Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
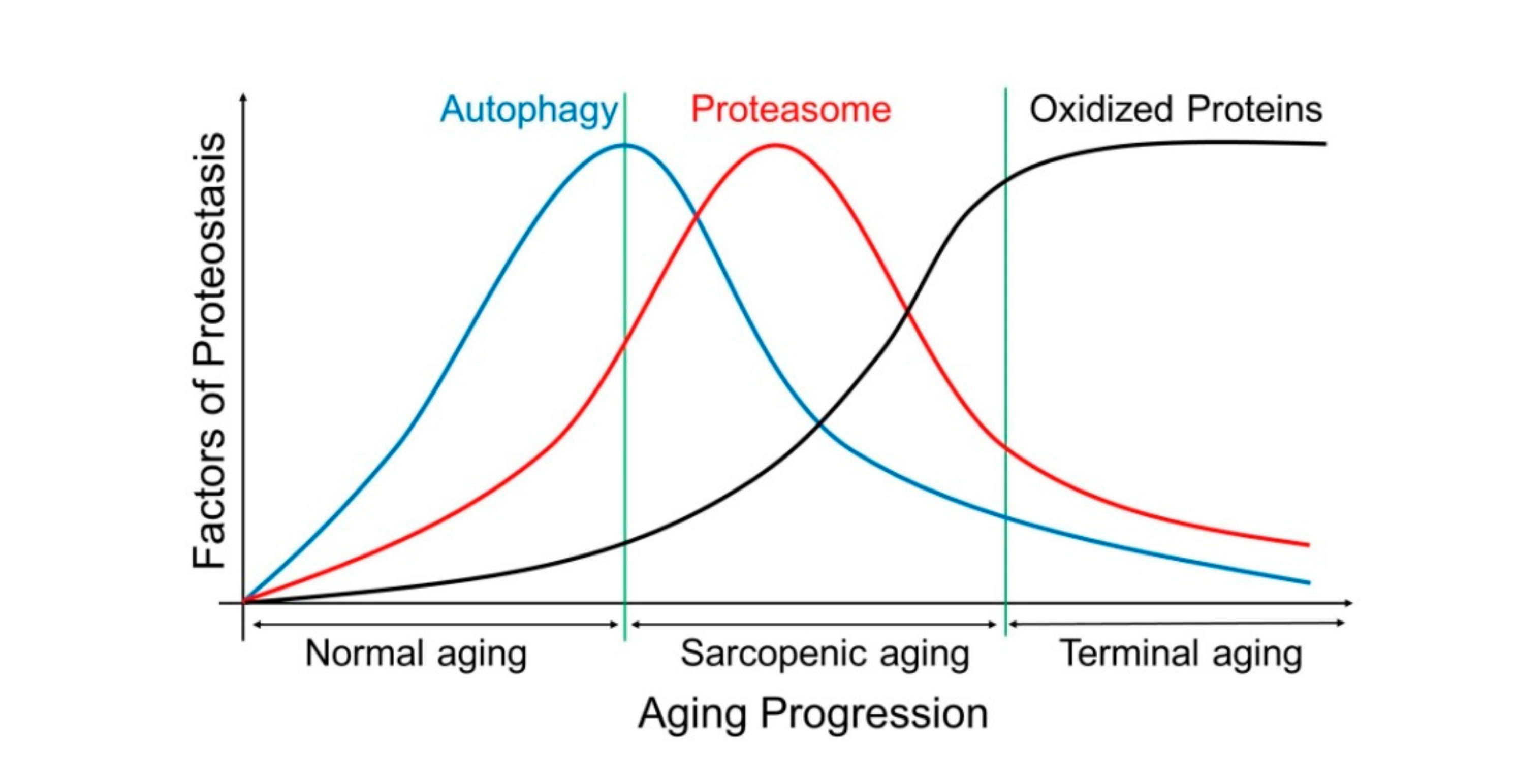
2. Molecular Mechanisms of Sarcopenia Development and Progression
2.1. General Consideration
2.2. Muscle Protein Catabolism
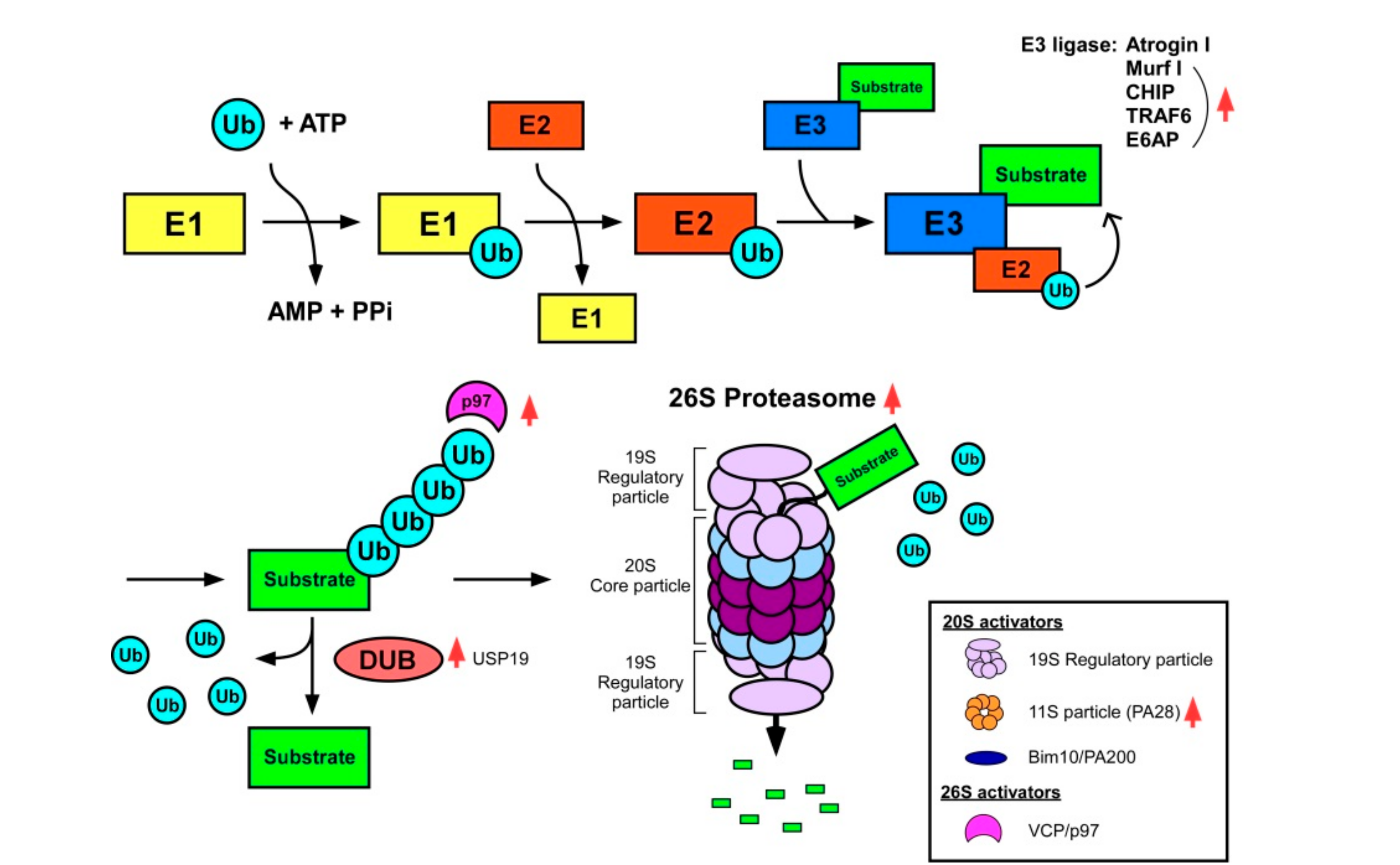
2.3. The Ubiquitin–Proteasome System (UPS)
2.4. The Autophagy–Lysosome System (ALS)
3. Therapeutic Potential of DHA in Sarcopenia by Modulating UPS and ALS
4. Conclusion and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Miljkovic, N.; Lim, J.Y.; Miljkovic, I.; Frontera, W.R. Aging of skeletal muscle fibers. Ann. Rehabil. Med. 2015, 39, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, I.H. Sarcopenia: Origins and clinical relevance. J. Nutr. 1997, 127, 990–991. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.J. Invited review: Aging and sarcopenia. J. Appl. Physiol. 2003, 95, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing. 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Shafiee, G.; Keshtkar, A.; Soltani, A.; Ahadi, Z.; Larijani, B.; Heshmat, R. Prevalence of sarcopenia in the world: A systematic review and meta- analysis of general population studies. J. Diabetes Metab. Disord. 2017, 16, 21. [Google Scholar] [CrossRef]
- United Nations. World Population Ageing 2017; United Nations: New York, NY, USA, 2017. [Google Scholar]
- Park, S.S.; Kwon, E.S.; Kwon, K.S. Molecular mechanisms and therapeutic interventions in sarcopenia. Osteoporos Sarcopenia 2017, 3, 117–122. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Walker, D.K.; Dickinson, J.M.; Timmerman, K.L.; Drummond, M.J.; Reidy, P.T.; Fry, C.S.; Gundermann, D.M.; Rasmussen, B.B. Exercise, amino acids, and aging in the control of human muscle protein synthesis. Med. Sci. Sports Exerc. 2011, 43, 2249–2258. [Google Scholar] [CrossRef]
- Yoo, S.Z.; No, M.H.; Heo, J.W.; Park, D.H.; Kang, J.H.; Kim, S.H.; Kwak, H.B. Role of exercise in age-related sarcopenia. J. Exerc. Rehabil. 2018, 14, 551–558. [Google Scholar] [CrossRef]
- Gray, S.R.; Da Boit, M. Fish oils and their potential in the treatment of sarcopenia. J. Frailty Aging 2013, 2, 211–216. [Google Scholar]
- Rondanelli, M.; Rigon, C.; Perna, S.; Gasparri, C.; Iannello, G.; Akber, R.; Alalwan, T.A.; Freije, A.M. Novel insights on intake of fish and prevention of sarcopenia: All reasons for an adequate consumption. Nutrients 2020, 12, 307. [Google Scholar] [CrossRef] [PubMed]
- De Spiegeleer, A.; Beckwee, D.; Bautmans, I.; Petrovic, M.; Sarcopenia Guidelines Development group of the Belgian Society of Gerontology and Geriatrics. Pharmacological interventions to improve muscle mass, muscle strength and physical performance in older people: An umbrella review of systematic reviews and meta-analyses. Drugs Aging 2018, 35, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Dupont, J.; Dedeyne, L.; Dalle, S.; Koppo, K.; Gielen, E. The role of omega-3 in the prevention and treatment of sarcopenia. Aging Clin. Exp. Res. 2019, 31, 825–836. [Google Scholar] [CrossRef] [PubMed]
- Jeromson, S.; Gallagher, I.J.; Galloway, S.D.; Hamilton, D.L. Omega-3 fatty acids and skeletal muscle health. Mar. Drugs. 2015, 13, 6977–7004. [Google Scholar] [CrossRef]
- Shin, S.K.; Kim, J.H.; Lee, J.H.; Son, Y.H.; Lee, M.W.; Kim, H.J.; Noh, S.A.; Kim, K.P.; Kim, I.G.; Lee, M.J. Docosahexaenoic acid-mediated protein aggregates may reduce proteasome activity and delay myotube degradation during muscle atrophy In Vitro. Exp. Mol. Med. 2017, 49, e287. [Google Scholar] [CrossRef]
- Jing, K.; Song, K.S.; Shin, S.; Kim, N.; Jeong, S.; Oh, H.R.; Park, J.H.; Seo, K.S.; Heo, J.Y.; Han, J.; et al. Docosahexaenoic acid induces autophagy through p53/AMPK/mTOR signaling and promotes apoptosis in human cancer cells harboring wild-type p53. Autophagy 2011, 7, 1348–1358. [Google Scholar] [CrossRef]
- Kim, E.; Park, S.; Lee, J.H.; Mun, J.Y.; Choi, W.H.; Yun, Y.; Lee, J.; Kim, J.H.; Kang, M.J.; Lee, M.J. Dual function of USP14 deubiquitinase in cellular proteasomal activity and autophagic flux. Cell Rep. 2018, 24, 732–743. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, S.; Kim, E.; Lee, M.J. Negative-feedback coordination between proteasomal activity and autophagic flux. Autophagy 2019, 15, 726–728. [Google Scholar] [CrossRef]
- Jiang, Y.; Lee, J.; Lee, J.H.; Lee, J.W.; Kim, J.H.; Choi, W.H.; Yoo, Y.D.; Cha-Molstad, H.; Kim, B.Y.; Kwon, Y.T.; et al. The arginylation branch of the N-end rule pathway positively regulates cellular autophagic flux and clearance of proteotoxic proteins. Autophagy 2016, 12, 2197–2212. [Google Scholar] [CrossRef]
- Scicchitano, B.M.; Dobrowolny, G.; Sica, G.; Musaro, A. Molecular insights into muscle homeostasis, atrophy and wasting. Curr. Genom. 2018, 19, 356–369. [Google Scholar] [CrossRef]
- Fernando, R.; Drescher, C.; Nowotny, K.; Grune, T.; Castro, J.P. Impaired proteostasis during skeletal muscle aging. Free Radic. Biol. Med. 2019, 132, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Arthur, S.T.; Cooley, I.D. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int. J. Biol. Sci. 2012, 8, 731–760. [Google Scholar] [CrossRef] [PubMed]
- Lecker, S.H.; Solomon, V.; Mitch, W.E.; Goldberg, A.L. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J. Nutr. 1999, 129, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The role of inflammation in age-related sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef]
- Marshall, S.; Agarwal, E. Comparing Characteristics of Malnutrition, Starvation, Sarcopenia and Cachexia in Older Adults; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Rolland, Y.; Czerwinski, S.; Abellan Van Kan, G.; Morley, J.E.; Cesari, M.; Onder, G.; Woo, J.; Baumgartner, R.; Pillard, F.; Boirie, Y.; et al. Sarcopenia: Its assessment, etiology, pathogenesis, consequences and future perspectives. J. Nutr. Health Aging 2008, 12, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Park, S.W.; Harris, T.B.; Kritchevsky, S.B.; Nevitt, M.; Schwartz, A.V.; Simonsick, E.M.; Tylavsky, F.A.; Visser, M.; Newman, A.B. The loss of skeletal muscle strength, mass, and quality in older adults: The health, aging and body composition study. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 1059–1064. [Google Scholar] [CrossRef]
- Bhanji, R.A.; Narayanan, P.; Allen, A.M.; Malhi, H.; Watt, K.D. Sarcopenia in hiding: The risk and consequence of underestimating muscle dysfunction in nonalcoholic steatohepatitis. Hepatology 2017, 66, 2055–2065. [Google Scholar] [CrossRef]
- Roh, E.; Choi, K.M. Health consequences of sarcopenic obesity: A narrative review. Front. Endocrinol. 2020, 11, 332. [Google Scholar] [CrossRef]
- Shou, J.; Chen PJ, Xiao, W. H. Mechanism of increased risk of insulin resistance in aging skeletal muscle. Diabetol. Metab. Syndr. 2020, 12, 14. [Google Scholar] [CrossRef]
- Barbalho, S.M.; Flato, U.A.P.; Tofano, R.J.; Goulart, R.A.; Guiguer, E.L.; Detregiachi, C.R.P.; Buchaim, D.V.; Araujo, A.C.; Buchaim, R.L.; Reina, F.T.R.; et al. Physical exercise and myokines: relationships with sarcopenia and cardiovascular complications. Int. J. Mol. Sci. 2020, 21, 3607. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.Y.; Kwon, K.S. Pharmacological interventions for treatment of sarcopenia: current status of drug development for sarcopenia. Ann. Geriatr. Med. Res. 2019, 23, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Buford, T.W.; Anton, S.D.; Judge, A.R.; Marzetti, E.; Wohlgemuth, S.E.; Carter, C.S.; Leeuwenburgh, C.; Pahor, M.; Manini, T.M. Models of accelerated sarcopenia: Critical pieces for solving the puzzle of age-related muscle atrophy. Ageing Res. Rev. 2010, 9, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef]
- Lee, C.E.; McArdle, A.; Griffiths, R.D. The role of hormones, cytokines and heat shock proteins during age-related muscle loss. Clin. Nutr. 2007, 26, 524–534. [Google Scholar] [CrossRef]
- Fan, J.; Kou, X.; Jia, S.; Yang, X.; Yang, Y.; Chen, N. Autophagy as a potential target for sarcopenia. J. Cell Physiol. 2016, 231, 1450–1459. [Google Scholar] [CrossRef]
- Sandri, M. Protein breakdown in muscle wasting: Role of autophagy-lysosome and ubiquitin-proteasome. Int. J. Biochem. Cell Biol. 2013, 45, 2121–2129. [Google Scholar] [CrossRef]
- Barclay, R.D.; Burd, N.A.; Tyler, C.; Tillin, N.A.; Mackenzie, R.W. The role of the IGF-1 signaling cascade in muscle protein synthesis and anabolic resistance in aging skeletal muscle. Front. Nutr. 2019, 6, 146. [Google Scholar] [CrossRef]
- Sakuma, K.; Yamaguchi, A. Recent advances in pharmacological, hormonal, and nutritional intervention for sarcopenia. Pflug. Arch. 2018, 470, 449–460. [Google Scholar] [CrossRef]
- Grounds, M.D. Reasons for the degeneration of ageing skeletal muscle: A central role for IGF-1 signalling. Biogerontology 2002, 3, 19–24. [Google Scholar] [CrossRef]
- Lavin, K.M.; Perkins, R.K.; Jemiolo, B.; Raue, U.; Trappe, S.W.; Trappe, T.A. Effects of aging and lifelong aerobic exercise on basal and exercise-induced inflammation. J. Appl. Physiol. 2020, 128, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Retamales, A.; Zuloaga, R.; Valenzuela, C.A.; Gallardo-Escarate, C.; Molina, A.; Valdes, J.A. Insulin-like growth factor-1 suppresses the Myostatin signaling pathway during myogenic differentiation. Biochem. Biophys. Res. Commun. 2015, 464, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Bacurau, A.V.; Jannig, P.R.; de Moraes, W.M.; Cunha, T.F.; Medeiros, A.; Barberi, L.; Coelho, M.A.; Bacurau, R.F.; Ugrinowitsch, C.; Musaro, A.; et al. Akt/mTOR pathway contributes to skeletal muscle anti-atrophic effect of aerobic exercise training in heart failure mice. Int. J. Cardiol. 2016, 214, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.; Tay, L.; Lim, J.P.; Leung, B.P.; Yeo, A.; Yew, S.; Ding, Y.Y.; Lim, W.S. Serum myostatin and IGF-1 as gender-specific biomarkers of frailty and low muscle mass in community-dwelling older adults. J. Nutr. Health Aging 2019, 23, 979–986. [Google Scholar] [CrossRef]
- Taylor, W.E.; Bhasin, S.; Artaza, J.; Byhower, F.; Azam, M.; Willard, D.H., Jr.; Kull, F.C., Jr.; Gonzalez-Cadavid, N. Myostatin inhibits cell proliferation and protein synthesis in C2C12 muscle cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, 221–228. [Google Scholar] [CrossRef]
- Leger, B.; Derave, W.; De Bock, K.; Hespel, P.; Russell, A.P. Human sarcopenia reveals an increase in SOCS-3 and myostatin and a reduced efficiency of Akt phosphorylation. Rejuvenation Res. 2008, 11, 163–175. [Google Scholar] [CrossRef]
- Yarasheski, K.E.; Bhasin, S.; Sinha-Hikim, I.; Pak-Loduca, J.; Gonzalez-Cadavid, N.F. Serum myostatin-immunoreactive protein is increased in 60–92 year old women and men with muscle wasting. J. Nutr. Health Aging 2002, 6, 343–348. [Google Scholar]
- Zimmers, T.A.; Davies, M.V.; Koniaris, L.G.; Haynes, P.; Esquela, A.F.; Tomkinson, K.N.; McPherron, A.C.; Wolfman, N.M.; Lee, S.J. Induction of cachexia in mice by systemically administered myostatin. Science 2002, 296, 1486–1488. [Google Scholar] [CrossRef]
- Murphy, K.T.; Koopman, R.; Naim, T.; Leger, B.; Trieu, J.; Ibebunjo, C.; Lynch, G.S. Antibody-directed myostatin inhibition in 21-mo-old mice reveals novel roles for myostatin signaling in skeletal muscle structure and function. FASEB J. 2010, 24, 4433–4442. [Google Scholar] [CrossRef]
- Whittemore, L.A.; Song, K.; Li, X.; Aghajanian, J.; Davies, M.; Girgenrath, S.; Hill, J.J.; Jalenak, M.; Kelley, P.; Knight, A.; et al. Inhibition of myostatin in adult mice increases skeletal muscle mass and strength. Biochem. Biophys. Res. Commun. 2003, 300, 965–971. [Google Scholar] [CrossRef]
- Kikis, E.A. The intrinsic and extrinsic factors that contribute to proteostasis decline and pathological protein misfolding. Adv. Protein Chem. Struct. Biol. 2019, 118, 145–161. [Google Scholar] [PubMed]
- Deutz, N.E.; Bauer, J.M.; Barazzoni, R.; Biolo, G.; Boirie, Y.; Bosy-Westphal, A.; Cederholm, T.; Cruz-Jentoft, A.; Krznaric, Z.; Nair, K.S.; et al. Protein intake and exercise for optimal muscle function with aging: Recommendations from the ESPEN Expert Group. Clin. Nutr. 2014, 33, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Bowen, T.S.; Schuler, G.; Adams, V. Skeletal muscle wasting in cachexia and sarcopenia: Molecular pathophysiology and impact of exercise training. J. Cachexia Sarcopenia Muscle 2015, 6, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kostek, M.C.; Delmonico, M.J.; Reichel, J.B.; Roth, S.M.; Douglass, L.; Ferrell, R.E.; Hurley, B.F. Muscle strength response to strength training is influenced by insulin-like growth factor 1 genotype in older adults. J. Appl. Physiol. (1985) 2005, 98, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.I.; Priego, T.; Lopez-Calderon, A. Hormones and muscle atrophy. Adv. Exp. Med. Biol. 2018, 1088, 207–233. [Google Scholar] [PubMed]
- Vinciguerra, M.; Musaro, A.; Rosenthal, N. Regulation of muscle atrophy in aging and disease. Adv. Exp. Med. Biol. 2010, 694, 211–233. [Google Scholar]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflug. Arch. 2017, 469, 573–591. [Google Scholar] [CrossRef]
- Combaret, L.; Dardevet, D.; Bechet, D.; Taillandier, D.; Mosoni, L.; Attaix, D. Skeletal muscle proteolysis in aging. Curr. Opin. Clin. Nutr. Metab. Care 2009, 12, 37–41. [Google Scholar] [CrossRef]
- Piccirillo, R.; Demontis, F.; Perrimon, N.; Goldberg, A.L. Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn. 2014, 243, 201–215. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Finley, D.; Ciechanover, A.; Varshavsky, A. Ubiquitin as a central cellular regulator. Cell 2004, 116, 29–32. [Google Scholar] [CrossRef]
- Choi, W.H.; de Poot, S.A.; Lee, J.H.; Kim, J.H.; Han, D.H.; Kim, Y.K.; Finley, D.; Lee, M.J. Open-gate mutants of the mammalian proteasome show enhanced ubiquitin-conjugate degradation. Nat. Commun. 2016, 7, 10963. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Rubinsztein, D.C.; Walker, D.W. Autophagy as a promoter of longevity: Insights from model organisms. Nat. Rev. Mol. Cell Biol. 2018, 19, 579–593. [Google Scholar] [CrossRef] [PubMed]
- Strucksberg, K.H.; Tangavelou, K.; Schroder, R.; Clemen, C.S. Proteasomal activity in skeletal muscle: A matter of assay design, muscle type, and age. Anal. Biochem. 2010, 399, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Hwee, D.T.; Baehr, L.M.; Philp, A.; Baar, K.; Bodine, S.C. Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell 2014, 13, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Husom, A.D.; Peters, E.A.; Kolling, E.A.; Fugere, N.A.; Thompson, L.V.; Ferrington, D.A. Altered proteasome function and subunit composition in aged muscle. Arch. Biochem. Biophys. 2004, 421, 67–76. [Google Scholar] [CrossRef]
- Radak, Z.; Takahashi, R.; Kumiyama, A.; Nakamoto, H.; Ohno, H.; Ookawara, T.; Goto, S. Effect of aging and late onset dietary restriction on antioxidant enzymes and proteasome activities, and protein carbonylation of rat skeletal muscle and tendon. Exp. Gerontol. 2002, 37, 1423–1430. [Google Scholar] [CrossRef]
- Mitch, W.E.; Goldberg, A.L. Mechanisms of muscle wasting. The role of the ubiquitin-proteasome pathway. N. Engl. J. Med. 1996, 335, 1897–1905. [Google Scholar] [CrossRef]
- Mitch, W.E.; Medina, R.; Grieber, S.; May, R.C.; England, B.K.; Price, S.R.; Bailey, J.L.; Goldberg, A.L. Metabolic acidosis stimulates muscle protein degradation by activating the adenosine triphosphate-dependent pathway involving ubiquitin and proteasomes. J. Clin. Invest. 1994, 93, 2127–2133. [Google Scholar] [CrossRef]
- Altun, M.; Besche, H.C.; Overkleeft, H.S.; Piccirillo, R.; Edelmann, M.J.; Kessler, B.M.; Goldberg, A.L.; Ulfhake, B. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J. Biol. Chem. 2010, 285, 39597–39608. [Google Scholar] [CrossRef]
- Hepple, R.T.; Qin, M.; Nakamoto, H.; Goto, S. Caloric restriction optimizes the proteasome pathway with aging in rat plantaris muscle: Implications for sarcopenia. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, 1231–2317. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Ferrington, D.A.; Baumann, C.W.; Thompson, L.V. Denervation-induced activation of the standard proteasome and immunoproteasome. PLoS ONE 2016, 11, e0166831. [Google Scholar] [CrossRef] [PubMed]
- Attaix, D.; Mosoni, L.; Dardevet, D.; Combaret, L.; Mirand, P.P.; Grizard, J. Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. Int. J. Biochem. Cell Biol. 2005, 37, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 644–646. [Google Scholar] [CrossRef]
- Selsby, J.T.; Judge, A.R.; Yimlamai, T.; Leeuwenburgh, C.; Dodd, S.L. Life long calorie restriction increases heat shock proteins and proteasome activity in soleus muscles of Fisher 344 rats. Exp. Gerontol. 2005, 40, 37–42. [Google Scholar] [CrossRef]
- Bardag-Gorce, F.; Farout, L.; Veyrat-Durebex, C.; Briand, Y.; Briand, M. Changes in 20S proteasome activity during ageing of the LOU rat. Mol. Biol. Rep. 1999, 26, 89–93. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef]
- Price, S.R.; Bailey, J.L.; Wang, X.; Jurkovitz, C.; England, B.K.; Ding, X.; Phillips, L.S.; Mitch, W.E. Muscle wasting in insulinopenic rats results from activation of the ATP-dependent, ubiquitin-proteasome proteolytic pathway by a mechanism including gene transcription. J. Clin. Invest. 1996, 98, 1703–1708. [Google Scholar] [CrossRef]
- Gumucio, J.P.; Mendias, C.L. Atrogin-1, MuRF-1, and sarcopenia. Endocrine 2013, 43, 12–21. [Google Scholar] [CrossRef]
- Gomes, M.D.; Lecker, S.H.; Jagoe, R.T.; Navon, A.; Goldberg, A.L. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc. Natl. Acad. Sci. USA 2001, 98, 14440–14445. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Hyatt, J.P.; Raffaello, A.; Jagoe, R.T.; Roy, R.R.; Edgerton, V.R.; Lecker, S.H.; Goldberg, A.L. Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J. 2007, 21, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.V.; Waddell, D.S.; Siu, R.; Stein, M.; Dewey, S.; Furlow, J.D.; Bodine, S.C. Upregulation of proteasome activity in muscle RING finger 1-null mice following denervation. FASEB J. 2012, 26, 2986–2999. [Google Scholar] [CrossRef] [PubMed]
- Labeit, S.; Kohl, C.H.; Witt, C.C.; Labeit, D.; Jung, J.; Granzier, H. Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO mice. J. Biomed. Biotechnol. 2010, 2010, 693741. [Google Scholar] [CrossRef] [PubMed]
- Baehr, L.M.; Furlow, J.D.; Bodine, S.C. Muscle sparing in muscle RING finger 1 null mice: Response to synthetic glucocorticoids. J. Physiol. 2011, 589, 4759–4776. [Google Scholar] [CrossRef]
- Bilodeau, P.A.; Coyne, E.S.; Wing, S.S. The ubiquitin proteasome system in atrophying skeletal muscle: Roles and regulation. Am. J. Physiol Cell Physiol. 2016, 311, 392–403. [Google Scholar] [CrossRef]
- Lagirand-Cantaloube, J.; Offner, N.; Csibi, A.; Leibovitch, M.P.; Batonnet-Pichon, S.; Tintignac, L.A.; Segura, C.T.; Leibovitch, S.A. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008, 27, 1266–1276. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Stitt, T.N.; Drujan, D.; Clarke, B.A.; Panaro, F.; Timofeyva, Y.; Kline, W.O.; Gonzalez, M.; Yancopoulos, G.D.; Glass, D.J. The IGF-1/PI3K/Akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting FOXO transcription factors. Mol. Cell 2004, 14, 395–403. [Google Scholar] [CrossRef]
- Lokireddy, S.; Wijesoma, I.W.; Sze, S.K.; McFarlane, C.; Kambadur, R.; Sharma, M. Identification of atrogin-1-targeted proteins during the myostatin-induced skeletal muscle wasting. Am. J. Physiol. Cell Physiol. 2012, 303, 512–529. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yi, X.; Yao, Z.; Chakkalakal, J.V.; Xing, L.; Boyce, B.F. TNF receptor-associated factor 6 mediates TNFalpha-induced skeletal muscle atrophy in mice during aging. J. Bone Miner Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Ichihara, A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem. Biophys. Res. Commun. 1989, 159, 1309–1315. [Google Scholar] [CrossRef]
- Russell, S.J.; Steger, K.A.; Johnston, S.A. Subcellular localization, stoichiometry, and protein levels of 26 S proteasome subunits in yeast. J. Biol. Chem. 1999, 274, 21943–21952. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, Y.; Tashiro, Y.; Suzuki, N.; Warita, H.; Kato, M.; Tateyama, M.; Ando, R.; Izumi, R.; Yamazaki, M.; Abe, M. Proteasome dysfunction induces muscle growth defects and protein aggregation. J. Cell Sci. 2014, 127, 5204–5217. [Google Scholar] [CrossRef]
- Chondrogianni, N.; Voutetakis, K.; Kapetanou, M.; Delitsikou, V.; Papaevgeniou, N.; Sakellari, M.; Lefaki, M.; Filippopoulou, K.; Gonos, E.S. Proteasome activation: An innovative promising approach for delaying aging and retarding age-related diseases. Ageing Res. Rev. 2015, 23, 37–55. [Google Scholar] [CrossRef]
- Piccirillo, R.; Goldberg, A.L. The p97/VCP ATPase is critical in muscle atrophy and the accelerated degradation of muscle proteins. EMBO J. 2012, 31, 3334–3350. [Google Scholar] [CrossRef]
- Besche, H.C.; Haas, W.; Gygi, S.P.; Goldberg, A.L. Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 2009, 48, 2538–2549. [Google Scholar] [CrossRef]
- Kloppsteck, P.; Ewens, C.A.; Forster, A.; Zhang, X.; Freemont, P.S. Regulation of p97 in the ubiquitin-proteasome system by the UBX protein-family. Biochim. Biophys. Acta 2012, 1823, 125–129. [Google Scholar] [CrossRef]
- Lowell, B.B.; Ruderman, N.B.; Goodman, M.N. Evidence that lysosomes are not involved in the degradation of myofibrillar proteins in rat skeletal muscle. Biochem. J. 1986, 234, 237–240. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef]
- Liang, J.; Zeng, Z.; Zhang, Y.; Chen, N. Regulatory role of exercise-induced autophagy for sarcopenia. Exp. Gerontol. 2020, 130, 110789. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Seo, Y.K.; Kwon, K.S. Sarcopenia targeting with autophagy mechanism by exercise. BMB Rep. 2019, 52, 64–69. [Google Scholar] [CrossRef]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.F.; Vainshtein, A.; Iqbal, S.; Ostojic, O.; Hood, D.A. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am. J. Physiol. Cell Physiol. 2013, 304, 422–430. [Google Scholar] [CrossRef]
- Sakuma, K.; Kinoshita, M.; Ito, Y.; Aizawa, M.; Aoi, W.; Yamaguchi, A. p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J. Cachexia Sarcopenia Muscle 2016, 7, 204–212. [Google Scholar] [CrossRef]
- McMullen, C.A.; Ferry, A.L.; Gamboa, J.L.; Andrade, F.H.; Dupont-Versteegden, E.E. Age-related changes of cell death pathways in rat extraocular muscle. Exp. Gerontol. 2009, 44, 420–425. [Google Scholar] [CrossRef]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1alpha expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef]
- Raben, N.; Schreiner, C.; Baum, R.; Takikita, S.; Xu, S.; Xie, T.; Myerowitz, R.; Komatsu, M.; Van der Meulen, J.H.; Nagaraju, K.; et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder--murine Pompe disease. Autophagy 2010, 6, 1078–1089. [Google Scholar] [CrossRef]
- Reznick, R.M.; Zong, H.; Li, J.; Morino, K.; Moore, I.K.; Yu, H.J.; Liu, Z.X.; Dong, J.; Mustard, K.J.; Hawley, S.A.; et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007, 5, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E. Treatment of sarcopenia: The road to the future. J. Cachexia Sarcopenia Muscle 2018, 9, 1196–1199. [Google Scholar] [CrossRef] [PubMed]
- Fiatarone, M.A.; O’Neill, E.F.; Ryan, N.D.; Clements, K.M.; Solares, G.R.; Nelson, M.E.; Roberts, S.B.; Kehayias, J.J.; Lipsitz, L.A.; Evans, W.J. Exercise training and nutritional supplementation for physical frailty in very elderly people. N. Engl. J. Med. 1994, 330, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Breen, L.; Phillips, S.M. Skeletal muscle protein metabolism in the elderly: Interventions to counteract the ‘anabolic resistance’ of ageing. Nutr. Metab. 2011, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Jamart, C.; Raymackers, J.M.; Li An, G.; Deldicque, L.; Francaux, M. Prevention of muscle disuse atrophy by MG132 proteasome inhibitor. Muscle Nerve 2011, 43, 708–716. [Google Scholar] [CrossRef]
- Caron, A.Z.; Haroun, S.; Leblanc, E.; Trensz, F.; Guindi, C.; Amrani, A.; Grenier, G. The proteasome inhibitor MG132 reduces immobilization-induced skeletal muscle atrophy in mice. BMC Musculoskelet Disord. 2011, 12, 185. [Google Scholar] [CrossRef]
- Enrico, O.; Gabriele, B.; Nadia, C.; Sara, G.; Daniele, V.; Giulia, C.; Antonio, S.; Mario, P. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br. J. Haematol. 2007, 138, 396–397. [Google Scholar]
- McGlory, C.; Calder, P.C.; Nunes, E.A. The influence of Omega-3 fatty acids on skeletal muscle protein turnover in health, disuse, and disease. Front. Nutr. 2019, 6, 144. [Google Scholar] [CrossRef]
- Brand, A.; Bauer, N.G.; Hallott, A.; Goldbaum, O.; Ghebremeskel, K.; Reifen, R.; Richter-Landsberg, C. Membrane lipid modification by polyunsaturated fatty acids sensitizes oligodendroglial OLN-93 cells against oxidative stress and promotes up-regulation of heme oxygenase-1 (HSP32). J. Neurochem. 2010, 113, 465–476. [Google Scholar] [CrossRef]
- Ministry of Agriculture FaF. Dietary intake of iodine and fatty acids. Food Inf. Surveill. Sheet 1997, 127, 1–10. [Google Scholar]
- Tachtsis, B.; Camera, D.; Lacham-Kaplan, O. Potential roles of n-3 PUFAs during skeletal muscle growth and regeneration. Nutrients 2018, 10, 309. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Jones, A.E.; Wootton, S.A. Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men*. Br. J. Nutr. 2002, 88, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamid, A.S.; Brown, T.J.; Brainard, J.S.; Biswas, P.; Thorpe, G.C.; Moore, H.J.; Deane, K.H.; AlAbdulghafoor, F.K.; Summerbell, C.D.; Worthington, H.V.; et al. Omega-3 fatty acids for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2018, 11. CD003177. [Google Scholar]
- Cornwell, D.G.; Huttner, J.J.; Milo, G.E.; Panganamala, R.V.; Sharma, H.M.; Geer, J.C. Polyunsaturated fatty acids, vitamin E, and the proliferation of aortic smooth muscle cells. Lipids 1979, 14, 194–207. [Google Scholar] [CrossRef]
- Huttner, J.J.; Gwebu, E.T.; Panganamala, R.V.; Milo, G.E.; Cornwell, D.C.; Sharma, H.M.; Geer, J.C. Fatty acids and their prostaglandin derivatives: Inhibitors of proliferation in aortic smooth muscle cells. Science 1977, 197, 289–291. [Google Scholar] [CrossRef]
- Stadtman, E.R. Protein oxidation and aging. Free Radic. Res. 2006, 40, 1250–1258. [Google Scholar] [CrossRef]
- Shin, S.; Jing, K.; Jeong, S.; Kim, N.; Song, K.S.; Heo, J.Y.; Park, J.H.; Seo, K.S.; Han, J.; Park, J.I.; et al. The omega-3 polyunsaturated fatty acid DHA induces simultaneous apoptosis and autophagy via mitochondrial ROS-mediated Akt-mTOR signaling in prostate cancer cells expressing mutant p53. Biomed. Res. Int. 2013, 2013, 568671. [Google Scholar] [CrossRef]
- Gabande-Rodriguez, E.; Gomez de Las Heras, M.M.; Mittelbrunn, M. Control of inflammation by calorie restriction mimetics: on the crossroad of autophagy and mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef]
- Mildenberger, J.; Johansson, I.; Sergin, I.; Kjobli, E.; Damas, J.K.; Razani, B.; Flo, T.H.; Bjorkoy, G. N-3 PUFAs induce inflammatory tolerance by formation of KEAP1-containing SQSTM1/p62-bodies and activation of NFE2L2. Autophagy 2017, 13, 1664–1678. [Google Scholar] [CrossRef]
- Kamolrat, T.; Gray, S.R. The effect of eicosapentaenoic and docosahexaenoic acid on protein synthesis and breakdown in murine C2C12 myotubes. Biochem. Biophys. Res. Commun. 2013, 432, 593–598. [Google Scholar] [CrossRef]
- Calder, P.C. Docosahexaenoic acid. Ann. Nutr. Metab. 2016, 69 (Suppl. 1), 7–21. [Google Scholar] [CrossRef] [PubMed]
- Cornish, S.M.; Chilibeck, P.D. Alpha-linolenic acid supplementation and resistance training in older adults. Appl. Physiol. Nutr. Metab. 2009, 34, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Da Boit, M.; Sibson, R.; Sivasubramaniam, S.; Meakin, J.R.; Greig, C.A.; Aspden, R.M.; Thies, F.; Jeromson, S.; Hamilton, D.L.; Speakman, J.R.; et al. Sex differences in the effect of fish-oil supplementation on the adaptive response to resistance exercise training in older people: A randomized controlled trial. Am. J. Clin. Nutr. 2017, 105, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Rodacki, C.L.; Rodacki, A.L.; Pereira, G.; Naliwaiko, K.; Coelho, I.; Pequito, D.; Fernandes, L.C. Fish-oil supplementation enhances the effects of strength training in elderly women. Am. J. Clin. Nutr. 2012, 95, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.Y.; Chan, R.; Kwok, T.; Cheng, K.C.; Ha, A.; Woo, J. Effects of exercise and nutrition supplementation in community-dwelling older Chinese people with sarcopenia: A randomized controlled trial. Age Ageing 2019, 48, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.I.; Atherton, P.; Reeds, D.N.; Mohammed, B.S.; Rankin, D.; Rennie, M.J.; Mittendorfer, B. Dietary omega-3 fatty acid supplementation increases the rate of muscle protein synthesis in older adults: A randomized controlled trial. Am. J. Clin. Nutr. 2011, 93, 402–412. [Google Scholar] [CrossRef]
- Smith, G.I.; Julliand, S.; Reeds, D.N.; Sinacore, D.R.; Klein, S.; Mittendorfer, B. Fish oil-derived n-3 PUFA therapy increases muscle mass and function in healthy older adults. Am. J. Clin. Nutr. 2015, 102, 115–122. [Google Scholar] [CrossRef]
- Hutchins-Wiese, H.L.; Kleppinger, A.; Annis, K.; Liva, E.; Lammi-Keefe, C.J.; Durham, H.A.; Kenny, A.M. The impact of supplemental n-3 long chain polyunsaturated fatty acids and dietary antioxidants on physical performance in postmenopausal women. J. Nutr. Health Aging 2013, 17, 76–80. [Google Scholar] [CrossRef]
- Yoshino, J.; Smith, G.I.; Kelly, S.C.; Julliand, S.; Reeds, D.N.; Mittendorfer, B. Effect of dietary n-3 PUFA supplementation on the muscle transcriptome in older adults. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef]
- Huang, F.; Wei, H.; Luo, H.; Jiang, S.; Peng, J. EPA inhibits the inhibitor of kappaBalpha (IkappaBalpha)/NF-kappaB/muscle RING finger 1 pathway in C2C12 myotubes in a PPARgamma-dependent manner. Br. J. Nutr. 2011, 105, 348–356. [Google Scholar] [CrossRef]
- Whitehouse, A.S.; Smith, H.J.; Drake, J.L.; Tisdale, M.J. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res. 2001, 61, 3604–3609. [Google Scholar] [PubMed]
- Whitehouse, A.S.; Tisdale, M.J. Downregulation of ubiquitin-dependent proteolysis by eicosapentaenoic acid in acute starvation. Biochem. Biophys. Res. Commun. 2001, 285, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.J.; Khal, J.; Tisdale, M.J. Downregulation of ubiquitin-dependent protein degradation in murine myotubes during hyperthermia by eicosapentaenoic acid. Biochem. Biophys. Res. Commun. 2005, 332, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Khal, J.; Tisdale, M.J. Downregulation of muscle protein degradation in sepsis by eicosapentaenoic acid (EPA). Biochem. Biophys. Res. Commun. 2008, 375, 238–240. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, Q.W.; Zheng, P.P.; Zhang, J.S.; Huang, F.R. DHA inhibits protein degradation more efficiently than EPA by regulating the PPARgamma/NFkappaB pathway in C2C12 myotubes. Biomed. Res. Int. 2013, 2013. 318981. [Google Scholar]
- Woodworth-Hobbs, M.E.; Hudson, M.B.; Rahnert, J.A.; Zheng, B.; Franch, H.A.; Price, S.R. Docosahexaenoic acid prevents palmitate-induced activation of proteolytic systems in C2C12 myotubes. J. Nutr. Biochem. 2014, 25, 868–874. [Google Scholar] [CrossRef]
- Deval, C.; Capel, F.; Laillet, B.; Polge, C.; Bechet, D.; Taillandier, D.; Attaix, D.; Combaret, L. Docosahexaenoic acid-supplementation prior to fasting prevents muscle atrophy in mice. J. Cachexia Sarcopenia Muscle 2016, 7, 587–603. [Google Scholar] [CrossRef]
- Kim, K.M.; Jang, H.C.; Lim, S. Differences among skeletal muscle mass indices derived from height-, weight-, and body mass index-adjusted models in assessing sarcopenia. Korean J. Intern. Med. 2016, 31, 643–650. [Google Scholar] [CrossRef]
- Smith, H.J.; Lorite, M.J.; Tisdale, M.J. Effect of a cancer cachectic factor on protein synthesis/degradation in murine C2C12 myoblasts: Modulation by eicosapentaenoic acid. Cancer Res. 1999, 59, 5507–5513. [Google Scholar]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef]
- Schiaffino, S.; Dyar, K.A.; Ciciliot, S.; Blaauw, B.; Sandri, M. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 2013, 280, 4294–4314. [Google Scholar] [CrossRef] [PubMed]
- Izumiya, Y.; Hopkins, T.; Morris, C.; Sato, K.; Zeng, L.; Viereck, J.; Hamilton, J.A.; Ouchi, N.; LeBrasseur, N.K.; Walsh, K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008, 7, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Pallafacchina, G.; Calabria, E.; Serrano, A.L.; Kalhovde, J.M.; Schiaffino, S. A protein kinase B-dependent and rapamycin-sensitive pathway controls skeletal muscle growth but not fiber type specification. Proc. Natl. Acad. Sci. USA 2002, 99, 9213–9218. [Google Scholar] [CrossRef] [PubMed]
- Falcon, L.J.; Harris-Love, M.O. Sarcopenia and the new ICD-10-CM code: Screening, staging, and diagnosis considerations. Fed. Pract. 2017, 34, 24–32. [Google Scholar]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, J.H.; Jeon, J.H.; Lee, M.J. Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems. Nutrients 2020, 12, 2597. https://doi.org/10.3390/nu12092597
Lee JH, Jeon JH, Lee MJ. Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems. Nutrients. 2020; 12(9):2597. https://doi.org/10.3390/nu12092597
Chicago/Turabian StyleLee, Jung Hoon, Jun Hyoung Jeon, and Min Jae Lee. 2020. "Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems" Nutrients 12, no. 9: 2597. https://doi.org/10.3390/nu12092597
APA StyleLee, J. H., Jeon, J. H., & Lee, M. J. (2020). Docosahexaenoic Acid, a Potential Treatment for Sarcopenia, Modulates the Ubiquitin–Proteasome and the Autophagy–Lysosome Systems. Nutrients, 12(9), 2597. https://doi.org/10.3390/nu12092597