid Profiles at Different Time Point of Ketogenic Diet Therapy and Their Association of Ketogenic Diet Effectiveness

 ,
,
Abstract
1. Introduction
2. Materials and Methods
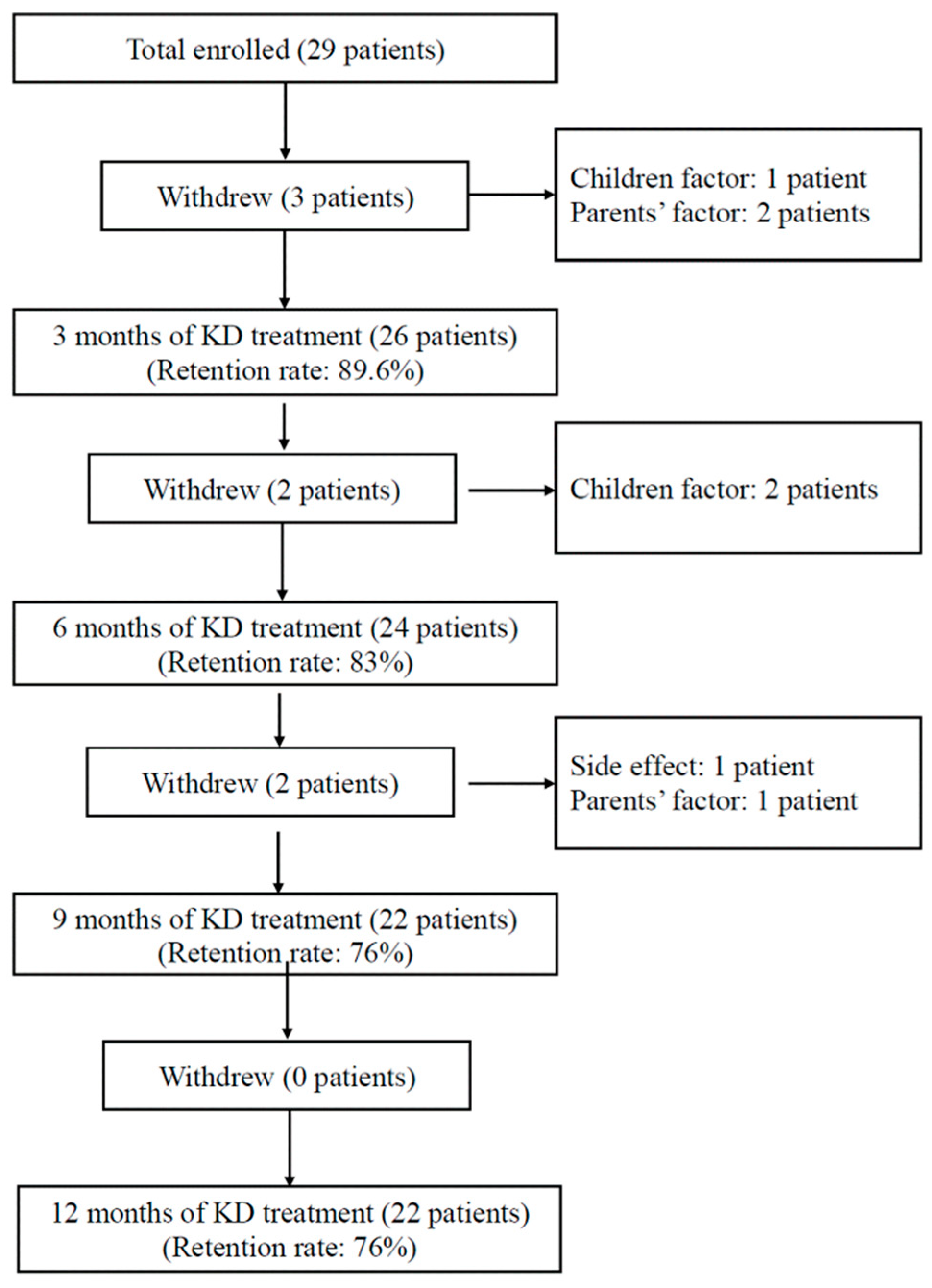
2.1. Participants
2.2. Study Protocol
2.3. Determination of Response to KDT
2.4. Tandem Mass Spectrometry
2.5. Statistical Analysis
3. Results
3.1. Clinical Characteristics
3.2. Metabolic Changes after KDT
3.3. Short-Chain Acylcarnitines after KDT
3.4. Medium-Chain Acylcarnitines after KDT
3.5. Long-Chain Acylcarnitnes after KDT
3.6. Amino Acids after KDT
3.7. Free Carnitine after KDT
3.8. Differences in Metabolic Parameters between Responders and Non-Responders
3.9. Free Carnitine and βHB Levels in Responders
3.10. Short-Chain Acylcarnitines in Responders
3.11. Medium-Chain Acylcarnitines in Responders
3.12. Long-Chain Acylcarnitines in Responders
3.13. Amino Acids in Responders
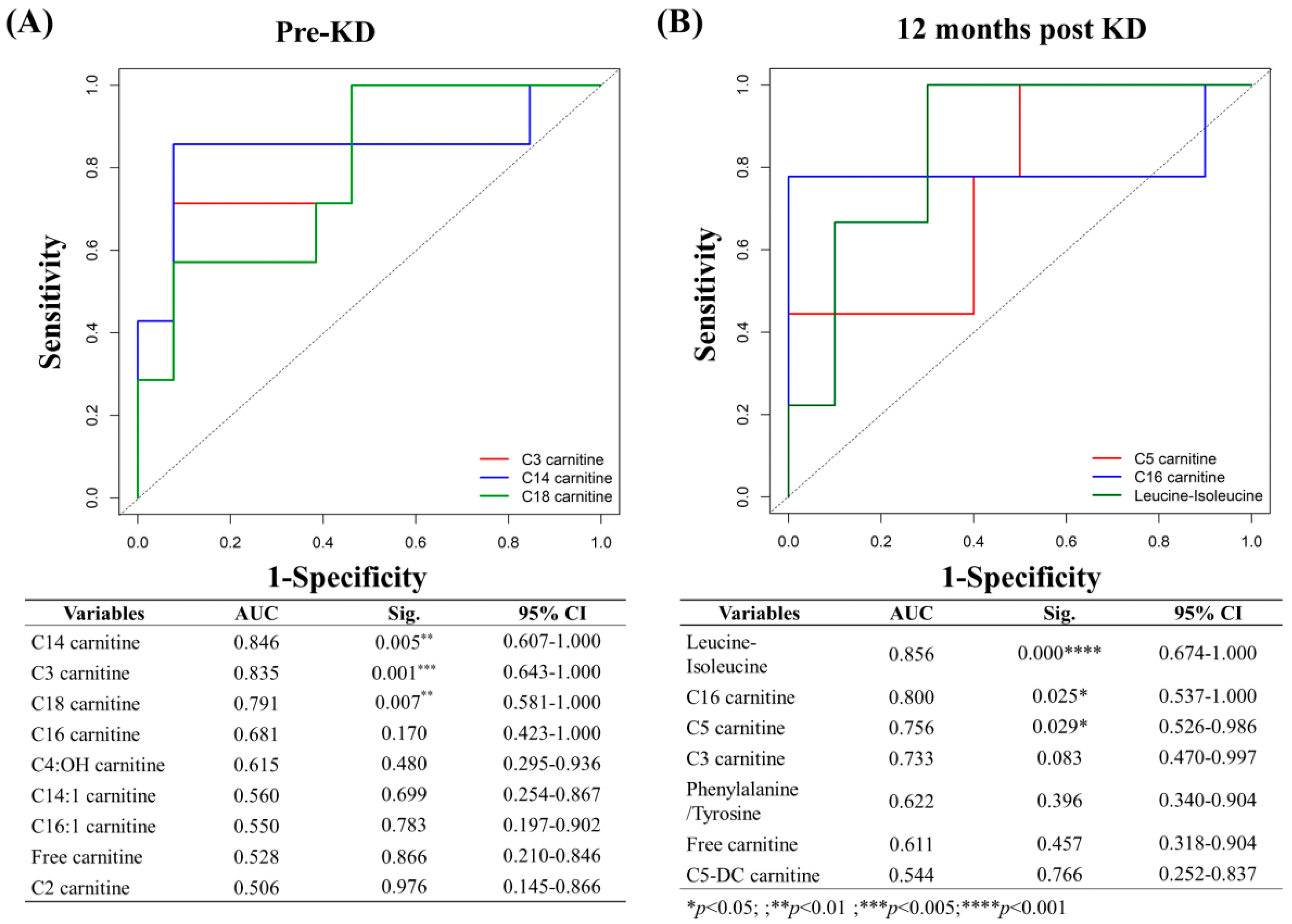
3.14. Predictors and Associated Factors of KD Effectiveness
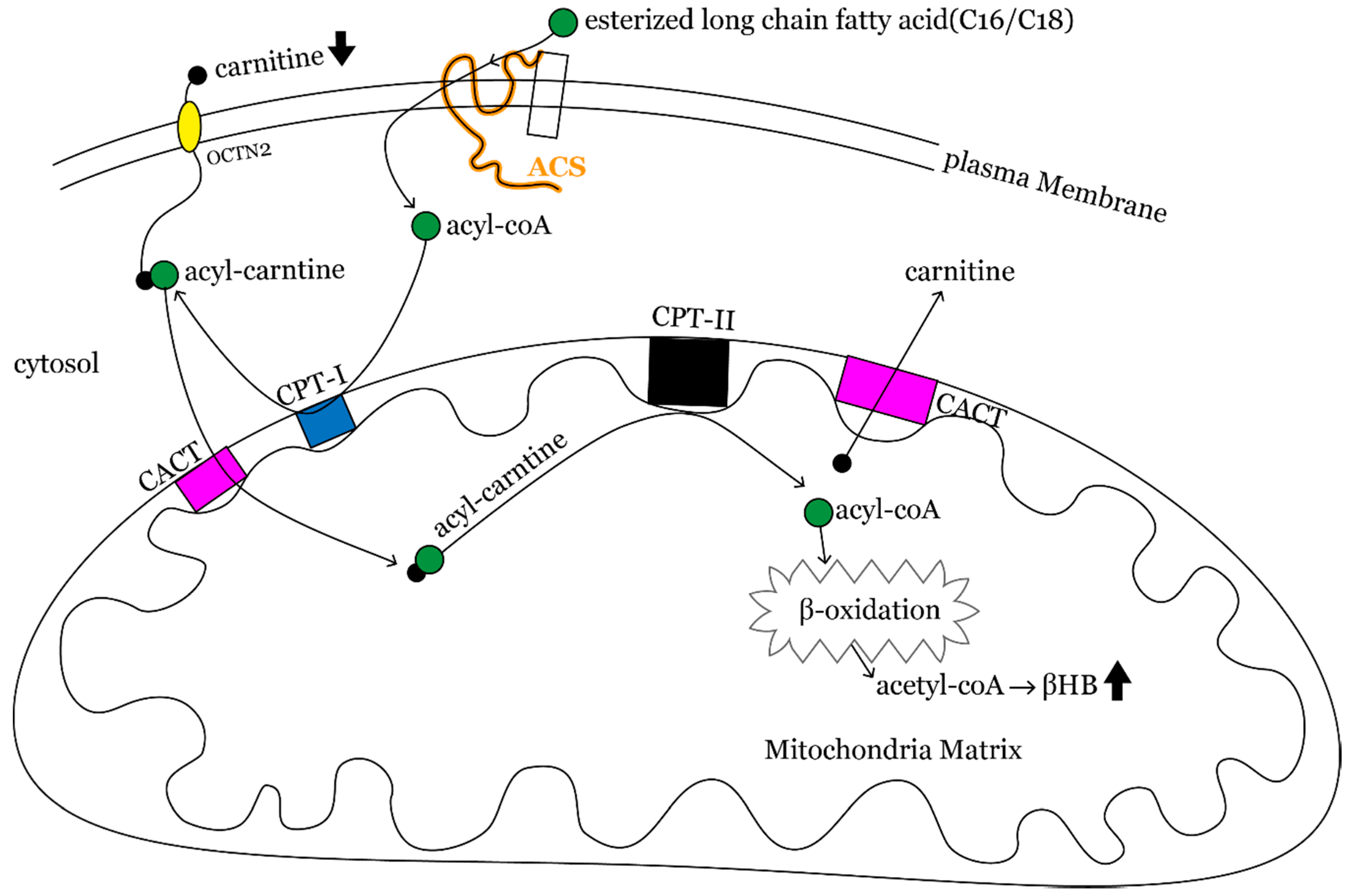
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vining, E.P.; Freeman, J.M.; Ballaban-Gil, K.; Camfield, C.S.; Camfield, P.R.; Holmes, G.L.; Shinnar, S.; Shuman, R.; Trevathan, E.; Wheless, J.W. A multicenter study of the efficacy of the ketogenic diet. Arch. Neurol. 1998, 55, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.M.; Vining, E.P.; Pillas, D.J.; Pyzik, P.L.; Casey, J.C.; Kelly, L.M. The efficacy of the ketogenic diet-1998: A prospective evaluation of intervention in 150 children. Pediatrics 1998, 102, 1358–1363. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.M.; Keene, D.L.; Whiting, S.E.; Jacob, P.J.; Champagne, J.R.; Humphreys, P. Ketogenic diet in the treatment of refractory epilepsy in childhood. Pediatr. Neurol. 1999, 21, 548–552. [Google Scholar] [CrossRef]
- Katyal, N.G.; Koehler, A.N.; McGhee, B.; Foley, C.M.; Crumrine, P.K. The ketogenic diet in refractory epilepsy: The experience of Children’s Hospital of Pittsburgh. Clin. Pediatr. 2000, 39, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Wlodarek, D. Role of Ketogenic Diets in Neurodegenerative Diseases (Alzheimer’s Disease and Parkinson’s Disease). Nutrients 2019, 11, 169. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Zupec-Kania, B.A.; Amark, P.E.; Ballaban-Gil, K.R.; Bergqvist, A.G.C.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving the ketogenic diet: Recommendations of the International Ketogenic Diet Study Group. Epilepsia 2009, 50, 304–317. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Cross, J.H.; Sander, J.W.; Sisodiya, S.M. Can we predict a favourable response to Ketogenic Diet Therapies for drug-resistant epilepsy? Epilepsy Res. 2013, 106, 1–16. [Google Scholar] [CrossRef]
- Meidenbauer, J.J.; Mukherjee, P.; Seyfried, T.N. The glucose ketone index calculator: A simple tool to monitor therapeutic efficacy for metabolic management of brain cancer. Nutr. Metab. 2015, 12, 12. [Google Scholar] [CrossRef]
- Schoeler, N.E.; Bell, G.; Yuen, A.; Kapelner, A.D.; Heales, S.J.R.; Cross, J.H.; Sisodiya, S. An examination of biochemical parameters and their association with response to ketogenic dietary therapies. Epilepsia 2017, 58, 893–900. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Booth, G.; Sanchez, A.C.; Woodbury-Kolb, J. Carnitine levels and the ketogenic diet. Epilepsia 2001, 42, 1445–1451. [Google Scholar] [CrossRef]
- Boison, D. New insights into the mechanisms of the ketogenic diet. Curr. Opin. Neurol. 2017, 30, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.M.; Kossoff, E.H.; Hartman, A.L. The ketogenic diet: One decade later. Pediatrics 2007, 119, 535–543. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea Meira, I.; Romao, T.T.; Pires do Prado, H.J.; Kruger, L.T.; Pires, M.E.P.; da Conceicao, P.O. Ketogenic Diet and Epilepsy: What We Know So Far. Front. Neurosci. 2019, 13, 5. [Google Scholar] [CrossRef] [PubMed]
- Sariego-Jamardo, A.; Garcia-Cazorla, A.; Artuch, R.; Castejón, E.; García-Arenas, D.; Molero-Luis, M.; Ormazábal, A.; Sanmartí, F.X.; Molero, M. Efficacy of the Ketogenic Diet for the Treatment of Refractory Childhood Epilepsy: Cerebrospinal Fluid Neurotransmitters and Amino Acid Levels. Pediatr. Neurol. 2015, 53, 422–426. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Melo, T.M.; Nissim, I.; Sonnewald, U.; Nissim, I. The ketogenic diet and brain metabolism of amino acids: Relationship to the anticonvulsant effect. Annu. Rev. Nutr. 2007, 27, 415–430. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Lazarow, A.; Nissim, I. Ketogenic diet, brain glutamate metabolism and seizure control. Prostaglandins Leukot Essent. Fat. Acids 2004, 70, 277–285. [Google Scholar] [CrossRef]
- Jirapinyo, P.; Kankirawatana, P.; Densupsoontorn, N.; Thamonsiri, N.; Wongarn, R. High plasma branched-chain amino acids:aromatic amino acids ratio in children on the ketogenic diet: A mechanism in controlling epilepsy. J. Med. Assoc. Thai. 2004, 87, 432–437. [Google Scholar]
- Kwan, P.; Arzimanoglou, A.; Berg, A.T.; Brodie, M.J.; Hauser, W.A.; Mathern, G.; Moshé, S.L.; Perucca, E.; Wiebe, S.; French, J. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010, 51, 1069–1077. [Google Scholar] [CrossRef]
- Yala, A.; Lehman, C.; Schuster, T.; Portnoi, T.; Barzilay, R. A Deep Learning Mammography-based Model for Improved Breast Cancer Risk Prediction. Radiology 2019, 292, 60–66. [Google Scholar] [CrossRef]
- Soeters, M.R.; Serlie, M.J.; Sauerwein, H.P.; Duran, M.; Ruiter, J.P.; Kulik, W.; Ackermans, M.T.; Minkler, P.E.; Hoppel, C.L.; Wanders, R.J.A.; et al. Characterization of D-3-hydroxybutyrylcarnitine (ketocarnitine): An identified ketosis-induced metabolite. Metabolism 2012, 61, 966–973. [Google Scholar] [CrossRef]
- Rinaldo, P.; Cowan, T.M.; Matern, D. Acylcarnitine profile analysis. Genet. Med. 2008, 10, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; Delany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, C.; St-Pierre, V.; Pierotti, T.; Fortier, M.; Castellano, C.A.; Cunnane, S.C. Tricaprylin Alone Increases Plasma Ketone Response More Than Coconut Oil or Other Medium-Chain Triglycerides: An Acute Crossover Study in Healthy Adults. Curr. Dev. Nutr. 2017, 1, e000257. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, D.C.; Bohan, T.P.; Coulter, D.L.; Dreifuss, F.E.; Greenwood, R.S.; Nordli, D.R.; Shields, W.D.; Stafstrom, C.E.; Tein, I. L-carnitine supplementation in childhood epilepsy: Current perspectives. Epilepsia 1998, 39, 1216–1225. [Google Scholar] [CrossRef]
- Longo, N.; Amat di San Filippo, C.; Pasquali, M. Disorders of carnitine transport and the carnitine cycle. Am. J. Med. Genet. C Semin. Med. Genet. 2006, 142C, 77–85. [Google Scholar] [CrossRef]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef]
- Swink, T.D.; Vining, E.P.; Freeman, J.M. The ketogenic diet: 1997. Adv. Pediatr. 1997, 44, 297–329. [Google Scholar]
- Couce, M.L.; Sánchez-Pintos, P.; Diogo, L.; Leão-Teles, E.; Martins, E.; Santos, H.; Couce, M.L.; Delgado-Pecellín, C.; Castiñeiras, D.; Cocho, J.A.; et al. Newborn screening for medium-chain acyl-CoA dehydrogenase deficiency: Regional experience and high incidence of carnitine deficiency. Orphanet J. Rare Dis. 2013, 8, 102. [Google Scholar] [CrossRef]
- Costa, C.C.; de Almeida, I.T.; Jakobs, C.; Poll-The, B.T.; Duran, M. Dynamic changes of plasma acylcarnitine levels induced by fasting and sunflower oil challenge test in children. Pediatr. Res. 1999, 46, 440–444. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Genuth, S.M. Carnitine metabolism in normal-weight and obese human subjects during fasting. Am. J. Physiol. 1980, 238, E409–E415. [Google Scholar] [CrossRef]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Lazarow, A.; Nissim, I. Ketogenic diet, amino acid metabolism, and seizure control. J. Neurosci. Res. 2001, 66, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Yudkoff, M.; Daikhin, Y.; Nissim, I.; Horyn, O.; Lazarow, A.; Luhovyy, B.; Wehrli, S. Response of brain amino acid metabolism to ketosis. Neurochem. Int. 2005, 47, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Millichap, J.G. CSF Amino Acids, Pterins and Mechanism of the Ketogenic Diet. Pediatr. Neurol. Briefs 2015, 29, 79. [Google Scholar] [CrossRef][Green Version]
- Lima, P.A.; Sampaio, L.P.; Damasceno, N.R. Neurobiochemical mechanisms of a ketogenic diet in refractory epilepsy. Clinics 2014, 69, 699–705. [Google Scholar] [CrossRef]
- Szot, P.; Weinshenker, D.; Rho, J.M.; Storey, T.W.; Schwartzkroin, P.A. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Brain Res. Dev. Brain Res. 2001, 129, 211–214. [Google Scholar] [CrossRef]
- Weinshenker, D. The contribution of norepinephrine and orexigenic neuropeptides to the anticonvulsant effect of the ketogenic diet. Epilepsia 2008, 49 (Suppl. 8), 104–107. [Google Scholar] [CrossRef]
- Liebhaber, G.; Pascher, B.; Gempel, K.; Baumeister, F.A. Asymptomatic carnitine depletion on ketogenic diet in patients with pharmacoresistant epilepsies. Klin. Padiatr. 2006, 218, 260–263. [Google Scholar] [CrossRef]
- Kava, M.P.; Robertson, A.; Greed, L.; Balasubramaniam, S. Ketogenic diet, a potentially valuable therapeutic option for the management of refractory epilepsy in classical neonatal nonketotic hyperglycinemia: A case report. Eur. J. Clin. Nutr. 2019, 73, 961–965. [Google Scholar] [CrossRef]
- Cusmai, R.; Martinelli, D.; Moavero, R.; Vici, C.D.; Vigevano, F.; Castana, C.; Elia, M.; Bernabei, S.; Bevivino, E. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur. J. Paediatr. Neurol. 2012, 16, 509–513. [Google Scholar] [CrossRef]
- Lheureux, P.E.; Penaloza, A.; Zahir, S.; Gris, M. Science review: Carnitine in the treatment of valproic acid-induced toxicity—What is the evidence? Crit. Care 2005, 9, 431–440. [Google Scholar] [CrossRef][Green Version]
- Silva, M.F.; Jakobs, C.; Duran, M.; de Almeida, I.T.; Wanders, R.J. Valproate induces in vitro accumulation of long-chain fatty acylcarnitines. Mol. Genet. Metab. 2001, 73, 358–361. [Google Scholar] [CrossRef]
- Nakajima, Y.; Ito, T.; Maeda, Y.; Ichiki, S.; Kobayashi, S.; Ando, N.; Hussein, M.H.; Kurono, Y.; Sugiyama, N.; Togari, H. Evaluation of valproate effects on acylcarnitine in epileptic children by LC-MS/MS. Brain Dev. 2011, 33, 816–823. [Google Scholar] [CrossRef]
- Riva, R.; Albani, F.; Gobbi, G.; Santucci, M.; Baruzzi, A. Carnitine disposition before and during valproate therapy in patients with epilepsy. Epilepsia 1993, 34, 184–187. [Google Scholar] [CrossRef]
- Coulter, D.L. Carnitine deficiency in epilepsy: Risk factors and treatment. J. Child Neurol. 1995, 10, S32–S39. [Google Scholar] [CrossRef]
- Hug, G.; McGraw, C.A.; Bates, S.R.; Landrigan, E.A. Reduction of serum carnitine concentrations during anticonvulsant therapy with phenobarbital, valproic acid, phenytoin, and carbamazepine in children. J. Pediatr. 1991, 119, 799–802. [Google Scholar] [CrossRef]
- Opala, G.; Winter, S.; Vance, C.; Vance, H.; Hutchison, H.T.; Linn, L.S. The effect of valproic acid on plasma carnitine levels. Am. J. Dis. Child 1991, 145, 999–1001. [Google Scholar] [CrossRef]
- Hiraoka, A.; Arato, T.; Tominaga, I. Reduction in blood free carnitine levels in association with changes in sodium valproate (VPA) disposition in epileptic patients treated with VPA and other anti-epileptic drugs. Biol. Pharm. Bull. 1997, 20, 91–93. [Google Scholar] [CrossRef][Green Version]
- Beghi, E.; Bizzi, A.; Codegoni, A.M.; Trevisan, D.; Torri, W. Valproate, carnitine metabolism, and biochemical indicators of liver function. Collaborative Group for the Study of Epilepsy. Epilepsia 1990, 31, 346–352. [Google Scholar] [CrossRef]
- Auro, K.; Joensuu, A.; Fischer, K.; Kettunen, J.; Salo, P.; Mattsson, H.; Niironen, M.; Kaprio, J.; Eriksson, J.G.; Lehtimäki, T.; et al. A metabolic view on menopause and ageing. Nat. Commun. 2014, 5, 4708. [Google Scholar] [CrossRef] [PubMed]
- Kochhar, S.; Jacobs, D.M.; Ramadan, Z.; Berruex, F.; Fuerholz, A.; Fay, L.B. Probing gender-specific metabolism differences in humans by nuclear magnetic resonance-based metabonomics. Anal. Biochem. 2006, 352, 274–281. [Google Scholar] [CrossRef]
- Mittelstrass, K.; Ried, J.S.; Yu, Z.; Krumsiek, J.; Gieger, C.; Prehn, C.; Roemisch-Margl, W.; Polonikov, A.; Peters, A.; Theis, F.J.; et al. Discovery of sexual dimorphisms in metabolic and genetic biomarkers. PLoS Genet. 2011, 7, e1002215. [Google Scholar] [CrossRef]
- Slupsky, C.M.; Rankin, K.N.; Wagner, J.; Fu, H.; Chang, D.; Weljie, A.M.; Saude, E.J.; Lix, B.; Adamko, D.J.; Shah, S.; et al. Investigations of the effects of gender, diurnal variation, and age in human urinary metabolomic profiles. Anal. Chem. 2007, 79, 6995–7004. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Fatty Acid, n = 22 | |||||||||||||||||||||
| Free Carnitine | C2 Carnitine | C3 Carnitine | C4:OH Carnitine | C4-DC Carnitine | C5 Carnitine | C5:OH Carnitine | |||||||||||||||
| Months | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p |
| 0 | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - |
| 3 | −3.08 | 2.07 | 0.136 | 7.29 | 2.40 | 0.002 *** | −0.64 | 0.24 | 0.006 ** | 0.25 | 0.04 | 0.000 **** | −0.25 | 0.04 | 0.000 **** | −0.02 | 0.01 | 0.253 | −0.03 | 0.02 | 0.093 |
| 6 | −3.91 | 2.06 | 0.057 | 8.15 | 2.80 | 0.004 *** | −0.80 | 0.19 | 0.000 **** | 0.26 | 0.04 | 0.000 **** | −0.26 | 0.05 | 0.000 **** | −0.02 | 0.01 | 0.063 | −0.05 | 0.02 | 0.005 ** |
| 9 | −5.10 | 1.69 | 0.003 *** | 5.12 | 2.46 | 0.037 * | −0.93 | 0.16 | 0.000 **** | 0.23 | 0.04 | 0.000 **** | −0.30 | 0.04 | 0.000 **** | −0.02 | 0.01 | 0.044 * | −0.05 | 0.02 | 0.008 ** |
| 12 | −4.07 | 1.97 | 0.039 * | 7.14 | 3.12 | 0.022 * | −0.79 | 0.17 | 0.000 **** | 0.24 | 0.05 | 0.000 **** | −0.31 | 0.04 | 0.000 **** | −0.02 | 0.01 | 0.022 * | −0.04 | 0.02 | 0.039 * |
| C8/C10 Carnitine | C14 Carnitine | C14:1 Carnitine | C14:2 Carnitine | C16 Carnitine | C16:1 Carnitine | ||||||||||||||||
| Months | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | |||
| 0 | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | |||
| 3 | −0.05 | 0.07 | 0.531 | 0.02 | 0.03 | 0.521 | 0.03 | 0.01 | 0.005 *** | 0.00 | 0.00 | 0.314 | −0.12 | 0.13 | 0.333 | −0.01 | 0.01 | 0.296 | |||
| 6 | −0.02 | 0.11 | 0.822 | −0.03 | 0.01 | 0.002 *** | 0.00 | 0.01 | 0.741 | 0.00 | 0.01 | 0.855 | −0.26 | 0.10 | 0.009 ** | −0.02 | 0.01 | 0.025 * | |||
| 9 | −0.04 | 0.07 | 0.591 | −0.02 | 0.01 | 0.040 * | 0.02 | 0.01 | 0.070 | 0.00 | 0.00 | 0.896 | −0.31 | 0.11 | 0.007 ** | −0.02 | 0.01 | 0.083 | |||
| 12 | −0.15 | 0.05 | 0.004 *** | −0.02 | 0.01 | 0.151 | 0.03 | 0.02 | 0.113 | 0.01 | 0.01 | 0.035 * | −0.38 | 0.12 | 0.001 *** | −0.03 | 0.01 | 0.027 * | |||
| Amino Acid, n=22 | |||||||||||||||||||||
| Phenylalanine | Tyrosine | Glycine | Proline | Leucine-Isoleucine | Methionine | Arginine | |||||||||||||||
| Months | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p | β | SE | p |
| 0 | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - |
| 3 | −6.61 | 2.00 | 0.001 **** | −12.15 | 3.29 | 0.000 **** | 78.21 | 22.39 | 0.000 **** | −26.54 | 17.18 | 0.122 | −35.61 | 19.59 | 0.069 | −3.64 | 1.40 | 0.009 ** | −0.27 | 0.46 | 0.552 |
| 6 | −7.92 | 2.37 | 0.001 **** | −14.75 | 2.91 | 0.000 **** | 91.01 | 37.92 | 0.016 * | −52.66 | 13.52 | 0.000 **** | −56.41 | 18.67 | 0.003 *** | −4.37 | 1.63 | 0.007 ** | −0.90 | 0.46 | 0.048 * |
| 9 | −9.28 | 1.81 | 0.000 **** | −11.77 | 2.72 | 0.000 **** | 74.71 | 23.49 | 0.001 *** | −49.16 | 14.48 | 0.001 **** | −52.51 | 13.75 | 0.000 **** | −5.28 | 1.48 | 0.000 **** | −0.55 | 0.45 | 0.221 |
| 12 | −6.63 | 1.89 | 0.000 **** | −13.79 | 2.94 | 0.000 **** | 49.82 | 17.93 | 0.005 ** | −29.88 | 16.82 | 0.076 | −48.35 | 14.31 | 0.001 **** | −5.48 | 1.19 | 0.000 **** | −0.53 | 0.49 | 0.278 |
| Valine | Citrulline | Homocysteine | |||||||||||||||||||
| Months | β | SE | p | β | SE | p | β | SE | p | ||||||||||||
| 0 | 0.00 | - | - | 0.00 | - | - | 0.00 | - | - | ||||||||||||
| 3 | −23.39 | 21.37 | 0.274 | 2.57 | 1.26 | 0.041 * | 1.17 | 0.52 | 0.025 * | ||||||||||||
| 6 | −37.94 | 22.61 | 0.093 | −1.00 | 1.47 | 0.497 | 0.54 | 0.63 | 0.387 | ||||||||||||
| 9 | −38.38 | 17.38 | 0.027 * | 1.06 | 1.35 | 0.432 | 0.34 | 0.49 | 0.486 | ||||||||||||
| 12 | −41.06 | 17.11 | 0.016 * | −0.51 | 1.50 | 0.732 | −0.23 | 0.47 | 0.630 | ||||||||||||
| Time (Month) | β | SE | p | Time (Month) | β | SE | p | ||
|---|---|---|---|---|---|---|---|---|---|
| BHB | 0 | 0.37 | 0.87 | 0.673 | C6-DC carnitine | 0 | 0.01 | 0.01 | 0.102 |
| 3 | 0.31 | 0.57 | 0.588 | 3 | 0.00 | 0.00 | 0.324 | ||
| 6 | 1.43 | 0.64 | 0.025 * | 6 | −0.01 | 0.01 | 0.031 * | ||
| 9 | −0.65 | 0.74 | 0.384 | 9 | −0.01 | 0.00 | 0.275 | ||
| 12 | −0.06 | 0.77 | 0.934 | 12 | 0.00 | 0.00 | 0.465 | ||
| Free carnitine | 0 | −7.85 | 3.73 | 0.035 * | C8 carnitine | 0 | −0.01 | 0.03 | 0.657 |
| 3 | −11.84 | 4.05 | 0.003 *** | 3 | −0.07 | 0.07 | 0.333 | ||
| 6 | −15.94 | 4.18 | 0.000 **** | 6 | 0.01 | 0.01 | 0.405 | ||
| 9 | −6.77 | 3.58 | 0.059 | 9 | −0.06 | 0.01 | 0.000 **** | ||
| 12 | −8.59 | 3.11 | 0.006 ** | 12 | −0.01 | 0.01 | 0.230 | ||
| C2 carnitine | 0 | −8.95 | 3.29 | 0.006 ** | C10 carnitine | 0 | −0.02 | 0.04 | 0.688 |
| 3 | −9.67 | 3.84 | 0.012 * | 3 | −0.06 | 0.05 | 0.240 | ||
| 6 | −2.65 | 4.50 | 0.556 | 6 | 0.02 | 0.02 | 0.333 | ||
| 9 | −1.60 | 4.96 | 0.747 | 9 | −0.07 | 0.02 | 0.000 **** | ||
| 12 | 1.464 | 5.3079 | 0.783 | 12 | −0.03 | 0.02 | 0.112 | ||
| C3 carnitine | 0 | −1.28 | 0.60 | 0.032 * | C10:1 carnitine | 0 | 0.01 | 0.03 | 0.664 |
| 3 | −1.68 | 0.34 | 0.000 **** | 3 | 0.02 | 0.02 | 0.355 | ||
| 6 | −1.80 | 0.30 | 0.000 **** | 6 | 0.02 | 0.02 | 0.144 | ||
| 9 | −1.12 | 0.51 | 0.027 * | 9 | −0.08 | 0.01 | 0.000 **** | ||
| 12 | −0.98 | 0.43 | 0.022 * | 12 | −0.02 | 0.02 | 0.253 | ||
| C4 carnitine | 0 | −0.05 | 0.06 | 0.456 | C8/C10 carnitine | 0 | −0.04 | 0.17 | 0.817 |
| 3 | −0.21 | 0.06 | 0.000 **** | 3 | 0.07 | 0.15 | 0.628 | ||
| 6 | −0.14 | 0.06 | 0.028 * | 6 | −0.02 | 0.11 | 0.846 | ||
| 9 | −0.10 | 0.06 | 0.084 | 9 | 0.07 | 0.13 | 0.575 | ||
| 12 | −0.03 | 0.05 | 0.547 | 12 | −0.01 | 0.12 | 0.949 | ||
| C4:OH carnitine | 0 | −0.07 | 0.03 | 0.033 * | C12 carnitine | 0 | −0.02 | 0.02 | 0.337 |
| 3 | −0.04 | 0.08 | 0.617 | 3 | −0.07 | 0.03 | 0.027 * | ||
| 6 | 0.06 | 0.07 | 0.404 | 6 | −0.02 | 0.01 | 0.151 | ||
| 9 | −0.07 | 0.08 | 0.379 | 9 | −0.03 | 0.02 | 0.055 | ||
| 12 | 0.09 | 0.11 | 0.409 | 12 | −0.01 | 0.02 | 0.560 | ||
| C4-DC carnitine | 0 | −0.15 | 0.09 | 0.108 | C14 carnitine | 0 | −0.04 | 0.02 | 0.022 * |
| 3 | −0.16 | 0.08 | 0.043 * | 3 | −0.11 | 0.08 | 0.146 | ||
| 6 | −0.06 | 0.10 | 0.500 | 6 | −0.04 | 0.01 | 0.007 ** | ||
| 9 | 0.05 | 0.08 | 0.550 | 9 | −0.04 | 0.02 | 0.016 * | ||
| 12 | 0.04 | 0.11 | 0.712 | 12 | 0.00 | 0.01 | 0.979 | ||
| C5 carnitine | 0 | −0.02 | 0.04 | 0.592 | C14:1 carnitine | 0 | −0.04 | 0.01 | 0.000 **** |
| 3 | −0.09 | 0.02 | 0.000 **** | 3 | −0.05 | 0.02 | 0.022 * | ||
| 6 | −0.10 | 0.01 | 0.000 **** | 6 | 0.00 | 0.02 | 0.911 | ||
| 9 | −0.07 | 0.02 | 0.002 *** | 9 | −0.05 | 0.02 | 0.009 ** | ||
| 12 | −0.04 | 0.01 | 0.002 *** | 12 | −0.01 | 0.02 | 0.564 | ||
| C5:1 carnitine | 0 | 0.00 | 0.00 | 0.275 | C14:2 carnitine | 0 | 0.00 | 0.01 | 0.896 |
| 3 | −0.01 | 0.00 | 0.013 * | 3 | −0.01 | 0.01 | 0.153 | ||
| 6 | 0.00 | 0.00 | 0.401 | 6 | 0.00 | 0.01 | 0.831 | ||
| 9 | −0.01 | 0.00 | 0.001 *** | 9 | −0.03 | 0.01 | 0.003 *** | ||
| 12 | 0.00 | 0.00 | 0.504 | 12 | 0.00 | 0.01 | 0.761 | ||
| C5:OH carnitine | 0 | −0.08 | 0.09 | 0.371 | C16 carnitine | 0 | −0.70 | 0.25 | 0.005 ** |
| 3 | −0.12 | 0.04 | 0.006 ** | 3 | −1.08 | 0.35 | 0.002 *** | ||
| 6 | −0.10 | 0.05 | 0.062 | 6 | −0.83 | 0.23 | 0.000 **** | ||
| 9 | −0.11 | 0.04 | 0.012 * | 9 | −0.62 | 0.21 | 0.003 *** | ||
| 12 | −0.11 | 0.09 | 0.224 | 12 | −0.39 | 0.16 | 0.013 * | ||
| C5-DC carnitine | 0 | 0.01 | 0.01 | 0.055 | C16:1 carnitine | 0 | −0.07 | 0.03 | 0.011 * |
| 3 | −0.01 | 0.00 | 0.003 *** | 3 | −0.07 | 0.01 | 0.000 **** | ||
| 6 | 0.00 | 0.00 | 0.232 | 6 | −0.05 | 0.02 | 0.014 * | ||
| 9 | −0.02 | 0.01 | 0.011 * | 9 | −0.05 | 0.03 | 0.059 | ||
| 12 | −0.01 | 0.00 | 0.005 *** | 12 | −0.01 | 0.01 | 0.235 | ||
| C6 carnitine | 0 | −0.01 | 0.02 | 0.620 | C18 carnitine | 0 | −0.47 | 0.08 | 0.000 **** |
| 3 | −0.09 | 0.06 | 0.152 | 3 | −0.50 | 0.30 | 0.093 | ||
| 6 | −0.02 | 0.00 | 0.000 **** | 6 | −0.51 | 0.18 | 0.004 *** | ||
| 9 | −0.02 | 0.01 | 0.031 * | 9 | −0.29 | 0.11 | 0.006 ** | ||
| 12 | 0.00 | 0.01 | 0.836 | 12 | −0.22 | 0.16 | 0.168 |
| Time (month) | β | SE | p | Time (month) | β | SE | p | ||
|---|---|---|---|---|---|---|---|---|---|
| Phenylalanine | 0 | 0.18 | 3.99 | 0.964 | Arginine | 0 | 1.52 | 1.06 | 0.154 |
| 3 | −3.35 | 1.78 | 0.060 | 3 | 0.63 | 1.08 | 0.560 | ||
| 6 | −11.94 | 3.74 | 0.001 *** | 6 | 0.83 | 0.32 | 0.010 ** | ||
| 9 | −3.81 | 1.63 | 0.019 * | 9 | 0.83 | 0.76 | 0.272 | ||
| 12 | 0.21 | 3.00 | 0.943 | 12 | 0.74 | 0.46 | 0.107 | ||
| Tyrosine | 0 | 4.84 | 7.65 | 0.527 | Phenylalanine | 0 | −0.04 | 0.09 | 0.618 |
| 3 | −4.09 | 5.68 | 0.472 | /Tyrosine | 3 | −0.06 | 0.18 | 0.719 | |
| 6 | −0.99 | 2.38 | 0.677 | 6 | −0.30 | 0.06 | 0.000 **** | ||
| 9 | 1.38 | 4.20 | 0.743 | 9 | −0.12 | 0.08 | 0.144 | ||
| 12 | −7.21 | 3.77 | 0.056 | 12 | 0.21 | 0.06 | 0.000 **** | ||
| Glycine | 0 | 38.49 | 25.29 | 0.128 | Alanine | 0 | −27.17 | 44.74 | 0.544 |
| 3 | 92.61 | 25.09 | 0.000 **** | 3 | −13.96 | 38.93 | 0.720 | ||
| 6 | 154.21 | 58.87 | 0.009 ** | 6 | −13.97 | 32.42 | 0.666 | ||
| 9 | 77.63 | 38.82 | 0.046* | 9 | 15.19 | 21.28 | 0.475 | ||
| 12 | 53.38 | 33.84 | 0.115 | 12 | 28.74 | 25.08 | 0.252 | ||
| Proline | 0 | −1.84 | 33.52 | 0.956 | Valine | 0 | −8.54 | 29.85 | 0.775 |
| 3 | −0.04 | 31.20 | 0.999 | 3 | −23.79 | 20.00 | 0.234 | ||
| 6 | −7.89 | 21.46 | 0.713 | 6 | −12.92 | 15.43 | 0.402 | ||
| 9 | −23.27 | 15.74 | 0.139 | 9 | −21.57 | 10.83 | 0.046 * | ||
| 12 | −37.73 | 30.29 | 0.213 | 12 | −21.74 | 18.87 | 0.249 | ||
| Leucine | 0 | −9.71 | 22.42 | 0.665 | Ornithine | 0 | 9.72 | 4.70 | 0.039 * |
| -Isoleucine | 3 | −58.47 | 8.09 | 0.000 **** | 3 | −7.56 | 2.82 | 0.007 ** | |
| 6 | −15.30 | 10.54 | 0.147 | 6 | −5.79 | 2.71 | 0.033 * | ||
| 9 | −37.16 | 8.81 | 0.000 **** | 9 | −6.91 | 9.64 | 0.473 | ||
| 12 | −36.19 | 13.60 | 0.008 ** | 12 | 3.15 | 4.83 | 0.515 | ||
| Methionine | 0 | −2.34 | 3.26 | 0.473 | Citrulline | 0 | −3.28 | 3.06 | 0.284 |
| 3 | −4.81 | 2.82 | 0.088 | 3 | −3.58 | 2.18 | 0.101 | ||
| 6 | −1.38 | 2.90 | 0.635 | 6 | −2.28 | 2.35 | 0.332 | ||
| 9 | 0.57 | 1.19 | 0.629 | 9 | −1.91 | 3.02 | 0.528 | ||
| 12 | −2.49 | 1.64 | 0.129 | 12 | 1.17 | 2.26 | 0.603 | ||
| Glutamic acid | 0 | −3.61 | 12.51 | 0.773 | Homocysteine | 0 | −0.03 | 0.48 | 0.946 |
| 3 | −20.97 | 14.72 | 0.154 | 3 | −0.44 | 0.94 | 0.640 | ||
| 6 | 5.27 | 10.47 | 0.615 | 6 | 0.55 | 1.18 | 0.643 | ||
| 9 | 6.36 | 11.81 | 0.590 | 9 | −1.42 | 0.64 | 0.027 * | ||
| 12 | 17.12 | 10.88 | 0.116 | 12 | −0.69 | 1.09 | 0.524 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hung, P.-L.; Lin, J.-L.; Chen, C.; Hung, K.-Y.; Hsieh, T.-Y.; Hsu, M.-H.; Kuo, H.-C.; Lin, Y.-J. An Examination of Serum Acylcarnitine and Amino Acid Profiles at Different Time Point of Ketogenic Diet Therapy and Their Association of Ketogenic Diet Effectiveness. Nutrients 2021, 13, 21. https://doi.org/10.3390/nu13010021
Hung P-L, Lin J-L, Chen C, Hung K-Y, Hsieh T-Y, Hsu M-H, Kuo H-C, Lin Y-J. An Examination of Serum Acylcarnitine and Amino Acid Profiles at Different Time Point of Ketogenic Diet Therapy and Their Association of Ketogenic Diet Effectiveness. Nutrients. 2021; 13(1):21. https://doi.org/10.3390/nu13010021
Chicago/Turabian StyleHung, Pi-Lien, Ju-Li Lin, Chien Chen, Kai-Yin Hung, Tzu-Yun Hsieh, Mei-Hsin Hsu, Hsuan-Chang Kuo, and Ying-Jui Lin. 2021. "An Examination of Serum Acylcarnitine and Amino Acid Profiles at Different Time Point of Ketogenic Diet Therapy and Their Association of Ketogenic Diet Effectiveness" Nutrients 13, no. 1: 21. https://doi.org/10.3390/nu13010021
APA StyleHung, P.-L., Lin, J.-L., Chen, C., Hung, K.-Y., Hsieh, T.-Y., Hsu, M.-H., Kuo, H.-C., & Lin, Y.-J. (2021). An Examination of Serum Acylcarnitine and Amino Acid Profiles at Different Time Point of Ketogenic Diet Therapy and Their Association of Ketogenic Diet Effectiveness. Nutrients, 13(1), 21. https://doi.org/10.3390/nu13010021