Gluten Degrading Enzymes for Treatment of Celiac Disease

 ,
,
Abstract
1. Introduction
2. Origin and Properties of Gluten
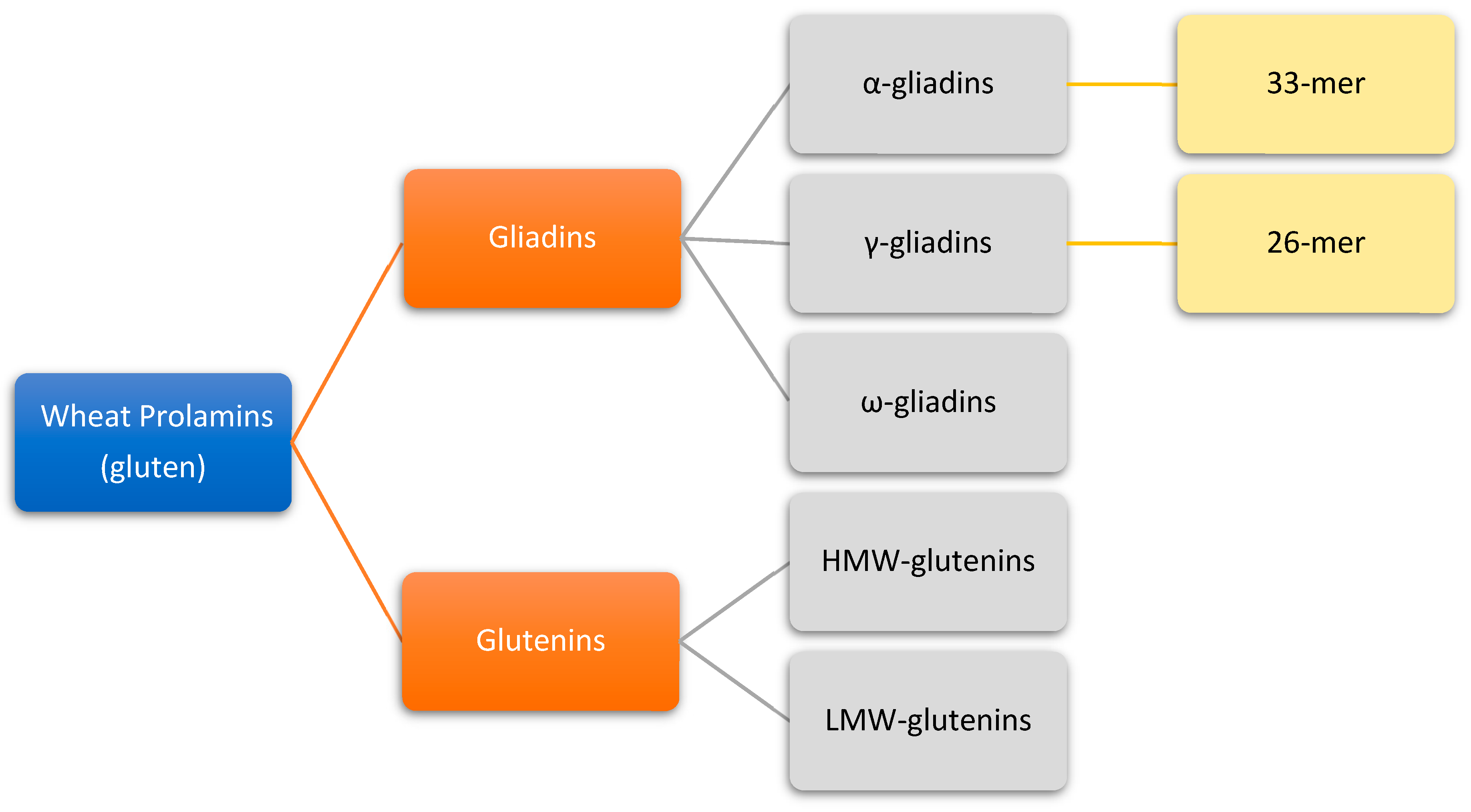
3. Gluten Structure
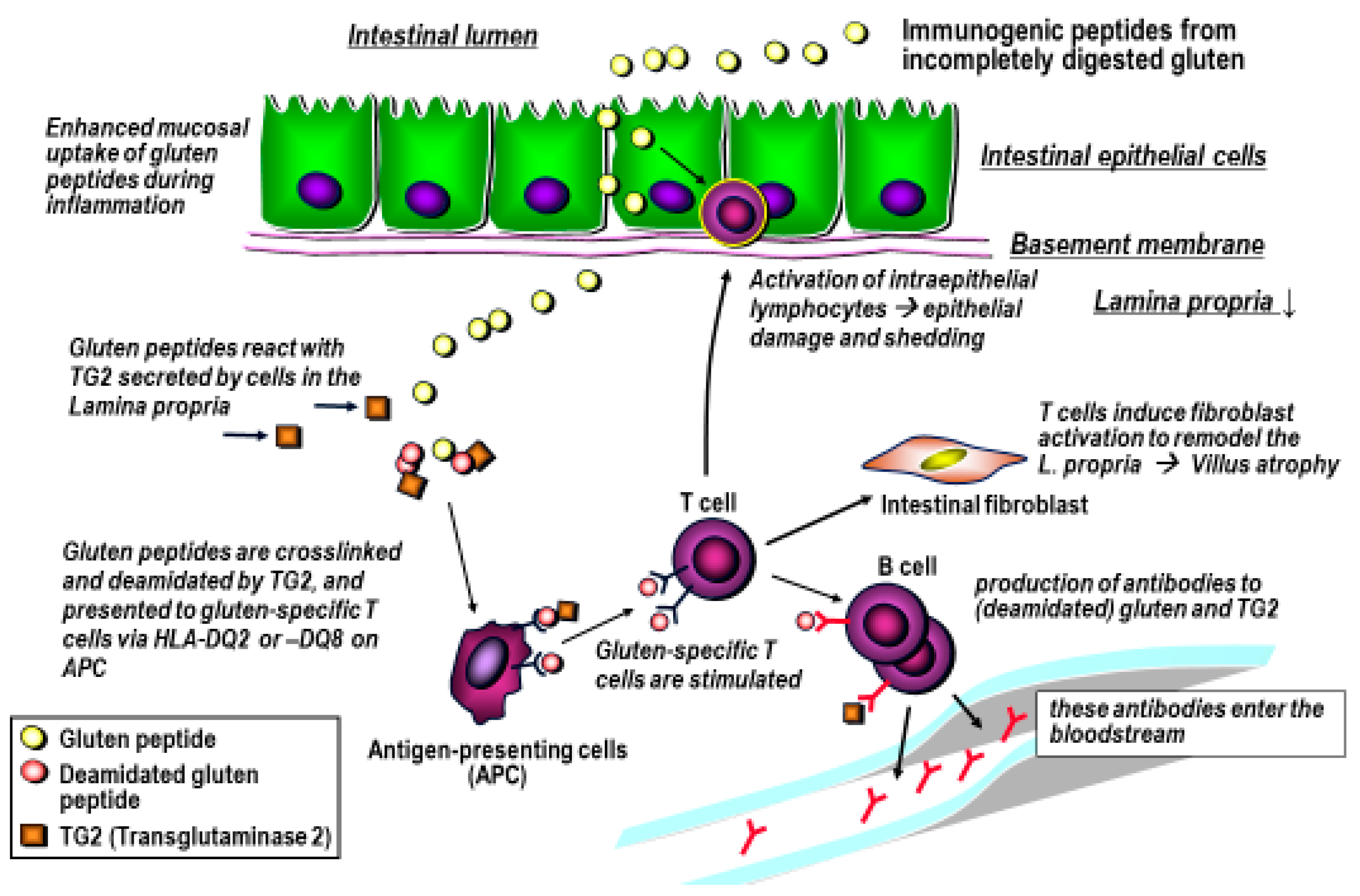
4. Gluten Immunogenic Peptides as Drivers of CeD
5. The Approach for the Use of Gluten-Degrading Enzymes in CeD
6. Classification and Origin of Gluten-Degrading Enzymes
6.1. Prolyl Endopeptidases (PEP)
6.2. Glutamine-Specific Cysteine Endoprotease (EP-B2)
6.3. Gluten-Degrading Enzymes in Human Saliva
7. Challenge of Glutenase Therapy for Celiac Disease
8. Novel Strategies for Enzyme Therapies
9. Enzyme Combinations
10. Molecular Modeling
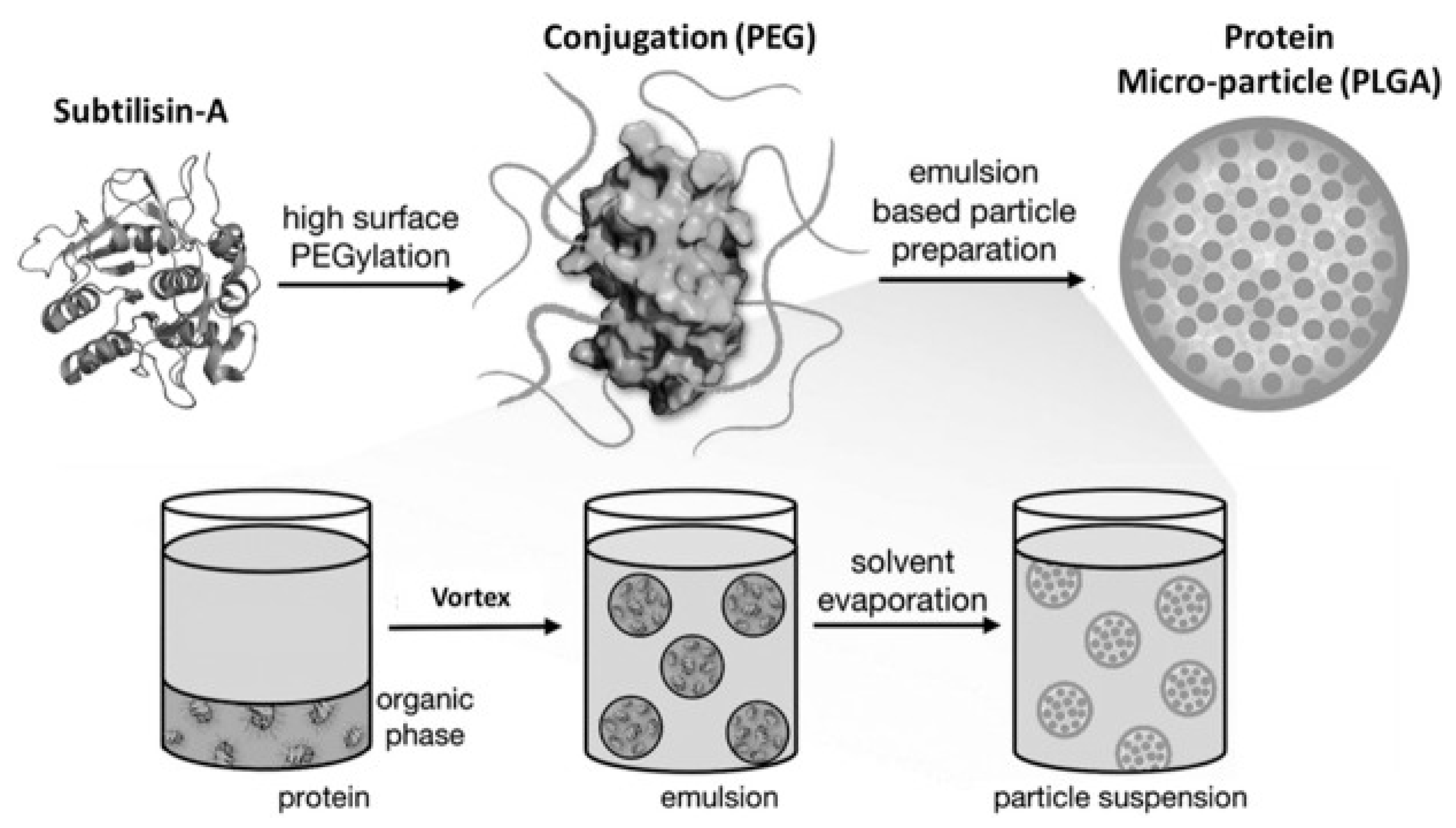
11. Pharmaceutical Enzyme Modification by PEGylation and Microencapsulation
12. Summary
Grant support
Funding
Conflicts of Interest
References
- Leffler, D.A.; Schuppan, D. Update on serologic testing in celiac disease. Am. J. Gastroenterol. 2010, 105, 2520–2524. [Google Scholar] [CrossRef] [PubMed]
- Werkstetter, K.J.; Korponay-Szabo, I.R.; Popp, A.; Villanacci, V.; Salemme, M.; Heilig, G.; Lillevang, S.T.; Mearin, M.L.; Ribes-Koninckx, C.; Thomas, A.; et al. Accuracy in diagnosis of celiac disease without biopsies in clinical practice. Gastroenterology 2017, 153, 924–935. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Junker, Y.; Barisani, D. Celiac disease: From pathogenesis to novel therapies. Gastroenterology 2009, 137, 1912–1933. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, B.; Ludvigsson, J.F.; Green, P.H. Celiac disease and non-celiac gluten sensitivity. BMJ 2015, 351, h4347. [Google Scholar] [CrossRef]
- Samasca, G.; Lerner, A.; Girbovan, A.; Sur, G.; Lupan, I.; Makovicky, P.; Matthias, T.; Freeman, H.J. Challenges in gluten-free diet in coeliac disease: Prague consensus. Eur. J. Clin. Investig. 2017, 47, 394–397. [Google Scholar] [CrossRef]
- Lundin, K.E.; Sollid, L.M. Advances in coeliac disease. Curr. Opin. Gastroenterol. 2014, 30, 154–162. [Google Scholar] [CrossRef]
- Serena, G.; Kelly, C.P.; Fasano, A. Nondietary therapies for celiac disease. Gastroenterol. Clin. N. Am. 2019, 48, 145–163. [Google Scholar] [CrossRef]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef]
- Acevedo-Pacheco, L.; Serna-Saldivar, S.O. In vivo protein quality of selected cereal-based staple foods enriched with soybean proteins. Food Nutr. Res. 2016, 60, 31382. [Google Scholar] [CrossRef]
- Godfrey, D.; Hawkesford, M.J.; Powers, S.J.; Millar, S.; Shewry, P.R. Effects of crop nutrition on wheat grain composition and end use quality. J. Agric. Food Chem. 2010, 58, 3012–3021. [Google Scholar] [CrossRef]
- Shewry, P.R.; Casey, R. Seed Proteins; Springer: Dordrecht, The Netherlands, 1999. [Google Scholar]
- Osborne, T.B. The Proteins of the Wheat Kernel; Carnegie Institution: Washington, DC, USA, 1907; p. 119. [Google Scholar]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Scherf, K.A.; Catassi, C.; Chirdo, F.; Ciclitira, P.J.; Feighery, C.; Gianfrani, C.; Koning, F.; Lundin, K.E.A.; Schuppan, D.; Smulders, M.J.M.; et al. Recent progress and recommendations on celiac disease from the working group on prolamin analysis and toxicity. Front. Nutr. 2020, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Koning, F.; Gilissen, L.; Wijmenga, C. Gluten: A two-edged sword. Immunopathogenesis of celiac disease. Springer Semin Immunopathol. 2005, 27, 217–232. [Google Scholar] [CrossRef]
- Hausch, F.; Shan, L.; Santiago, N.A.; Gray, G.M.; Khosla, C. Intestinal digestive resistance of immunodominant gliadin peptides. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G996–G1003. [Google Scholar] [CrossRef] [PubMed]
- Piper, J.L.; Gray, G.M.; Khosla, C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J. Pharmacol. Exp. Ther. 2004, 311, 213–219. [Google Scholar] [CrossRef]
- Barak, S.; Mudgil, D.; Khatkar, B.S. Biochemical and functional properties of wheat gliadins: A review. Crit Rev. Food Sci. Nutr. 2015, 55, 357–368. [Google Scholar] [CrossRef]
- Shan, L.; Molberg, O.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef]
- Shan, L.; Qiao, S.W.; Arentz-Hansen, H.; Molberg, O.; Gray, G.M.; Sollid, L.M.; Khosla, C. Identification and analysis of multivalent proteolytically resistant peptides from gluten: Implications for celiac sprue. J. Proteome Res. 2005, 4, 1732–1741. [Google Scholar] [CrossRef]
- Sollid, L.M.; Khosla, C. Novel therapies for coeliac disease. J. Intern. Med. 2011, 269, 604–613. [Google Scholar] [CrossRef]
- Sollid, L.M. Molecular basis of celiac disease. Annu. Rev. Immunol. 2000, 18, 53–81. [Google Scholar] [CrossRef]
- Dieterich, W.; Laag, E.; Schopper, H.; Volta, U.; Ferguson, A.; Gillett, H.; Riecken, E.O.; Schuppan, D. Autoantibodies to tissue transglutaminase as predictors of celiac disease. Gastroenterology 1998, 115, 1317–1321. [Google Scholar] [CrossRef]
- Dieterich, W.; Ehnis, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Molberg, O.; McAdam, S.N.; Korner, R.; Quarsten, H.; Kristiansen, C.; Madsen, L.; Fugger, L.; Scott, H.; Noren, O.; Roepstorff, P.; et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat. Med. 1998, 4, 713–717. [Google Scholar] [CrossRef] [PubMed]
- van de Wal, Y.; Kooy, Y.; van Veelen, P.; Pena, S.; Mearin, L.; Papadopoulos, G.; Koning, F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J. Immunol. 1998, 161, 1585–1588. [Google Scholar] [PubMed]
- Schuppan, D.; Gisbert-Schuppan, K. Wheat Syndromes: How Wheat, Gluten and ATI Cause Inflammation, IBS and Autoimmune Diseases; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Shan, L.; Marti, T.; Sollid, L.M.; Gray, G.M.; Khosla, C. Comparative biochemical analysis of three bacterial prolyl endopeptidases: Implications for coeliac sprue. Biochem. J. 2004, 383, 311–318. [Google Scholar] [CrossRef]
- Vader, L.W.; Stepniak, D.T.; Bunnik, E.M.; Kooy, Y.M.; de Haan, W.; Drijfhout, J.W.; Van Veelen, P.A.; Koning, F. Characterization of cereal toxicity for celiac disease patients based on protein homology in grains. Gastroenterology 2003, 125, 1105–1113. [Google Scholar] [CrossRef]
- Bethune, M.T.; Khosla, C. Oral enzyme therapy for celiac sprue. Methods Enzymol. 2012, 502, 241–271. [Google Scholar] [CrossRef]
- White, L.E.; Bannerman, E.; Gillett, P.M. Coeliac disease and the gluten-free diet: A review of the burdens; factors associated with adherence and impact on health-related quality of life, with specific focus on adolescence. J. Hum. Nutr. Diet. 2016, 29, 593–606. [Google Scholar] [CrossRef]
- Catassi, C.; Fabiani, E.; Iacono, G.; D’Agate, C.; Francavilla, R.; Biagi, F.; Volta, U.; Accomando, S.; Picarelli, A.; De Vitis, I.; et al. A prospective, double-blind, placebo-controlled trial to establish a safe gluten threshold for patients with celiac disease. Am. J. Clin. Nutr. 2007, 85, 160–166. [Google Scholar] [CrossRef]
- Khosla, C. Celiac disease: Lessons for and from chemical biology. ACS Chem. Biol. 2017, 12, 1455–1459. [Google Scholar] [CrossRef]
- Ehren, J.; Moron, B.; Martin, E.; Bethune, M.T.; Gray, G.M.; Khosla, C. A food-grade enzyme preparation with modest gluten detoxification properties. PLoS ONE 2009, 4, e6313. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Bethune, M.T.; Siegel, M.; Spencer, A.; Khosla, C. Combination enzyme therapy for gastric digestion of dietary gluten in patients with celiac sprue. Gastroenterology 2007, 133, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Khosla, C. Prolyl endopeptidases. Cell Mol. Life Sci. 2007, 64, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Krishnareddy, S.; Stier, K.; Recanati, M.; Lebwohl, B.; Green, P.H. Commercially available glutenases: A potential hazard in coeliac disease. Ther. Adv. Gastroenterol. 2017, 10, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Alhassan, E.; Yadav, A.; Kelly, C.P.; Mukherjee, R. Novel nondietary therapies for celiac disease. Cell Mol. Gastroenterol. Hepatol. 2019, 8, 335–345. [Google Scholar] [CrossRef]
- Cerf-Bensussan, N.; Matysiak-Budnik, T.; Cellier, C.; Heyman, M. Oral proteases: A new approach to managing coeliac disease. Gut 2007, 56, 157–160. [Google Scholar] [CrossRef][Green Version]
- Ludvigsson, J.F.; Ciacci, C.; Green, P.H.; Kaukinen, K.; Korponay-Szabo, I.R.; Kurppa, K.; Murray, J.A.; Lundin, K.E.A.; Maki, M.J.; Popp, A.; et al. Outcome measures in coeliac disease trials: The Tampere recommendations. Gut 2018, 67, 1410–1424. [Google Scholar] [CrossRef]
- Siegel, M.; Bethune, M.T.; Gass, J.; Ehren, J.; Xia, J.; Johannsen, A.; Stuge, T.B.; Gray, G.M.; Lee, P.P.; Khosla, C. Rational design of combination enzyme therapy for celiac sprue. Chem. Biol. 2006, 13, 649–658. [Google Scholar] [CrossRef]
- Zamakhchari, M.; Wei, G.; Dewhirst, F.; Lee, J.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. Identification of Rothia bacteria as gluten-degrading natural colonizers of the upper gastro-intestinal tract. PLoS ONE 2011, 6, e24455. [Google Scholar] [CrossRef]
- Helmerhorst, E.J.; Zamakhchari, M.; Schuppan, D.; Oppenheim, F.G. Discovery of a novel and rich source of gluten-degrading microbial enzymes in the oral cavity. PLoS ONE 2010, 5, e13264. [Google Scholar] [CrossRef] [PubMed]
- Stepniak, D.; Spaenij-Dekking, L.; Mitea, C.; Moester, M.; de Ru, A.; Baak-Pablo, R.; van Veelen, P.; Edens, L.; Koning, F. Highly efficient gluten degradation with a newly identified prolyl endoprotease: Implications for celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 291, G621–G629. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, S.; Goeltz, P.; Thibault, P.; Banville, D.; Gagnon, J. Characterization of a prolyl endopeptidase from Flavobacterium meningosepticum. Complete sequence and localization of the active-site serine. J. Biol. Chem. 1992, 267, 8192–8199. [Google Scholar] [PubMed]
- Diefenthal, T.; Dargatz, H.; Witte, V.; Reipen, G.; Svendsen, I. Cloning of proline-specific endopeptidase gene from Flavobacterium meningosepticum: Expression in Escherichia coli and purification of the heterologous protein. Appl. Microbiol. Biotechnol. 1993, 40, 90–97. [Google Scholar] [CrossRef]
- Kabashima, T.; Fujii, M.; Meng, Y.; Ito, K.; Yoshimoto, T. Prolyl endopeptidase from Sphingomonas capsulata: Isolation and characterization of the enzyme and nucleotide sequence of the gene. Arch. Biochem. Biophys. 1998, 358, 141–148. [Google Scholar] [CrossRef]
- Mitea, C.; Havenaar, R.; Drijfhout, J.W.; Edens, L.; Dekking, L.; Koning, F. Efficient degradation of gluten by a prolyl endoprotease in a gastrointestinal model: Implications for coeliac disease. Gut 2008, 57, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Edens, L.; Dekker, P.; van der Hoeven, R.; Deen, F.; de Roos, A.; Floris, R. Extracellular prolyl endoprotease from Aspergillus niger and its use in the debittering of protein hydrolysates. J. Agric. Food Chem. 2005, 53, 7950–7957. [Google Scholar] [CrossRef]
- Tack, G.J.; van de Water, J.M.; Bruins, M.J.; Kooy-Winkelaar, E.M.; van Bergen, J.; Bonnet, P.; Vreugdenhil, A.C.; Korponay-Szabo, I.; Edens, L.; von Blomberg, B.M.; et al. Consumption of gluten with gluten-degrading enzyme by celiac patients: A pilot-study. World. J. Gastroenterol. 2013, 19, 5837–5847. [Google Scholar] [CrossRef]
- Salden, B.N.; Monserrat, V.; Troost, F.J.; Bruins, M.J.; Edens, L.; Bartholome, R.; Haenen, G.R.; Winkens, B.; Koning, F.; Masclee, A.A. Randomised clinical study: Aspergillus niger-derived enzyme digests gluten in the stomach of healthy volunteers. Aliment. Pharmacol. Ther. 2015, 42, 273–285. [Google Scholar] [CrossRef]
- Konig, J.; Holster, S.; Bruins, M.J.; Brummer, R.J. Randomized clinical trial: Effective gluten degradation by Aspergillus niger-derived enzyme in a complex meal setting. Sci. Rep. 2017, 7, 13100. [Google Scholar] [CrossRef]
- Bethune, M.T.; Strop, P.; Tang, Y.; Sollid, L.M.; Khosla, C. Heterologous expression, purification, refolding, and structural-functional characterization of EP-B2, a self-activating barley cysteine endoprotease. Chem. Biol. 2006, 13, 637–647. [Google Scholar] [CrossRef]
- Gass, J.; Vora, H.; Bethune, M.T.; Gray, G.M.; Khosla, C. Effect of barley endoprotease EP-B2 on gluten digestion in the intact rat. J. Pharmacol. Exp. Ther. 2006, 318, 1178–1186. [Google Scholar] [CrossRef]
- Bethune, M.T.; Ribka, E.; Khosla, C.; Sestak, K. Transepithelial transport and enzymatic detoxification of gluten in gluten-sensitive rhesus macaques. PLoS ONE 2008, 3, e1857. [Google Scholar] [CrossRef]
- Vora, H.; McIntire, J.; Kumar, P.; Deshpande, M.; Khosla, C. A scaleable manufacturing process for pro-EP-B2, a cysteine protease from barley indicated for celiac sprue. Biotechnol. Bioeng. 2007, 98, 177–185. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Chen, T.; Izard, J.; Paster, B.J.; Tanner, A.C.; Yu, W.H.; Lakshmanan, A.; Wade, W.G. The human oral microbiome. J. Bacteriol. 2010, 192, 5002–5017. [Google Scholar] [CrossRef]
- Helmerhorst, E.J.; Sun, X.; Salih, E.; Oppenheim, F.G. Identification of Lys-Pro-Gln as a novel cleavage site specificity of saliva-associated proteases. J. Biol. Chem. 2008, 283, 19957–19966. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Feo, M.; Wei, G.; Blumenkranz, G.; Dewhirst, F.E.; Schuppan, D.; Oppenheim, F.G.; Helmerhorst, E.J. The cultivable human oral gluten-degrading microbiome and its potential implications in coeliac disease and gluten sensitivity. Clin. Microbiol. Infect. 2013, 19, E386–E394. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Faller, L.; Leffler, D.A.; Kelly, C.P.; Hansen, J.; Bosch, J.A.; Wei, G.; Paster, B.J.; Schuppan, D.; Helmerhorst, E.J. Salivary gluten degradation and oral microbial profiles in healthy individuals and celiac disease patients. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef]
- Tian, N.; Wei, G.; Schuppan, D.; Helmerhorst, E.J. Effect of Rothia mucilaginosa enzymes on gliadin (gluten) structure, deamidation, and immunogenic epitopes relevant to celiac disease. Am. J. Physiol Gastrointest. Liver Physiol. 2014, 307, G769–G776. [Google Scholar] [CrossRef]
- Wei, G.; Tian, N.; Siezen, R.; Schuppan, D.; Helmerhorst, E.J. Identification of food-grade subtilisins as gluten-degrading enzymes to treat celiac disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G571–G580. [Google Scholar] [CrossRef]
- Wei, G.; Tian, N.; Valery, A.C.; Zhong, Y.; Schuppan, D.; Helmerhorst, E.J. Identification of Pseudolysin (lasB) as an aciduric gluten-degrading enzyme with high therapeutic potential for celiac disease. Am. J. Gastroenterol. 2015, 110, 899–908. [Google Scholar] [CrossRef]
- Helmerhorst, E.J.; Wei, G. Experimental Strategy to Discover Microbes with Gluten-degrading Enzyme Activities. In Proceedings of the SPIE—The International Society for Optical Engineering, Baltimore, MD, USA, 5 May 2014; p. 9112:91120D. [Google Scholar] [CrossRef]
- Darwish, G.; Helmerhorst, E.J.; Schuppan, D.; Oppenheim, F.G.; Wei, G. Pharmaceutically modified subtilisins withstand acidic conditions and effectively degrade gluten in vivo. Sci Rep. 2019, 9, 7505. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann, G.; Leroux, J.C. In vivo fluorescence imaging of exogenous enzyme activity in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2011, 108, 9032–9037. [Google Scholar] [CrossRef] [PubMed]
- Tye-Din, J.A.; Anderson, R.P.; Ffrench, R.A.; Brown, G.J.; Hodsman, P.; Siegel, M.; Botwick, W.; Shreeniwas, R. The effects of ALV003 pre-digestion of gluten on immune response and symptoms in celiac disease in vivo. Clin. Immunol. 2010, 134, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Lahdeaho, M.L.; Kaukinen, K.; Laurila, K.; Vuotikka, P.; Koivurova, O.P.; Karja-Lahdensuu, T.; Marcantonio, A.; Adelman, D.C.; Maki, M. Glutenase ALV003 attenuates gluten-induced mucosal injury in patients with celiac disease. Gastroenterology 2014, 146, 1649–1658. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.A.; Kelly, C.P.; Green, P.H.R.; Marcantonio, A.; Wu, T.T.; Maki, M.; Adelman, D.C.; CeliAction Study Group of Investigators. No difference between latiglutenase and placebo in reducing villous atrophy or improving symptoms in patients with symptomatic celiac disease. Gastroenterology 2017, 152, 787–798.e2. [Google Scholar] [CrossRef]
- Syage, J.A.; Green, P.H.R.; Khosla, C.; Adelman, D.C.; Sealey-Voyksner, J.A.; Murray, J.A. Latiglutenase treatment for celiac disease: Symptom and quality of life improvement for seropositive patients on a gluten-free diet. GastroHep 2019, 1, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Stanley, E.J.; Wolf, S.; Toland, A.; Wu, S.J.; Hadidi, D.; Mills, J.H.; Baker, D.; Pultz, I.S.; Siegel, J.B. Computational design of an alpha-gliadin peptidase. J. Am. Chem. Soc. 2012, 134, 20513–20520. [Google Scholar] [CrossRef]
- Wolf, C.; Siegel, J.B.; Tinberg, C.; Camarca, A.; Gianfrani, C.; Paski, S.; Guan, R.; Montelione, G.; Baker, D.; Pultz, I.S. Engineering of Kuma030: A Gliadin Peptidase that rapidly degrades immunogenic gliadin peptides in gastric conditions. J. Am. Chem. Soc. 2015, 137, 13106–13113. [Google Scholar] [CrossRef]
- Castellanos, I.J.; Al-Azzam, W.; Griebenow, K. Effect of the covalent modification with poly(ethylene glycol) on alpha-chymotrypsin stability upon encapsulation in poly(lactic-co-glycolic) microspheres. J. Pharm. Sci. 2005, 94, 327–340. [Google Scholar] [CrossRef]
- Abuchowski, A.; van Es, T.; Palczuk, N.C.; Davis, F.F. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. J. Biol. Chem. 1977, 252, 3578–3581. [Google Scholar] [PubMed]
- Vinogradov, S.; Batrakova, E.; Kabanov, A. Poly(ethylene glycol)-polyethyleneimine NanoGel(TM) particles: Novel drug delivery systems for antisense oligonucleotides. Colloids Surf. B Biointerfaces 1999, 16, 14. [Google Scholar] [CrossRef]
- Gombotz, W.R.; Pettit, D.K. Biodegradable polymers for protein and peptide drug delivery. Bioconjug. Chem. 1995, 6, 332–351. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H. How Enzymes Work: From Structure to Function, 2nd ed.; Jenny Stanford Publishing: Singapore, 2020; p. 270. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence | HLA | tTG |
|---|---|---|---|
| Gliadins: | |||
| Glia α (206–217) | SGQGSFQPSQQN | DQ8 | (+) |
| Glia-α2 (62–75) | PQPQLPYPQPQLPY | DQ2 | (+++) |
| Glia-α2 33mer (56–88) | LQLQPFPQPQLPYPQPQLPYPQPQLPYPQPQPF | DQ2 | (+++) |
| Glia-α9 (57–68) | QLQPFPQPQLPY | DQ2 | (+++) |
| Glia-α20 (93–106) | PFRPQQPYPQPQPQ | DQ2 | (+++) |
| Glia- γ 1 (138–153) | QPQQPQQSFPQQQRPF | DQ2 | (+++) |
| Glia- γ(5) 26mer (26–51) | FLQPQQPFPQQPQQPYPQQPQQPFPQ | DQ2 | (+++) |
| Glia- γ 30 (222–236) | VQGQGIIQPQQPAQL | DQ2 | (-) |
| Glutenins: | DQ2 | (+++) | |
| LMW-Glt-156 (40–59) | QPPFSQQQQSPFSQ | DQ2 | (+++) |
| LMW-Glt-17 (46–60) | QQPFSQQQQQPLPQ | DQ2 | (+++) |
| LMW-Glt (723–735) | QQGYYPTSPQQSG | DQ2 | (+++) |
| DQ2 | (+++) | ||
| Glu-5 | QQQXPQQPQQF | DQ2 | (+++) |
| Glu-21 | PQQSEQSQQPFQPQ | DQ2 | (---) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, G.; Helmerhorst, E.J.; Darwish, G.; Blumenkranz, G.; Schuppan, D. Gluten Degrading Enzymes for Treatment of Celiac Disease. Nutrients 2020, 12, 2095. https://doi.org/10.3390/nu12072095
Wei G, Helmerhorst EJ, Darwish G, Blumenkranz G, Schuppan D. Gluten Degrading Enzymes for Treatment of Celiac Disease. Nutrients. 2020; 12(7):2095. https://doi.org/10.3390/nu12072095
Chicago/Turabian StyleWei, Guoxian, Eva J. Helmerhorst, Ghassan Darwish, Gabriel Blumenkranz, and Detlef Schuppan. 2020. "Gluten Degrading Enzymes for Treatment of Celiac Disease" Nutrients 12, no. 7: 2095. https://doi.org/10.3390/nu12072095
APA StyleWei, G., Helmerhorst, E. J., Darwish, G., Blumenkranz, G., & Schuppan, D. (2020). Gluten Degrading Enzymes for Treatment of Celiac Disease. Nutrients, 12(7), 2095. https://doi.org/10.3390/nu12072095