Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epidemiological Evidence for the Association Between Dietary Fat and the Risk of Obesity and Cardiovascular Disease
3. Fat vs. Carbohydrate Intake and Cardiometabolic Risk
4. Role of Dietary Fat in Regulating Food and Caloric Intake
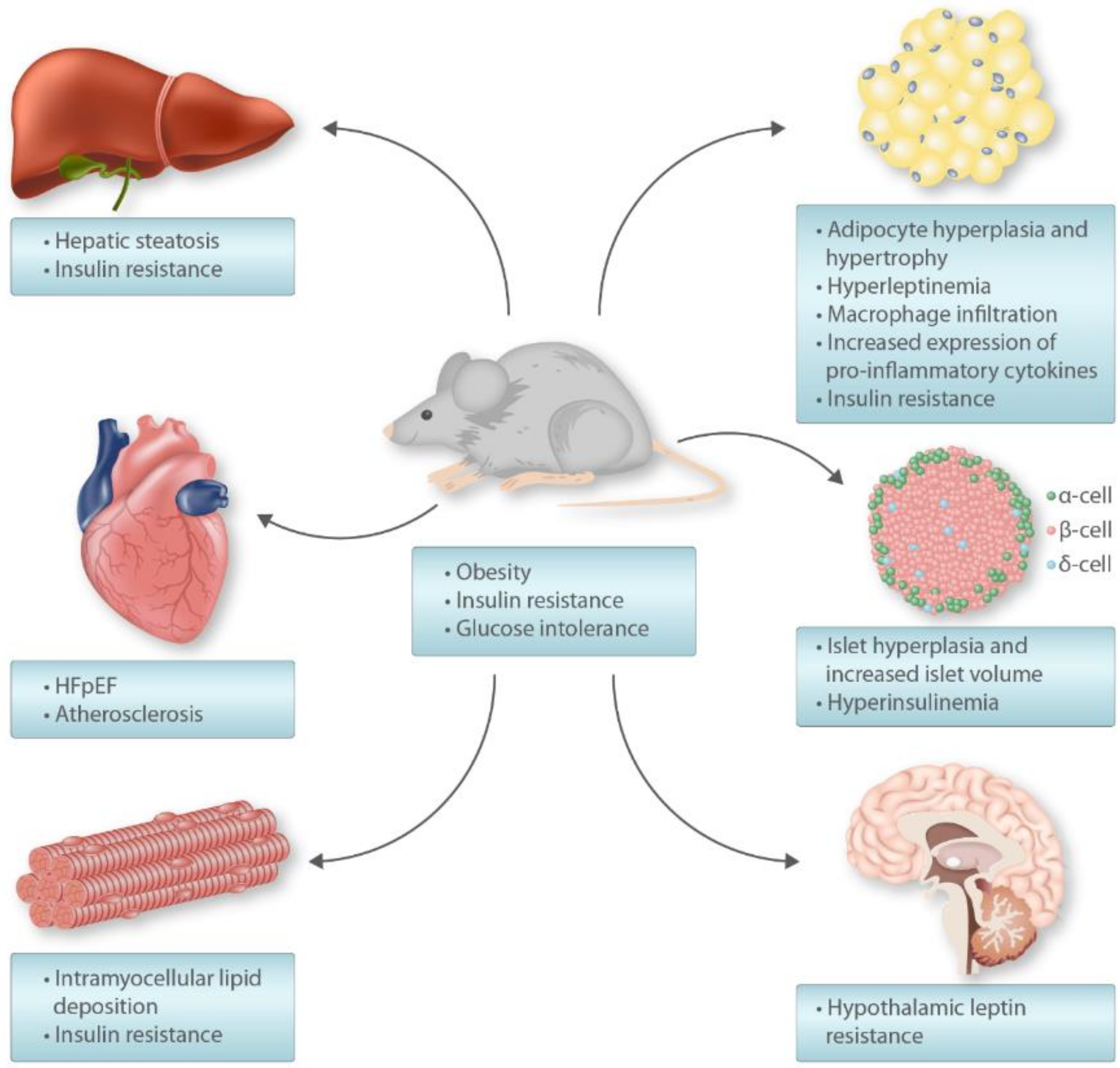
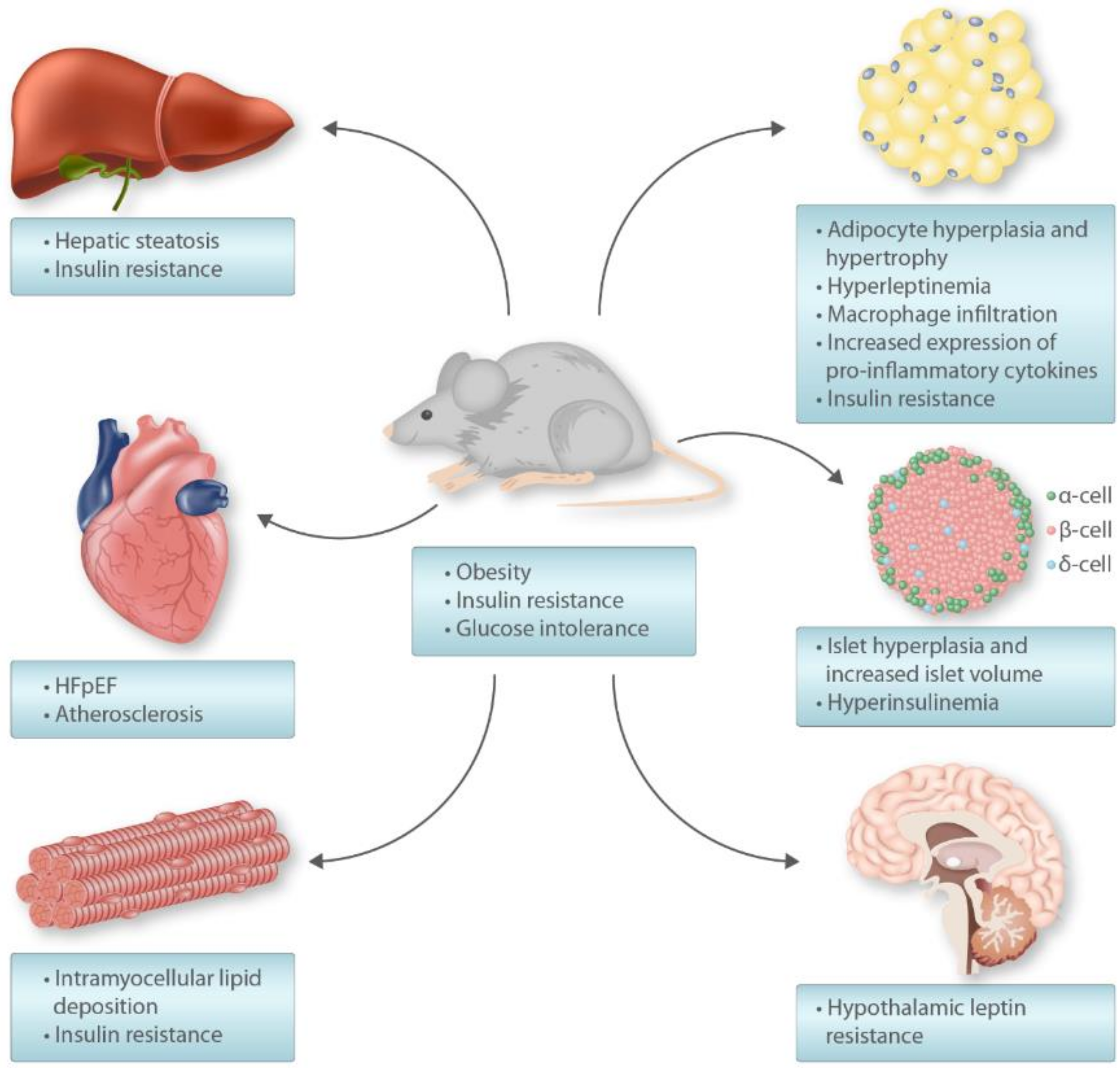
5. High-Fat Diet Rodent Model
6. High-Fat Diet-Induced Insulin Resistance
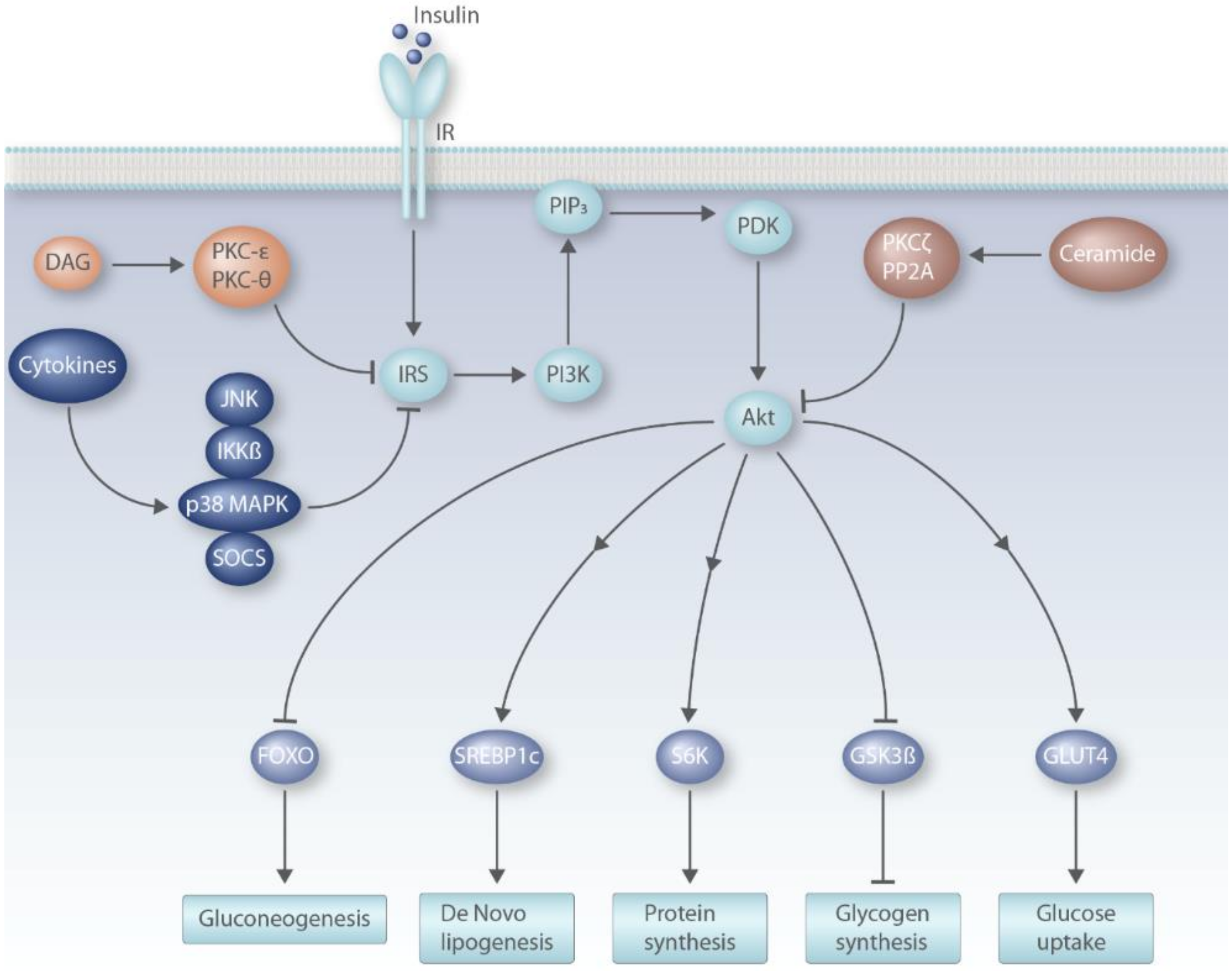
6.1. Insulin Signaling Pathway
6.2. DAG Model of Insulin Resistance
6.3. Ceramide Model of Insulin Resistance
6.4. Pro-Inflammatory Cytokines
6.5. Selective Hepatic Insulin Resistance
7. High-Fat Diet and the Heart
Funding
Conflicts of Interest
References
- Alwan, A. Global Status Report on Noncommunicable Diseases 2010; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Makinen, V.P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segre, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration (NCD-RisC). Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19 2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar]
- Cho, N.H.; Shaw, J.E.; Karuranga, S.; Huang, Y.; da Rocha Fernandes, J.D.; Ohlrogge, A.W.; Malanda, B. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res. Clin. Pract. 2018, 138, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Z. An Evolutionary Perspective on the Obesity Epidemic. Trends Endocrinol. Metab. 2018, 29, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Elia, M.; Cummings, J. Physiological aspects of energy metabolism and gastrointestinal effects of carbohydrates. Eur. J. Clin. Nutr. 2007, 61, S40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckel, R.H.; Jakicic, J.M.; Ard, J.D.; de Jesus, J.M.; Houston Miller, N.; Hubbard, V.S.; Lee, I.M.; Lichtenstein, A.H.; Loria, C.M.; Millen, B.E.; et al. 2013 AHA/ACC guideline on lifestyle management to reduce cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S76–S99. [Google Scholar] [CrossRef] [Green Version]
- EFSA Panel on Dietetic Products, Nutrition, and Allergies (NDA). Scientific Opinion on Dietary Reference Values for fats, including saturated fatty acids, polyunsaturated fatty acids, monounsaturated fatty acids, trans fatty acids, and cholesterol. EFSA J. 2010, 8, 1461. [Google Scholar]
- Nettleton, J.A.; Lovegrove, J.A.; Mensink, R.P.; Schwab, U. Dietary Fatty Acids: Is it Time to Change the Recommendations? Ann. Nutr. Metab. 2016, 68, 249–257. [Google Scholar] [CrossRef]
- Chiu, S.; Williams, P.T.; Krauss, R.M. Effects of a very high saturated fat diet on LDL particles in adults with atherogenic dyslipidemia: A randomized controlled trial. PLoS ONE 2017, 12, e0170664. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Roden, M.; Shulman, G.I. The integrative biology of type 2 diabetes. Nature 2019, 576, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smyth, S.; Heron, A. Diabetes and obesity: The twin epidemics. Nat. Med. 2006, 12, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Popkin, B.M. Dietary fat intake does affect obesity! Am. J. Clin. Nutr. 1998, 68, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Martin, N.; Jimoh, O.F.; Kirk, C.; Foster, E.; Abdelhamid, A.S. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst. Rev. 2020, 5. [Google Scholar] [CrossRef]
- de Souza, R.J.; Mente, A.; Maroleanu, A.; Cozma, A.I.; Ha, V.; Kishibe, T.; Uleryk, E.; Budylowski, P.; Schunemann, H.; Beyene, J.; et al. Intake of saturated and trans unsaturated fatty acids and risk of all cause mortality, cardiovascular disease, and type 2 diabetes: Systematic review and meta-analysis of observational studies. BMJ 2015, 351, h3978. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.A.; Johnson, L.; Franco, O.H.; Butterworth, A.S.; Forouhi, N.G.; Thompson, S.G.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef]
- Siri-Tarino, P.W.; Sun, Q.; Hu, F.B.; Krauss, R.M. Meta-analysis of prospective cohort studies evaluating the association of saturated fat with cardiovascular disease. Am. J. Clin. Nutr. 2010, 91, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Richard, D.; Bausero, P.; Schneider, C.; Visioli, F. Polyunsaturated fatty acids and cardiovascular disease. Cell. Mol. Life Sci. 2009, 66, 3277–3288. [Google Scholar] [CrossRef]
- Hu, F.B. Are refined carbohydrates worse than saturated fat? Am. J. Clin. Nutr. 2010, 91, 1541–1542. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Lopez-Otin, C.; Madeo, F.; de Cabo, R. Carbotoxicity-Noxious Effects of Carbohydrates. Cell 2018, 175, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary fat: From foe to friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, M.; Mente, A.; Zhang, X.; Swaminathan, S.; Li, W.; Mohan, V.; Iqbal, R.; Kumar, R.; Wentzel-Viljoen, E.; Rosengren, A.; et al. Associations of fats and carbohydrate intake with cardiovascular disease and mortality in 18 countries from five continents (PURE): A prospective cohort study. Lancet 2017, 390, 2050–2062. [Google Scholar] [CrossRef] [Green Version]
- Mente, A.; Dehghan, M.; Rangarajan, S.; McQueen, M.; Dagenais, G.; Wielgosz, A.; Lear, S.; Li, W.; Chen, H.; Yi, S. Association of dietary nutrients with blood lipids and blood pressure in 18 countries: A cross-sectional analysis from the PURE study. Lancet Diabetes Endocrinol. 2017, 5, 774–787. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.-P.; Tan, C.-C.; Yu, J.-T. Associations of fats and carbohydrates with cardiovascular disease and mortality—PURE and simple? Lancet 2018, 391, 1679–1680. [Google Scholar] [CrossRef]
- Seidelmann, S.B.; Claggett, B.; Cheng, S.; Henglin, M.; Shah, A.; Steffen, L.M.; Folsom, A.R.; Rimm, E.B.; Willett, W.C.; Solomon, S.D. Dietary carbohydrate intake and mortality: A prospective cohort study and meta-analysis. Lancet Public Health 2018, 3, e419–e428. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S.J.; Raubenheimer, D. The Nature of Nutrition: A Unifying Framework form Animal Adaption to Human Obesity; Princeton University Press: Princeton, NJ, USA, 2012. [Google Scholar]
- Simpson, S.J.; Batley, R.; Raubenheimer, D. Geometric analysis of macronutrient intake in humans: The power of protein? Appetite 2003, 41, 123–140. [Google Scholar] [CrossRef]
- Sorensen, A.; Mayntz, D.; Simpson, S.J.; Raubenheimer, D. Dietary ratio of protein to carbohydrate induces plastic responses in the gastrointestinal tract of mice. J. Comp. Physiol. B 2010, 180, 259–266. [Google Scholar] [CrossRef]
- Solon-Biet, S.; McMahon, A.; Ballard, J.W.O.; Ruohonen, K.; Wu, L.; Cogger, V.; Warren, A.; Huang, X.; Pichaud, N.; Melvin, R.G.; et al. The ratio of macronutrients, not caloric intake, dictates cardiometabolic health, aging and longevity in ad libitum-fed mice. Cell Metab. 2014, 19, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Gosby, A.K.; Conigrave, A.D.; Lau, N.S.; Iglesias, M.A.; Hall, R.M.; Jebb, S.A.; Brand-Miller, J.; Caterson, I.D.; Raubenheimer, D.; Simpson, S.J. Testing protein leverage in lean humans: A randomised controlled experimental study. PLoS ONE 2011, 6, e25929. [Google Scholar] [CrossRef] [Green Version]
- Hewson-Hughes, A.K.; Colyer, A.; Simpson, S.J.; Raubenheimer, D. Balancing macronutrient intake in a mammalian carnivore: Disentangling the influences of flavour and nutrition. R. Soc. Open Sci. 2016, 3, 160081. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.; Simpson, S.J.; Nielsen, V.H.; Hunt, J.; Raubenheimer, D.; Mayntz, D. Nutrient-specific compensatory feeding in a mammalian carnivore, the mink, Neovison vison. Br. J. Nutr. 2014, 112, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Raubenheimer, D.; Simpson, S.J. Protein leverage: Theoretical foundations and ten points of clarification. Obesity 2019, 27, 1225–1238. [Google Scholar] [CrossRef] [PubMed]
- Mayntz, D.; Raubenheimer, D.; Salomon, M.; Toft, S.; Simpson, S.J. Nutrient-specific foraging in invertebrate predators. Science 2005, 307, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Mayntz, D.; Toft, S.; Clissold, F.J.; Hunt, J.; Raubenheimer, D.; Simpson, S.J. Optimal foraging for specific nutrients in predatory beetles. Proc. Biol. Sci. 2012, 279, 2212–2218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, S.J.; Le Couteur, D.G.; Raubenheimer, D. Putting the balance back in diet. Cell 2015, 161, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Martinez Steele, E.; Raubenheimer, D.; Simpson, S.J.; Baraldi, L.G.; Monteiro, C.A. Ultra-processed foods, protein leverage and energy intake in the USA. Public Health Nutr. 2018, 21, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Hall, K.D.; Ayuketah, A.; Brychta, R.; Cai, H.; Cassimatis, T.; Chen, K.Y.; Chung, S.T.; Costa, E.; Courville, A.; Darcey, V.; et al. Ultra-Processed Diets Cause Excess Calorie Intake and Weight Gain: An Inpatient Randomized Controlled Trial of Ad Libitum Food Intake. Cell Metab. 2019, 30, 226. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Scholmerich, J.; Bollheimer, L.C. High-fat diets: Modeling the metabolic disorders of human obesity in rodents. Obesity 2007, 15, 798–808. [Google Scholar] [CrossRef]
- Fontaine, D.A.; Davis, D.B. Attention to Background Strain Is Essential for Metabolic Research: C57BL/6 and the International Knockout Mouse Consortium. Diabetes 2016, 65, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Reuter, T.Y. Diet-induced models for obesity and type 2 diabetes. Drug Discov. Today Dis. Model. 2007, 4, 3–8. [Google Scholar] [CrossRef]
- Small, L.; Brandon, A.E.; Turner, N.; Cooney, G.J. Modeling insulin resistance in rodents by alterations in diet: What have high-fat and high-calorie diets revealed? Am. J. Physiol. Endocrinol. Metab. 2018, 314, E251–E265. [Google Scholar] [CrossRef]
- Nicholson, A.; Reifsnyder, P.C.; Malcolm, R.D.; Lucas, C.A.; MacGregor, G.R.; Zhang, W.; Leiter, E.H. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity 2010, 18, 1902–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariri, N.; Thibault, L. High-fat diet-induced obesity in animal models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersson, U.S.; Walden, T.B.; Carlsson, P.O.; Jansson, L.; Phillipson, M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PLoS ONE 2012, 7, e46057. [Google Scholar] [CrossRef] [PubMed]
- Burchfield, J.G.; Kebede, M.A.; Meoli, C.C.; Stockli, J.; Whitworth, P.T.; Wright, A.L.; Hoffman, N.J.; Minard, A.Y.; Ma, X.; Krycer, J.R.; et al. High dietary fat and sucrose results in an extensive and time-dependent deterioration in health of multiple physiological systems in mice. J. Biol. Chem. 2018, 293, 5731–5745. [Google Scholar] [CrossRef] [Green Version]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L.; Lê, K.-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [Green Version]
- Bortolin, R.C.; Vargas, A.R.; Gasparotto, J.; Chaves, P.R.; Schnorr, C.E.; Martinello, K.B.; Silveira, A.K.; Rabelo, T.K.; Gelain, D.P.; Moreira, J.C.F. A new animal diet based on human Western diet is a robust diet-induced obesity model: Comparison to high-fat and cafeteria diets in term of metabolic and gut microbiota disruption. Int. J. Obes. 2018, 42, 525–534. [Google Scholar] [CrossRef]
- Surwit, R.; Feinglos, M.; Rodin, J.; Sutherland, A.; Petro, A.; Opara, E.; Kuhn, C.; Rebuffe-Scrive, M. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and AJ mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse Models of Nonalcoholic Steatohepatitis: Toward Optimization of Their Relevance to Human Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.A.; McGraw, T.E.; Accili, D. Biochemical and cellular properties of insulin receptor signalling. Nat. Rev. Mol. Cell Biol. 2018, 19, 31–44. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 2014, 510, 84. [Google Scholar] [CrossRef] [PubMed]
- Petersen, M.C.; Madiraju, A.K.; Gassaway, B.M.; Marcel, M.; Nasiri, A.R.; Butrico, G.; Marcucci, M.J.; Zhang, D.; Abulizi, A.; Zhang, X.M.; et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Investig. 2016, 126, 4361–4371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, K.W.; Gilijamse, P.W.; Versteeg, R.I.; Ackermans, M.T.; Nederveen, A.J.; la Fleur, S.E.; Romijn, J.A.; Nieuwdorp, M.; Zhang, D.; Samuel, V.T.; et al. Hepatic Diacylglycerol-Associated Protein Kinase Cepsilon Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep. 2017, 19, 1997–2004. [Google Scholar] [CrossRef]
- Cantley, J.L.; Yoshimura, T.; Camporez, J.P.; Zhang, D.; Jornayvaz, F.R.; Kumashiro, N.; Guebre-Egziabher, F.; Jurczak, M.J.; Kahn, M.; Guigni, B.A.; et al. CGI-58 knockdown sequesters diacylglycerols in lipid droplets/ER-preventing diacylglycerol-mediated hepatic insulin resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 1869–1874. [Google Scholar] [CrossRef] [Green Version]
- Brandon, A.E.; Liao, B.M.; Diakanastasis, B.; Parker, B.L.; Raddatz, K.; McManus, S.A.; O’Reilly, L.; Kimber, E.; van der Kraan, A.G.; Hancock, D.; et al. Protein Kinase C Epsilon Deletion in Adipose Tissue, but Not in Liver, Improves Glucose Tolerance. Cell Metab. 2019, 29, 183–191. [Google Scholar] [CrossRef] [Green Version]
- Raddatz, K.; Turner, N.; Frangioudakis, G.; Liao, B.M.; Pedersen, D.J.; Cantley, J.; Wilks, D.; Preston, E.; Hegarty, B.D.; Leitges, M.; et al. Time-dependent effects of Prkce deletion on glucose homeostasis and hepatic lipid metabolism on dietary lipid oversupply in mice. Diabetologia 2011, 54, 1447–1456. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Petersen, M.C.; Gassaway, B.M.; Vatner, D.F.; Rinehart, J.; Shulman, G.I. Considering the Links Between Nonalcoholic Fatty Liver Disease and Insulin Resistance: Revisiting the Role of Protein Kinase C ε. Hepatology 2019, 70, 2217–2220. [Google Scholar] [CrossRef]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Brozinick, J.T.; Wang, L.P.; Hawkins, E.D.; Sargent, K.M.; Liu, Y.; Narra, K.; Hoehn, K.L.; Knotts, T.A.; Siesky, A.; et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 2007, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Chavez, J.A.; Summers, S.A. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch. Biochem. Biophys. 2003, 419, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Bronneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaurasia, B.; Tippetts, T.S.; Mayoral Monibas, R.; Liu, J.; Li, Y.; Wang, L.; Wilkerson, J.L.; Sweeney, C.R.; Pereira, R.F.; Sumida, D.H.; et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 2019, 365, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Stratford, S.; Hoehn, K.L.; Liu, F.; Summers, S.A. Regulation of insulin action by ceramide: Dual mechanisms linking ceramide accumulation to the inhibition of Akt/protein kinase B. J. Biol. Chem. 2004, 279, 36608–36615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Ceramides. N. Engl. J. Med. 2019, 381, 1866–1869. [Google Scholar] [CrossRef]
- Bonezzi, F.; Piccoli, M.; Dei Cas, M.; Paroni, R.; Mingione, A.; Monasky, M.M.; Caretti, A.; Riganti, C.; Ghidoni, R.; Pappone, C.; et al. Sphingolipid Synthesis Inhibition by Myriocin Administration Enhances Lipid Consumption and Ameliorates Lipid Response to Myocardial Ischemia Reperfusion Injury. Front. Physiol. 2019, 10, 986. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-Derived Sphingolipids Interact with Mff and Promote Mitochondrial Fragmentation in Obesity. Cell 2019, 177, 1536–1552. [Google Scholar] [CrossRef]
- Osborn, O.; Olefsky, J.M. The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 2012, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Mathis, D. Immunological goings-on in visceral adipose tissue. Cell Metab. 2013, 17, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, S.; Manabe, I.; Nagasaki, M.; Eto, K.; Yamashita, H.; Ohsugi, M.; Otsu, M.; Hara, K.; Ueki, K.; Sugiura, S.; et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat. Med. 2009, 15, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- O’Rourke, R.W.; Metcalf, M.D.; White, A.E.; Madala, A.; Winters, B.R.; Maizlin, I.I.; Jobe, B.A.; Roberts, C.T., Jr.; Slifka, M.K.; Marks, D.L. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int. J. Obes. 2009, 33, 978–990. [Google Scholar] [CrossRef] [Green Version]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Lanthier, N.; Molendi-Coste, O.; Horsmans, Y.; van Rooijen, N.; Cani, P.D.; Leclercq, I.A. Kupffer cell activation is a causal factor for hepatic insulin resistance. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G107–G116. [Google Scholar] [CrossRef] [Green Version]
- Arkan, M.C.; Hevener, A.L.; Greten, F.R.; Maeda, S.; Li, Z.W.; Long, J.M.; Wynshaw-Boris, A.; Poli, G.; Olefsky, J.; Karin, M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat. Med. 2005, 11, 191–198. [Google Scholar] [CrossRef]
- de Alvaro, C.; Teruel, T.; Hernandez, R.; Lorenzo, M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J. Biol. Chem. 2004, 279, 17070–17078. [Google Scholar] [CrossRef] [Green Version]
- Lebrun, P.; Van Obberghen, E. SOCS proteins causing trouble in insulin action. Acta Physiol. 2008, 192, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.M.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cepsilon prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Asseldonk, E.J.; Stienstra, R.; Koenen, T.B.; Joosten, L.A.; Netea, M.G.; Tack, C.J. Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: A randomized, double-blind, placebo-controlled study. J. Clin. Endocrinol. Metab. 2011, 96, 2119–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Everett, B.M.; Donath, M.Y.; Pradhan, A.D.; Thuren, T.; Pais, P.; Nicolau, J.C.; Glynn, R.J.; Libby, P.; Ridker, P.M. Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. J. Am. Coll. Cardiol. 2018, 71, 2392–2401. [Google Scholar] [CrossRef]
- Ruscitti, P.; Masedu, F.; Alvaro, S.; Airò, P.; Battafarano, N.; Cantarini, L.; Cantatore, F.P.; Carlino, G.; D’Abrosca, V.; Frassi, M.; et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PLoS Med. 2019, 16, e1002901. [Google Scholar] [CrossRef]
- Fleischman, A.; Shoelson, S.E.; Bernier, R.; Goldfine, A.B. Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes Care 2008, 31, 289–294. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, I.; Matsuda, M.; Hammer, R.E.; Bashmakov, Y.; Brown, M.S.; Goldstein, J.L. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol. Cell 2000, 6, 77–86. [Google Scholar] [CrossRef]
- Michael, M.D.; Kulkarni, R.N.; Postic, C.; Previs, S.F.; Shulman, G.I.; Magnuson, M.A.; Kahn, C.R. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell 2000, 6, 87–97. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Wahed, A.; Guilmeau, S.; Postic, C. Sweet Sixteenth for ChREBP: Established Roles and Future Goals. Cell Metab. 2017, 26, 324–341. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H. Sickeningly Sweet: Does Sugar Cause Type 2 Diabetes? Yes. Can. J. Diabetes 2016, 40, 282–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AC, I. Influence of animal food on the organism of rabbits. Izvest Imper Voennomed Akad St Petersburg 1908, 16, 154–173. [Google Scholar]
- Steinberg, D. Thematic review series: The pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: Part I. J. Lipid Res. 2004, 45, 1583–1593. [Google Scholar] [CrossRef] [Green Version]
- Ahrens, E.H., Jr.; Hirsch, J.; Insull, W., Jr.; Tsaltas, T.T.; Blomstrand, R.; Peterson, M.L. Dietary control of serum lipids in relation to atherosclerosis. J. Am. Med. Assoc. 1957, 164, 1905–1911. [Google Scholar] [CrossRef]
- Keys, A. Diet and the epidemiology of coronary heart disease. J. Am. Med. Assoc. 1957, 164, 1912–1919. [Google Scholar] [CrossRef]
- Keys, A. Seven Countries: A Multivariate Analysis of Death and Coronary Heart Diseas; Harvard University Press: Cambridge, MA, USA, 1980. [Google Scholar]
- The National Diet-Heart Study Research Group. The National Diet-Heart Study Final Report. Circulation 1968, 37, I1–I428. [Google Scholar]
- Ornish, D.; Brown, S.E.; Scherwitz, L.W.; Billings, J.H.; Armstrong, W.T.; Ports, T.A.; McLanahan, S.M.; Kirkeeide, R.L.; Brand, R.J.; Gould, K.L. Can lifestyle changes reverse coronary heart disease? The Lifestyle Heart Trial. Lancet 1990, 336, 129–133. [Google Scholar] [CrossRef]
- Ornish, D.; Scherwitz, L.W.; Billings, J.H.; Brown, S.E.; Gould, K.L.; Merritt, T.A.; Sparler, S.; Armstrong, W.T.; Ports, T.A.; Kirkeeide, R.L.; et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA 1998, 280, 2001–2007. [Google Scholar] [CrossRef]
- Multiple Risk Factor Intervention Trial Research Group. Multiple risk factor intervention trial. Risk factor changes and mortality results. Multiple Risk Factor Intervention Trial Research Group. JAMA 1982, 248, 1465–1477. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvado, J.; Covas, M.I.; Corella, D.; Aros, F.; Gomez-Gracia, E.; Ruiz-Gutierrez, V.; Fiol, M.; Lapetra, J.; et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N. Engl. J. Med. 2013, 368, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dehghan, M.; Mente, A.; Teo, K.K.; Gao, P.; Sleight, P.; Dagenais, G.; Avezum, A.; Probstfield, J.L.; Dans, T.; Yusuf, S.; et al. Relationship between healthy diet and risk of cardiovascular disease among patients on drug therapies for secondary prevention: A prospective cohort study of 31 546 high-risk individuals from 40 countries. Circulation 2012, 126, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Costacou, T.; Bamia, C.; Trichopoulos, D. Adherence to a Mediterranean diet and survival in a Greek population. N. Engl. J. Med. 2003, 348, 2599–2608. [Google Scholar] [CrossRef] [Green Version]
- Mente, A.; de Koning, L.; Shannon, H.S.; Anand, S.S. A systematic review of the evidence supporting a causal link between dietary factors and coronary heart disease. Arch. Intern. Med. 2009, 169, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Rimm, E.B. Fruit and vegetables-building a solid foundation. Am. J. Clin. Nutr. 2002, 76, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, R.; Anand, S.; Ounpuu, S.; Islam, S.; Zhang, X.; Rangarajan, S.; Chifamba, J.; Al-Hinai, A.; Keltai, M.; Yusuf, S.; et al. Dietary patterns and the risk of acute myocardial infarction in 52 countries: Results of the INTERHEART study. Circulation 2008, 118, 1929–1937. [Google Scholar] [CrossRef] [Green Version]
- Bazzano, L.A.; He, J.; Ogden, L.G.; Loria, C.M.; Vupputuri, S.; Myers, L.; Whelton, P.K. Fruit and vegetable intake and risk of cardiovascular disease in US adults: The first National Health and Nutrition Examination Survey Epidemiologic Follow-up Study. Am. J. Clin. Nutr. 2002, 76, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Joshipura, K.J.; Hu, F.B.; Manson, J.E.; Stampfer, M.J.; Rimm, E.B.; Speizer, F.E.; Colditz, G.; Ascherio, A.; Rosner, B.; Spiegelman, D.; et al. The effect of fruit and vegetable intake on risk for coronary heart disease. Ann. Intern. Med. 2001, 134, 1106–1114. [Google Scholar] [CrossRef]
- Hu, F.B.; Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Spiegelman, D.; Willett, W.C. Prospective study of major dietary patterns and risk of coronary heart disease in men. Am. J. Clin. Nutr. 2000, 72, 912–921. [Google Scholar] [CrossRef]
- Li, S.; Chiuve, S.E.; Flint, A.; Pai, J.K.; Forman, J.P.; Hu, F.B.; Willett, W.C.; Mukamal, K.J.; Rimm, E.B. Better diet quality and decreased mortality among myocardial infarction survivors. JAMA Intern. Med. 2013, 173, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Fehily, A.M.; Gilbert, J.F.; Rogers, S.; Holliday, R.M.; Sweetnam, P.M.; Elwood, P.C.; Deadman, N.M. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: Diet and reinfarction trial (DART). Lancet 1989, 2, 757–761. [Google Scholar] [CrossRef]
- de Lorgeril, M.; Renaud, S.; Mamelle, N.; Salen, P.; Martin, J.L.; Monjaud, I.; Guidollet, J.; Touboul, P.; Delaye, J. Mediterranean alpha-linolenic acid-rich diet in secondary prevention of coronary heart disease. Lancet 1994, 343, 1454–1459. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Di Raimondo, D.; Casuccio, A.; Velardo, M.; Salamone, G.; Cataldi, M.; Corpora, F.; Restivo, V.; Pecoraro, R.; Della Corte, V.; et al. Mediterranean diet adherence and congestive heart failure: Relationship with clinical severity and ischemic pathogenesis. Nutrition 2020, 70, 110584. [Google Scholar] [CrossRef]
- Metra, M.; Dinatolo, E.; Dasseni, N. The New Heart Failure Association Definition of Advanced Heart Failure. Card. Fail. Rev. 2019, 5, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Tsutsui, T.; Tsutamoto, T.; Wada, A.; Maeda, K.; Mabuchi, N.; Hayashi, M.; Ohnishi, M.; Kinoshita, M. Plasma oxidized low-density lipoprotein as a prognostic predictor in patients with chronic congestive heart failure. J. Am. Coll. Cardiol. 2002, 39, 957–962. [Google Scholar] [CrossRef] [Green Version]
- Fito, M.; Estruch, R.; Salas-Salvado, J.; Martinez-Gonzalez, M.A.; Aros, F.; Vila, J.; Corella, D.; Diaz, O.; Saez, G.; de la Torre, R.; et al. Effect of the Mediterranean diet on heart failure biomarkers: A randomized sample from the PREDIMED trial. Eur. J. Heart Fail. 2014, 16, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Tektonidis, T.G.; Akesson, A.; Gigante, B.; Wolk, A.; Larsson, S.C. A Mediterranean diet and risk of myocardial infarction, heart failure and stroke: A population-based cohort study. Atherosclerosis 2015, 243, 93–98. [Google Scholar] [CrossRef]
- Tektonidis, T.G.; Akesson, A.; Gigante, B.; Wolk, A.; Larsson, S.C. Adherence to a Mediterranean diet is associated with reduced risk of heart failure in men. Eur. J. Heart Fail. 2016, 18, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Sartori, M.; Conti, F.F.; Dias, D.D.S.; Dos Santos, F.; Machi, J.F.; Palomino, Z.; Casarini, D.E.; Rodrigues, B.; De Angelis, K.; Irigoyen, M.C. Association between Diastolic Dysfunction with Inflammation and Oxidative Stress in Females ob/ob Mice. Front. Physiol. 2017, 8, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Linthout, S.; Tschope, C. Inflammation-Cause or Consequence of Heart Failure or Both? Curr. Heart Fail. Rep. 2017, 14, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitan, E.B.; Lewis, C.E.; Tinker, L.F.; Eaton, C.B.; Ahmed, A.; Manson, J.E.; Snetselaar, L.G.; Martin, L.W.; Trevisan, M.; Howard, B.V.; et al. Mediterranean and DASH diet scores and mortality in women with heart failure: The Women’s Health Initiative. Circ. Heart Fail. 2013, 6, 1116–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabelli, M.; Chiefari, E.; Arcidiacono, B.; Corigliano, D.M.; Brunetti, F.S.; Maggisano, V.; Russo, D.; Foti, D.P.; Brunetti, A. Mediterranean Diet Nutrients to Turn the Tide against Insulin Resistance and Related Diseases. Nutrients 2020, 12, 1066. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wu, C.; Qiu, S.; Yuan, X.; Li, L. Effects of resveratrol on glucose control and insulin sensitivity in subjects with type 2 diabetes: Systematic review and meta-analysis. Nutr. Metab. 2017, 14, 60. [Google Scholar] [CrossRef]
- Wahl, D.; Bernier, M.; Simpson, S.J.; de Cabo, R.; Le Couteur, D.G. Future directions of resveratrol research. Nutr. Healthy Aging 2018, 4, 287–290. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Luo, J.; Huang, J.; Wen, Q. Flavonoids intake and risk of type 2 diabetes mellitus: A meta-analysis of prospective cohort studies. Medicine 2018, 97, e0686. [Google Scholar] [CrossRef]
- Summerhill, V.; Karagodin, V.; Grechko, A.; Myasoedova, V.; Orekhov, A. Vasculoprotective Role of Olive Oil Compounds via Modulation of Oxidative Stress in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 188. [Google Scholar] [CrossRef] [Green Version]
- Gogoi, B.; Chatterjee, P.; Mukherjee, S.; Buragohain, A.K.; Bhattacharya, S.; Dasgupta, S. A polyphenol rescues lipid induced insulin resistance in skeletal muscle cells and adipocytes. Biochem. Biophys. Res. Commun. 2014, 452, 382–388. [Google Scholar] [CrossRef]
- Lombardo, G.E.; Lepore, S.M.; Morittu, V.M.; Arcidiacono, B.; Colica, C.; Procopio, A.; Maggisano, V.; Bulotta, S.; Costa, N.; Mignogna, C.; et al. Effects of Oleacein on High-Fat Diet-Dependent Steatosis, Weight Gain, and Insulin Resistance in Mice. Front. Endocrinol. 2018, 9, 116. [Google Scholar] [CrossRef] [Green Version]
- Lepore, S.M.; Maggisano, V.; Bulotta, S.; Mignogna, C.; Arcidiacono, B.; Procopio, A.; Brunetti, A.; Russo, D.; Celano, M. Oleacein Prevents High Fat Diet-Induced Adiposity and Ameliorates Some Biochemical Parameters of Insulin Sensitivity in Mice. Nutrients 2019, 11, 1829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuttolomondo, A.; Simonetta, I.; Daidone, M.; Mogavero, A.; Ortello, A.; Pinto, A. Metabolic and Vascular Effect of the Mediterranean Diet. Int. J. Mol. Sci. 2019, 20, 4716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wali, J.A.; Jarzebska, N.; Raubenheimer, D.; Simpson, S.J.; Rodionov, R.N.; O’Sullivan, J.F. Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review. Nutrients 2020, 12, 1505. https://doi.org/10.3390/nu12051505
Wali JA, Jarzebska N, Raubenheimer D, Simpson SJ, Rodionov RN, O’Sullivan JF. Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review. Nutrients. 2020; 12(5):1505. https://doi.org/10.3390/nu12051505
Chicago/Turabian StyleWali, Jibran A., Natalia Jarzebska, David Raubenheimer, Stephen J. Simpson, Roman N. Rodionov, and John F. O’Sullivan. 2020. "Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review" Nutrients 12, no. 5: 1505. https://doi.org/10.3390/nu12051505
APA StyleWali, J. A., Jarzebska, N., Raubenheimer, D., Simpson, S. J., Rodionov, R. N., & O’Sullivan, J. F. (2020). Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review. Nutrients, 12(5), 1505. https://doi.org/10.3390/nu12051505