The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Treatment
2.2. Cardiac Function Assessment
2.3. Lipid Analysis
2.3.1. Total Lipid Extraction
2.3.2. Solid Phase Extraction
2.3.3. Fatty Acid Hydrolysis and Derivatization
2.3.4. GC–MS Analysis
2.4. Lipid Peroxidation
2.5. Free Cholesterol Measurement
2.6. Protein Carbonyl Groups Determination
2.7. Statistical Analysis
3. Results
3.1. Dietary-Induced Obesity
3.2. Cardiac Function Assessment
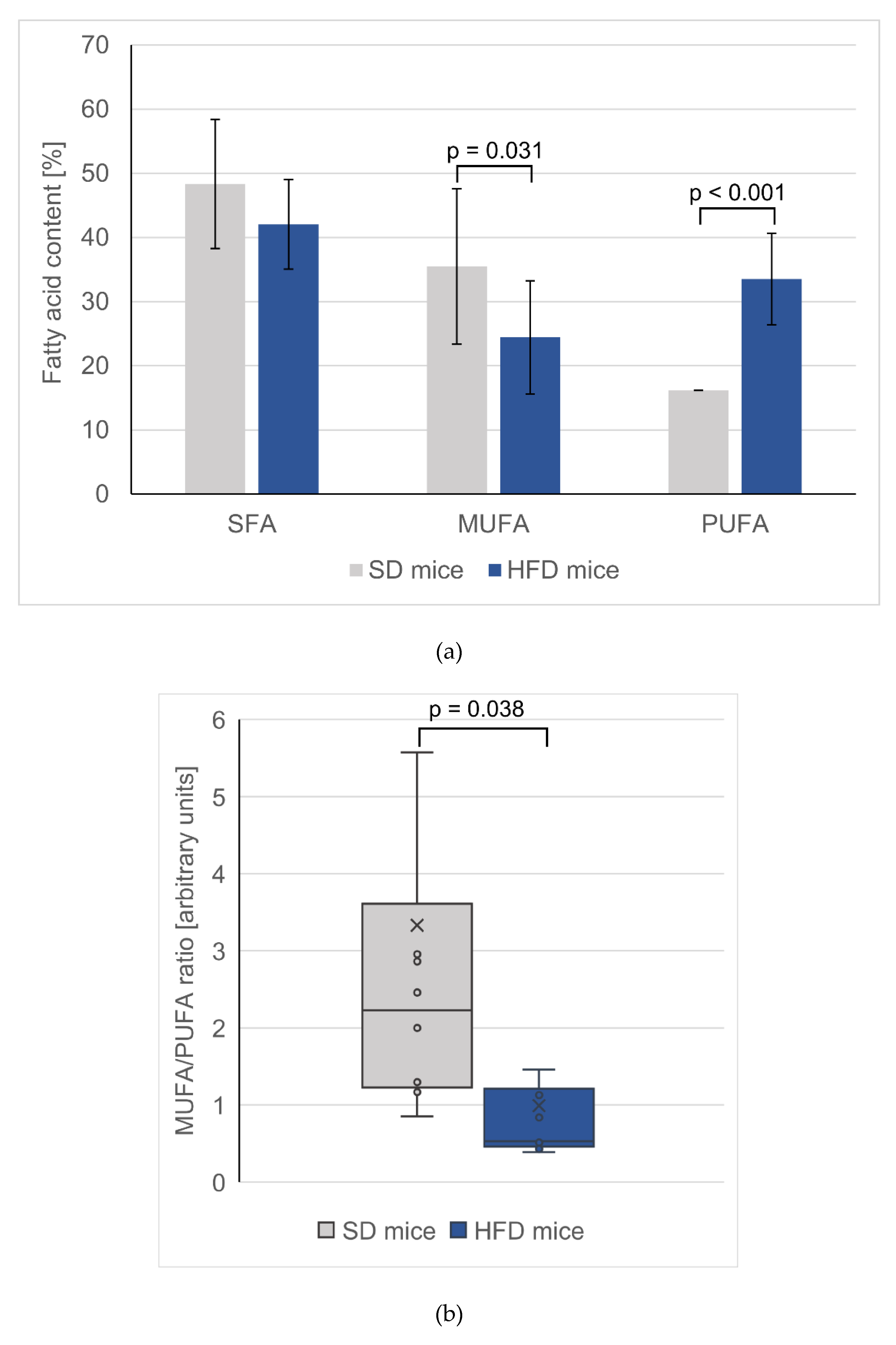
3.3. Changes in Total FA Profiles
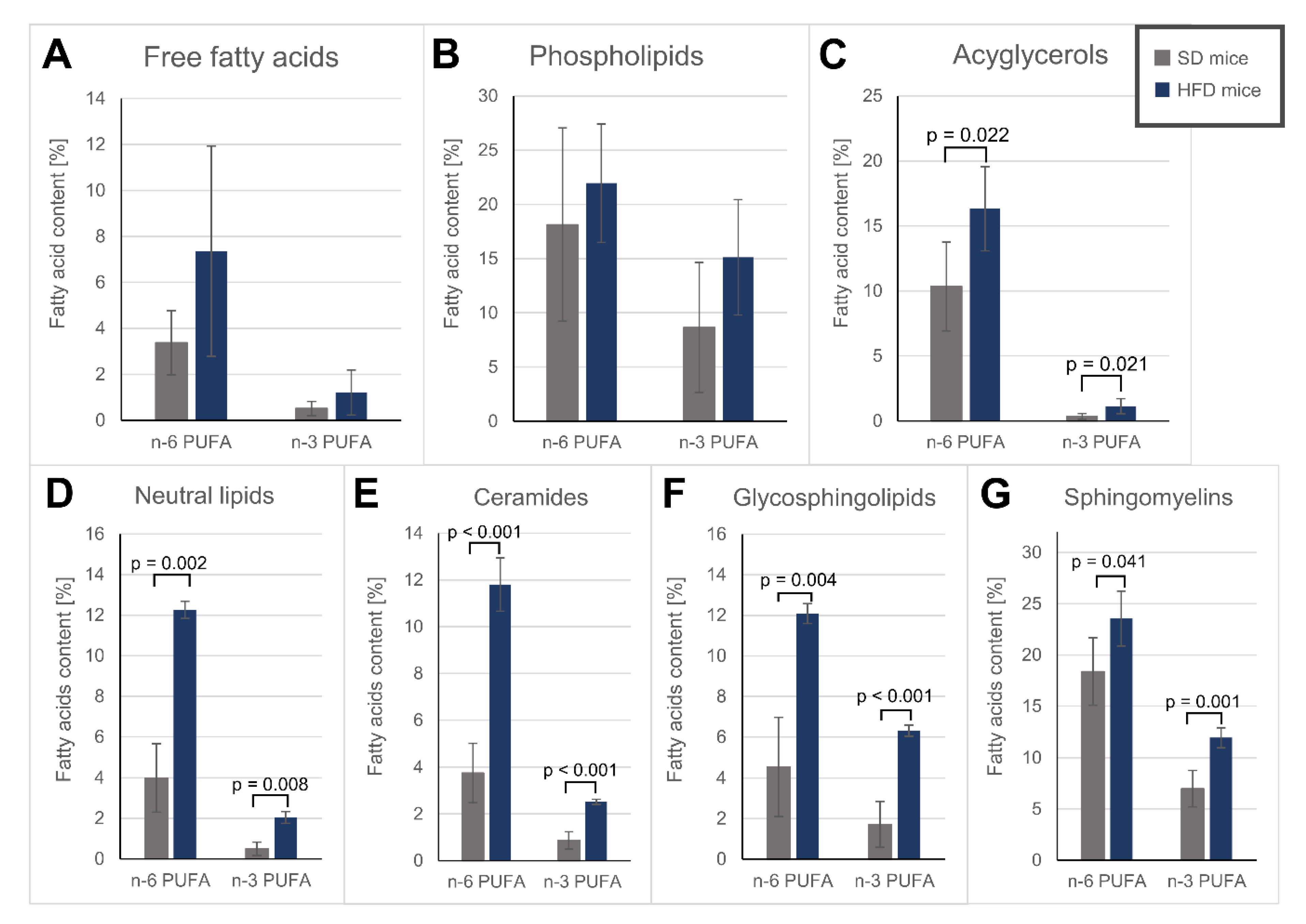
3.4. Fatty Acid Alterations in Specific Lipid Fractions of the Hearts
3.5. Fatty Acid Oxidation and Protein Carbonylation Content
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Psaltopoulou, T.; Hatzis, G.; Papageorgiou, N.; Androulakis, E.; Briasoulis, A.; Tousoulis, D. Socioeconomic status and risk factors for cardiovascular disease: Impact of dietary mediators. Hell. J. Cardiol. 2017, 58, 32–42. [Google Scholar] [CrossRef]
- Fung, T.T.; Stampfer, M.J.; Manson, J.E.; Rexrode, K.M.; Willett, W.C.; Hu, F.B. Prospective Study of Major Dietary Patterns and Stroke Risk in Women. Stroke 2004, 35, 2014–2019. [Google Scholar] [CrossRef]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Zinöcker, M.; Lindseth, I. The Western Diet–Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Amato, K.R.; Yeoman, C.J.; Cerda, G.; Schmitt, C.A.; Cramer, J.D.; Miller, M.E.B.; Gomez, A.; Turner, T.R.; Wilson, B.A.; Stumpf, R.M.; et al. Variable responses of human and non-human primate gut microbiomes to a Western diet. Microbiome 2015, 3, 53. [Google Scholar] [CrossRef] [PubMed]
- Martinez, K.B.; Leone, V.; Chang, E.B. Western diets, gut dysbiosis, and metabolic diseases: Are they linked? Gut Microbes 2017, 8, 130–142. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thévenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef]
- Hyoju, S.K.; Zaborin, A.; Keskey, R.; Sharma, A.; Arnold, W.; van den Berg, F.; Kim, S.M.; Gottel, N.; Bethel, C.; Charnot-Katsikas, A.; et al. Mice Fed an Obesogenic Western Diet, Administered Antibiotics, and Subjected to a Sterile Surgical Procedure Develop Lethal Septicemia with Multidrug-Resistant Pathobionts. mBio 2019, 10, 263–271. [Google Scholar] [CrossRef]
- Harris, K.L.; Pulliam, S.R.; Okoro, E.; Guo, Z.; Washington, M.K.; Adunyah, S.E.; Amos-Landgraf, J.M.; Ramesh, A. Western diet enhances benzo(a)pyrene-induced colon tumorigenesis in a polyposis in rat coli (PIRC) rat model of colon cancer. Oncotarget 2016, 7, 28947–28960. [Google Scholar] [CrossRef]
- Mustafi, R.; Dougherty, U.; Mustafi, D.; Ayaloglu-Butun, F.; Fletcher, M.; Adhikari, S.; Sadiq, F.; Meckel, K.; Haider, H.I.; Khalil, A.; et al. ADAM17 is a Tumor Promoter and Therapeutic Target in Western Diet–associated Colon Cancer. Clin. Cancer Res. 2017, 23, 549–561. [Google Scholar] [CrossRef]
- Laing, B.B.; Lim, A.G.; Ferguson, L.R. A Personalised Dietary Approach—A Way Forward to Manage Nutrient Deficiency, Effects of the Western Diet, and Food Intolerances in Inflammatory Bowel Disease. Nutrients 2019, 11, 1532. [Google Scholar] [CrossRef] [PubMed]
- Ranjit, S.; Dvornikov, A.; Dobrinskikh, E.; Wang, X.; Luo, Y.; Levi, M.; Gratton, E. Measuring the effect of a Western diet on liver tissue architecture by FLIM autofluorescence and harmonic generation microscopy. Biomed. Opt. Express 2017, 8, 3143–3154. [Google Scholar] [CrossRef]
- Gabbia, D.; Roverso, M.; Guido, M.; Sacchi, D.; Scaffidi, M.; Carrara, M.; Orso, G.; Russo, F.P.; Floreani, A.; Bogialli, S.; et al. Western Diet-Induced Metabolic Alterations Affect Circulating Markers of Liver Function before the Development of Steatosis. Nutrients 2019, 11, 1602. [Google Scholar] [CrossRef] [PubMed]
- Lytle, K.A.; Depner, C.M.; Wong, C.P.; Jump, D.B. Docosahexaenoic acid attenuates Western diet-induced hepatic fibrosis in Ldlr −/− mice by targeting the TGFβ-Smad3 pathway. J. Lipid Res. 2015, 56, 1936–1946. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Di Lucente, J.; Jin, L.-W.; Maezawa, I.; Wan, Y.-J.Y. Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet–induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB J. 2018, 32, 2866–2877. [Google Scholar] [CrossRef]
- Rutkowsky, J.M.; Lee, L.L.; Puchowicz, M.; Golub, M.S.; Befroy, D.E.; Wilson, D.W.; Anderson, S.; Cline, G.; Bini, J.; Borkowski, K.; et al. Reduced cognitive function, increased blood-brain-barrier transport and inflammatory responses, and altered brain metabolites in LDLr -/-and C57BL/6 mice fed a western diet. PLoS ONE 2018, 13, e0191909. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.S.; Dunaway, K.; Rutkowsky, J.; Rutledge, J.C.; Milenkovic, D. Chronic consumption of a western diet modifies the DNA methylation profile in the frontal cortex of mice. Food Funct. 2018, 9, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Kanoski, S.E.; Davidson, T.L. Western diet consumption and cognitive impairment: Links to hippocampal dysfunction and obesity. Physiol. Behav. 2011, 103, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Maesako, M.; Uemura, K.; Iwata, A.; Kubota, M.; Watanabe, K.; Uemura, M.; Noda, Y.; Asada-Utsugi, M.; Kihara, T.; Takahashi, R.; et al. Continuation of Exercise Is Necessary to Inhibit High Fat Diet-Induced β-Amyloid Deposition and Memory Deficit in Amyloid Precursor Protein Transgenic Mice. PLoS ONE 2013, 8, e72796. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Tangney, C.C. Dietary fat composition and dementia risk. Neurobiol. Aging 2014, 35, S59–S64. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.C.; Harder, J.M.; Soto, I.; de Vries, W.N.; John, S.W.M.; Howell, G.R. Chronic consumption of a western diet induces robust glial activation in aging mice and in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 21568. [Google Scholar] [CrossRef] [PubMed]
- Veniaminova, E.; Cespuglio, R.; Cheung, C.W.; Umriukhin, A.; Markova, N.; Shevtsova, E.; Lesch, K.-P.; Anthony, D.C.; Strekalova, T. Autism-Like Behaviours and Memory Deficits Result from a Western Diet in Mice. Neural Plast. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; de Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M. Mouse Models of Diet-Induced Nonalcoholic Steatohepatitis Reproduce the Heterogeneity of the Human Disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef] [PubMed]
- Napier, B.A.; Andres-Terre, M.; Massis, L.M.; Hryckowian, A.J.; Higginbottom, S.K.; Cumnock, K.; Casey, K.M.; Haileselassie, B.; Lugo, K.A.; Schneider, D.S.; et al. Western diet regulates immune status and the response to LPS-driven sepsis independent of diet-associated microbiome. Proc. Natl. Acad. Sci. USA 2019, 116, 3688–3694. [Google Scholar] [CrossRef]
- Manzel, A.; Muller, D.N.; Hafler, D.A.; Erdman, S.E.; Linker, R.A.; Kleinewietfeld, M. Role of “Western Diet” in Inflammatory Autoimmune Diseases. Curr. Allergy Asthma Rep. 2014, 14, 404. [Google Scholar] [CrossRef]
- Totsch, S.K.; Waite, M.E.; Tomkovich, A.; Quinn, T.L.; Gower, B.A.; Sorge, R.E. Total Western Diet Alters Mechanical and Thermal Sensitivity and Prolongs Hypersensitivity Following Complete Freund’s Adjuvant in Mice. J. Pain 2016, 17, 119–125. [Google Scholar] [CrossRef]
- Shively, C.A.; Register, T.C.; Appt, S.E.; Clarkson, T.B.; Uberseder, B.; Clear, K.Y.J.; Wilson, A.S.; Chiba, A.; Tooze, J.A.; Cook, K.L. Consumption of Mediterranean versus Western Diet Leads to Distinct Mammary Gland Microbiome Populations. Cell Rep. 2018, 25, 47–56.e3. [Google Scholar] [CrossRef]
- Bedja, D.; Yan, W.; Lad, V.; Iocco, D.; Sivakumar, N.; Bandaru, V.V.R.; Chatterjee, S. Inhibition of glycosphingolipid synthesis reverses skin inflammation and hair loss in ApoE−/− mice fed western diet. Sci. Rep. 2018, 8, 11463. [Google Scholar] [CrossRef]
- Lytle, K.A.; Jump, D.B. Is Western Diet-Induced Nonalcoholic Steatohepatitis in Ldlr-/- Mice Reversible? PLoS ONE 2016, 11, e0146942. [Google Scholar] [CrossRef]
- Myles, I.A. Fast food fever: Reviewing the impacts of the Western diet on immunity. Nutr. J. 2014, 13, 61. [Google Scholar] [CrossRef]
- Nazni, P. Association of western diet & lifestyle with decreased fertility. Indian J. Med. Res. 2014, 140, 78–81. [Google Scholar]
- Sinclair, K.J.; Friesen–Waldner, L.J.; McCurdy, C.M.; Wiens, C.N.; Wade, T.P.; de Vrijer, B.; Regnault, T.R.H.; McKenzie, C.A. Quantification of fetal organ volume and fat deposition following in utero exposure to maternal Western Diet using MRI. PLoS ONE 2018, 13, e0192900. [Google Scholar] [CrossRef] [PubMed]
- Perez, P.A.; DiPatrizio, N.V. Impact of maternal western diet-induced obesity on offspring mortality and peripheral endocannabinoid system in mice. PLoS ONE 2018, 13, e0205021. [Google Scholar] [CrossRef] [PubMed]
- Frihauf, J.B.; Fekete, É.M.; Nagy, T.R.; Levin, B.E.; Zorrilla, E.P. Maternal Western diet increases adiposity even in male offspring of obesity-resistant rat dams: Early endocrine risk markers. Am. J. Physiol. Integr. Comp. Physiol. 2016, 311, R1045–R1059. [Google Scholar] [CrossRef][Green Version]
- Varlamov, O. Western-style diet, sex steroids and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1147–1155. [Google Scholar] [CrossRef]
- Hariharan, D.; Vellanki, K.; Kramer, H. The Western Diet and Chronic Kidney Disease. Curr. Hypertens. Rep. 2015, 17, 16. [Google Scholar] [CrossRef]
- Odermatt, A. The Western-style diet: A major risk factor for impaired kidney function and chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2011, 301, F919–F931. [Google Scholar] [CrossRef]
- Houston, M.; Minich, D.; Sinatra, S.T.; Kahn, J.K.; Guarneri, M. Recent Science and Clinical Application of Nutrition to Coronary Heart Disease. J. Am. Coll. Nutr. 2018, 37, 169–187. [Google Scholar] [CrossRef]
- Heidemann, C.; Schulze, M.B.; Franco, O.H.; van Dam, R.M.; Mantzoros, C.S.; Hu, F.B. Dietary Patterns and Risk of Mortality From Cardiovascular Disease, Cancer, and All Causes in a Prospective Cohort of Women. Circulation 2008, 118, 230–237. [Google Scholar] [CrossRef]
- Hu, F.B.; Rimm, E.B.; Stampfer, M.J.; Ascherio, A.; Spiegelman, D.; Willett, W.C. Prospective study of major dietary patterns and risk of coronary heart disease in men. Am. J. Clin. Nutr. 2000, 72, 912–921. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics—2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e66. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Thomas, M.C.; Harcourt, B.E.; Coughlan, M.T.; Pete, J.; Bialkowski, K.; Tan, A.; Bierhaus, A.; Cooper, M.E.; Forbes, J.M. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am. J. Physiol. Metab. 2008, 295, E323–E330. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Habibi, J.; Bostick, B.P.; Ma, L.; DeMarco, V.G.; Aroor, A.R.; Hayden, M.R.; Whaley-Connell, A.T.; Sowers, J.R. Uric Acid Promotes Left Ventricular Diastolic Dysfunction in Mice Fed a Western Diet. Hypertension 2015, 65, 531–539. [Google Scholar] [CrossRef]
- Stanley, W.C.; Dabkowski, E.R.; Ribeiro, R.F.; O’Connell, K.A. Dietary fat and heart failure: Moving from lipotoxicity to lipoprotection. Circ. Res. 2012, 110, 764–776. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and Atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Garcia, E.; Schulze, M.B.; Fung, T.T.; Meigs, J.B.; Rifai, N.; Manson, J.E.; Hu, F.B. Major dietary patterns are related to plasma concentrations of markers of inflammation and endothelial dysfunction. Am. J. Clin. Nutr. 2004, 80, 1029–1035. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Marín-Aguilar, F.; Bullon, P.; Cordero, M.D. Cardiovascular diseases, NLRP3 inflammasome, and western dietary patterns. Pharmacol. Res. 2018, 131, 44–50. [Google Scholar] [CrossRef]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef]
- Calligaris, S.D.; Lecanda, M.; Solis, F.; Ezquer, M.; Gutiérrez, J.; Brandan, E.; Leiva, A.; Sobrevia, L.; Conget, P. Mice Long-Term High-Fat Diet Feeding Recapitulates Human Cardiovascular Alterations: An Animal Model to Study the Early Phases of Diabetic Cardiomyopathy. PLoS ONE 2013, 8, e60931. [Google Scholar] [CrossRef]
- Harmancey, R.; Wilson, C.R.; Wright, N.R.; Taegtmeyer, H. Western diet changes cardiac acyl-CoA composition in obese rats: A potential role for hepatic lipogenesis. J. Lipid Res. 2010, 51, 1380–1393. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Decrease in membrane phospholipids unsaturation correlates with myocardial diastolic dysfunction. PLoS ONE 2018, 13, e0208396. [Google Scholar] [CrossRef] [PubMed]
- Raher, M.J.; Thibault, H.B.; Buys, E.S.; Kuruppu, D.; Shimizu, N.; Brownell, A.-L.; Blake, S.L.; Rieusset, J.; Kaneki, M.; Derumeaux, G.; et al. A short duration of high-fat diet induces insulin resistance and predisposes to adverse left ventricular remodeling after pressure overload. Am. J. Physiol. Circ. Physiol. 2008, 295, H2495–H2502. [Google Scholar] [CrossRef]
- Park, S.-Y.; Cho, Y.-R.; Kim, H.-J.; Higashimori, T.; Danton, C.; Lee, M.-K.; Dey, A.; Rothermel, B.; Kim, Y.-B.; Kalinowski, A.; et al. Unraveling the Temporal Pattern of Diet-Induced Insulin Resistance in Individual Organs and Cardiac Dysfunction in C57BL/6 Mice. Diabetes 2005, 54, 3530–3540. [Google Scholar] [CrossRef] [PubMed]
- Mourmoura, E.; Chaté, V.; Couturier, K.; Laillet, B.; Vial, G.; Rigaudiere, J.-P.; Morio, B.; Malpuech-Brugère, C.; Azarnoush, K.; Demaison, L. Body adiposity dictates different mechanisms of increased coronary reactivity related to improved in vivo cardiac function. Cardiovasc. Diabetol. 2014, 13, 54. [Google Scholar] [CrossRef]
- Ballal, K.; Wilson, C.R.; Harmancey, R.; Taegtmeyer, H. Obesogenic high fat western diet induces oxidative stress and apoptosis in rat heart. Mol. Cell. Biochem. 2010, 344, 221–230. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; Theobald, H.; Sheng, X.; Wright, J.J.; Hu, X.X.; Aziz, S.; Johnson, J.I.; Bugger, H.; Zaha, V.G.; et al. Mitochondrial Energetics in the Heart in Obesity-Related Diabetes: Direct Evidence for Increased Uncoupled Respiration and Activation of Uncoupling Proteins. Diabetes 2007, 56, 2457–2466. [Google Scholar] [CrossRef]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. pNaKtide Attenuates Steatohepatitis and Atherosclerosis by Blocking Na/K-ATPase/ROS Amplification in C57Bl6 and ApoE Knockout Mice Fed a Western Diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef]
- Neves, F.A.; Cortez, E.; Bernardo, A.F.; Mattos, A.B.M.; Vieira, A.K.; de Malafaia, T.O.; Thole, A.A.; de Rodrigues-Cunha, S.A.C.; Garcia-Souza, É.P.; Sichieri, R.; et al. Heart energy metabolism impairment in Western-diet induced obese mice. J. Nutr. Biochem. 2014, 25, 50–57. [Google Scholar] [CrossRef]
- Kankaanpaa, M.; Lehto, H.-R.; Parkka, J.P.; Komu, M.; Viljanen, A.; Ferrannini, E.; Knuuti, J.; Nuutila, P.; Parkkola, R.; Iozzo, P. Myocardial Triglyceride Content and Epicardial Fat Mass in Human Obesity: Relationship to Left Ventricular Function and Serum Free Fatty Acid Levels. J. Clin. Endocrinol. Metab. 2006, 91, 4689–4695. [Google Scholar] [CrossRef]
- Püschel, G.P.; Henkel, J. Dietary cholesterol does not break your heart but kills your liver. Porto Biomed. J. 2018, 3, e12. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Kaluzny, M.A.; Duncan, L.A.; Merritt, M.V.; Epps, D.E. Rapid separation of lipid classes in high yield and purity using bonded phase columns. J. Lipid Res. 1985, 26, 135–140. [Google Scholar] [PubMed]
- Bodennec, J.; Koul, O.; Aguado, I.; Brichon, G.; Zwingelstein, G.; Portoukalian, J. A procedure for fractionation of sphingolipid classes by solid-phase extraction on aminopropyl cartridges. J. Lipid Res. 2000, 41, 1524–1531. [Google Scholar]
- Pakiet, A.; Jakubiak, A.; Czumaj, A.; Sledzinski, T.; Mika, A. The effect of western diet on mice brain lipid composition. Nutr. Metab. 2019, 16, 81. [Google Scholar] [CrossRef]
- Sullivan, E.M.; Fix, A.; Crouch, M.J.; Sparagna, G.C.; Zeczycki, T.N.; Brown, D.A.; Shaikh, S.R. Murine diet-induced obesity remodels cardiac and liver mitochondrial phospholipid acyl chains with differential effects on respiratory enzyme activity. J. Nutr. Biochem. 2017, 45, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.; Futerman, A.H.; Prieto, M. Lipid Raft Composition Modulates Sphingomyelinase Activity and Ceramide-Induced Membrane Physical Alterations. Biophys. J. 2009, 96, 3210–3222. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.A.G.; Carvalho, F.; Pearson, M.; Horton, J.D.; Browning, J.D.; Jones, J.G.; Burgess, S.C. A high-fat diet suppresses de novo lipogenesis and desaturation but not elongation and triglyceride synthesis in mice. J. Lipid Res. 2014, 55, 2541–2553. [Google Scholar] [CrossRef]
- Mika, A.; Sledzinski, T. Alterations of specific lipid groups in serum of obese humans: A review. Obes. Rev. 2017, 18, 247–272. [Google Scholar] [CrossRef]
- Karlstaedt, A.; Schiffer, W.; Taegtmeyer, H. Actionable Metabolic Pathways in Heart Failure and Cancer—Lessons From Cancer Cell Metabolism. Front. Cardiovasc. Med. 2018, 5, 71. [Google Scholar] [CrossRef]
- Rennison, J.H.; McElfresh, T.A.; Chen, X.; Anand, V.R.; Hoit, B.D.; Hoppel, C.L.; Chandler, M.P. Prolonged exposure to high dietary lipids is not associated with lipotoxicity in heart failure. J. Mol. Cell. Cardiol. 2009, 46, 883–890. [Google Scholar] [CrossRef][Green Version]
- Brainard, R.E.; Watson, L.J.; DeMartino, A.M.; Brittian, K.R.; Readnower, R.D.; Boakye, A.A.; Zhang, D.; Hoetker, J.D.; Bhatnagar, A.; Baba, S.P.; et al. High Fat Feeding in Mice Is Insufficient to Induce Cardiac Dysfunction and Does Not Exacerbate Heart Failure. PLoS ONE 2013, 8, e83174. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Shao, D.; Tomasi, L.C.; Braun, A.; de Mattos, A.B.M.; Choi, Y.S.; Villet, O.; Roe, N.; Halterman, C.R.; Tian, R.; et al. The effects of fatty acid composition on cardiac hypertrophy and function in mouse models of diet-induced obesity. J. Nutr. Biochem. 2017, 46, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Zukowska, P.; Kutryb-Zajac, B.; Jasztal, A.; Toczek, M.; Zabielska, M.; Borkowski, T.; Khalpey, Z.; Smolenski, R.T.; Slominska, E.M. Deletion of CD73 in mice leads to aortic valve dysfunction. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Drolet, M.-C.; Roussel, E.; Deshaies, Y.; Couet, J.; Arsenault, M. A High Fat/High Carbohydrate Diet Induces Aortic Valve Disease in C57BL/6J Mice. J. Am. Coll. Cardiol. 2006, 47, 850–855. [Google Scholar] [CrossRef]
- Hofmann, B.; Yakobus, Y.; Indrasari, M.; Nass, N.; Santos, A.N.; Kraus, F.B.; Silber, R.E.; Simm, A. RAGE influences the development of aortic valve stenosis in mice on a high fat diet. Exp. Gerontol. 2014, 59, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Rajabi, M.; Kassiotis, C.; Razeghi, P.; Taegtmeyer, H. Return to the fetal gene program protects the stressed heart: A strong hypothesis. Heart Fail. Rev. 2007, 12, 331–343. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid Metabolism and Toxicity in the Heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef]
- Kalaivanisailaja, J.; Manju, V.; Nalini, N. Lipid profile in mice fed a high-fat diet after exogenous leptin administration. Pol. J. Pharmacol. 2003, 55, 763–769. [Google Scholar]
- Park, T.-S.; Hu, Y.; Noh, H.-L.; Drosatos, K.; Okajima, K.; Buchanan, J.; Tuinei, J.; Homma, S.; Jiang, X.-C.; Abel, E.D.; et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 2008, 49, 2101–2112. [Google Scholar] [CrossRef]
- Drosatos, K.; Schulze, P.C. Cardiac Lipotoxicity: Molecular Pathways and Therapeutic Implications. Curr. Heart Fail. Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef]
- Schulze, P.C.; Drosatos, K.; Goldberg, I.J. Lipid Use and Misuse by the Heart. Circ. Res. 2016, 118, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.R.; Tran, M.K.; Salazar, K.L.; Young, M.E.; Taegtmeyer, H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem. J. 2007, 406, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Boini, K.M.; Xia, M.; Koka, S.; Gehr, T.W.B.; Li, P.L. Sphingolipids in obesity and related complications. Front. Biosci. 2017, 22, 96–116. [Google Scholar]
- Du, X.; Edelstein, D.; Obici, S.; Higham, N.; Zou, M.-H.; Brownlee, M. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J. Clin. Investig. 2006, 116, 1071–1080. [Google Scholar] [CrossRef]
- Leger, T.; Hininger-Favier, I.; Capel, F.; Geloen, A.; Rigaudière, J.-P.; Jouve, C.; Pitois, E.; Pineau, G.; Vaysse, C.; Chardigny, J.-M.; et al. Dietary canolol protects the heart against the deleterious effects induced by the association of rapeseed oil, vitamin E and coenzyme Q10 in the context of a high-fat diet. Nutr. Metab. 2018, 15, 15. [Google Scholar] [CrossRef]
- Tepšic, V.; Ristic, V.; Ristic, D.; Vasiljevic, N.; Pecelj-Gec, M. Heart phospholipid content and fatty acid composition in the rat after feeding different lipid supplemented diets. Physiol. Res. 1998, 47, 413–418. [Google Scholar] [PubMed]
- Duivenvoorde, L.P.M.; Van Schothorst, E.M.; Swarts, H.M.; Kuda, O.; Steenbergh, E.; Termeulen, S.; Kopecky, J.; Keijer, J. A Difference in fatty acid composition of isocaloric high-fat diets alters metabolic flexibility in male C57BL/6JOlaHsd mice. PLoS ONE 2015, 10, e0128515. [Google Scholar] [CrossRef]
- Chen, B.; Huang, Y.; Zheng, D.; Ni, R.; Bernards, M.A. Dietary fatty acids alter lipid profiles and induce myocardial dysfunction without causing metabolic disorders in mice. Nutrients 2018, 10, 106. [Google Scholar] [CrossRef]
- Watkins, S.M.; Lin, T.Y.; Davis, R.M.; Ching, J.R.; DePeters, E.J.; Halpern, G.M.; Walzem, R.L.; German, J.B. Unique phospholipid metabolism in mouse heart in response to dietary docosahexaenoic or α-linolenic acids. Lipids 2001, 36, 247–254. [Google Scholar] [CrossRef]
- Tosi, F.; Sartori, F.; Guarini, P.; Olivieri, O.; Martinelli, N. Delta-5 and Delta-6 Desaturases: Crucial Enzymes in Polyunsaturated Fatty Acid-Related Pathways with Pleiotropic Influences in Health and Disease. In Oxidative Stress and Inflammation in Non-communicable Diseases—Molecular Mechanisms and Perspectives in Therapeutics; Camps, J., Ed.; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2014; Volume 824, pp. 61–81. [Google Scholar]
- DeLany, J.P.; Windhauser, M.M.; Champagne, C.M.; Bray, G.A. Differential oxidation of individual dietary fatty acids in humans. Am. J. Clin. Nutr. 2000, 72, 905–911. [Google Scholar] [CrossRef]
- Leyton, J.; Drury, P.J.; Crawford, M.A. Differential oxidation of saturated and unsaturated fatty acids in vivo in the rat. Br. J. Nutr. 1987, 57, 383–393. [Google Scholar] [CrossRef]
- Reubsaet, F.A.G.; Veerkamp, J.H.; Trijbels, J.M.F.; Monnens, L.A.H. Total and peroxisomal oxidation of various saturated and unsaturated fatty acids in rat liver, heart and m. quadriceps. Lipids 1989, 24, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Marventano, S.; Kolacz, P.; Castellano, S.; Galvano, F.; Buscemi, S.; Mistretta, A.; Grosso, G. A review of recent evidence in human studies of n-3 and n-6 PUFA intake on cardiovascular disease, cancer, and depressive disorders: Does the ratio really matter? Int. J. Food Sci. Nutr. 2015, 66, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.S.; Shearer, G.C. Omega-6 Fatty Acids and Cardiovascular Disease. Circulation 2014, 130, 1562–1564. [Google Scholar] [CrossRef] [PubMed]
- Shindou, H.; Shimizu, T. Acyl-CoA:Lysophospholipid Acyltransferases. J. Biol. Chem. 2009, 284, 1–5. [Google Scholar] [CrossRef]
- Sonnweber, T.; Pizzini, A.; Nairz, M.; Weiss, G.; Tancevski, I. Arachidonic Acid Metabolites in Cardiovascular and Metabolic Diseases. Int. J. Mol. Sci. 2018, 19, 3285. [Google Scholar] [CrossRef]
- Hatch, G.M. Cell biology of cardiac mitochondrial phospholipids. Biochem. Cell Biol. 2004, 82, 99–112. [Google Scholar] [CrossRef]
- Stubbs, C.D.; Smith, A.D. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim. Biophys. Acta Rev. Biomembr. 1984, 779, 89–137. [Google Scholar] [CrossRef]
- Shchepinov, M.S.; Roginsky, V.A.; Brenna, J.T.; Molinari, R.J.; To, R.; Tsui, H.; Clarke, C.F.; Manning-Boğ, A.B. Deuterium Protection of Polyunsaturated Fatty Acids against Lipid Peroxidation. In Omega-3 Fatty Acids in Brain and Neurological Health; Academic Press: Cambridge, MA, USA, 2014; pp. 373–383. [Google Scholar]
- Salmon, A.; Dodd, S.W.; Williams, G.D.; Beach, J.M.; Brown, M.F. Configurational Statistics of Acyl Chains in Polyunsaturated Lipid Bilayers from 2H NMR. J. Am. Chem. Soc. 1987, 109, 2600–2609. [Google Scholar] [CrossRef]
- Stulnig, T.M.; Huber, J.; Leitinger, N.; Imre, E.-M.; Angelisová, P.; Nowotny, P.; Waldhäusl, W. Polyunsaturated Eicosapentaenoic Acid Displaces Proteins from Membrane Rafts by Altering Raft Lipid Composition. J. Biol. Chem. 2001, 276, 37335–37340. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Yang, K.; Yang, J.; Cheng, H.; Gross, R.W. Shotgun lipidomics of cardiolipin molecular species in lipid extracts of biological samples. J. Lipid Res. 2006, 47, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin Remodeling in the Heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Cortie, C.; Else, P. Dietary Docosahexaenoic Acid (22:6) Incorporates into Cardiolipin at the Expense of Linoleic Acid (18:2): Analysis and Potential Implications. Int. J. Mol. Sci. 2012, 13, 15447–15463. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, S.; Urade, R.; Kito, M. Mitochondrial Function in Rats Is Affected by Modification of Membrane Phospholipids with Dietary Sardine Oil. J. Nutr. 1988, 118, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Eldho, N.V.; Feller, S.E.; Tristram-Nagle, S.; Polozov, I.V.; Gawrisch, K. Polyunsaturated docosahexaenoic vs docosapentaenoic acid—Differences in lipid matrix properties from the loss of one double bond. J. Am. Chem. Soc. 2003, 125, 6409–6421. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Parameter | SD | HFD | p-value |
|---|---|---|---|
| Left ventricular end-systolic volume (LVESV) (µL) | 8.80 ± 2.00 | 10.8 ± 2.40 | 0.144 |
| Left ventricular end-diastolic volume (LVEDV) (µL) | 29.8 ± 4.60 | 31.1 ± 3.85 | 0.613 |
| Stroke Volume (SV) (µL) | 21.0 ± 2.70 | 20.3 ± 2.50 | 0.626 |
| Cardiac Output (CO) (mL × min-1) | 6.15 ± 0.99 | 6.10 ± 0.67 | 0.922 |
| Left Ventricular Ejection Fraction (LVEF) (%) | 70.8 ± 4.32 | 65.4 ± 5.50 | 0.090 |
| Aortic Valve Area (AVA) (mm2) | 1.62 ± 0.45 | 1.34 ± 0.44 | 0.049 |
| Vmax (m/s) | 0.76 ± 0.27 | 0.98 ± 0.30 | 0.056 |
| Chow (mg/g) | Serum (mg/L) | Heart (%) | Skeletal muscle (%) | |||||
|---|---|---|---|---|---|---|---|---|
| 10% | 60% | SD | HFD | SD | HFD | SD | HFD | |
| 16:0 | 9.73 ± 0.46 | 74.3 ± 4.89 ** | 719 ± 210 | 1106 ± 244 * | 21.01 ± 2.82 | 17.56 ± 2.77 * | 23.17 ± 1.78 | 20.79 ± 1.15 |
| 18:0 | 4.59 ± 0.10 | 39.1 ± 19.2 ** | 228 ± 26.3 | 548 ± 90.4 # | 23.88 ± 11.67 | 21.49 ± 6.64 | 6.07 ± 2.44 | 5.10 ± 0.68 ** |
| Other SFAs | 3.55 ± 0.10 | 22.8 ± 1.22 ** | 101 ± 51.3 | 201 ± 70.5 * | 3.46 ± 0.98 | 3.01 ± 1.12 | 5.54 ± 0.35 | 5.04 ± 0.39 |
| SFAs | 17.9 ± 0.64 | 136 ± 8.00** | 1049 ± 286 | 1855 ± 397** | 48.4 ± 10.1 | 42.1 ± 6.97 | 34.8 ± 3.50 | 31.0 ± 1.63 |
| 16:1 | 0.75 ± 0.01 | 5.93 ± 0.33 ** | 222 ± 64.9 | 135 ± 23.7 * | 4.07 ± 2.21 | 1.74 ± 0.87 ** | 10.8 ± 1.01 | 9.39 ± 1.02 |
| 18:1 | 9.63 ± 0.14 | 79.7 ± 3.65 # | 920 ± 313 | 1127 ± 256 | 30.3 ± 9.60 | 21.9 ± 7.69 * | 45.0 ± 3.03 | 45.6 ± 3.20 |
| Other MUFAs | 0.29 ± 0.01 | 2.28 ± 0.19 ** | 21.9 ± 12.0 | 27.5 ± 8.83 | 1.13 ± 0.48 | 0.77 ± 0.32 | 1.70 ± 0.28 | 1.03 ± 0.17 ** |
| MUFAs | 10.7 ± 0.12 | 87.9 ± 4.15 # | 1164 ± 389 | 1289 ± 274 | 35.5 ± 12.1 | 24.4 ± 8.83 * | 57.5 ± 4.20 | 56.0 ± 2.65 |
| 18:2 n-6 (LA) | 2.04 ± 0.04 | 18.6 ± 0.70 # | 356 ± 29.1 | 915 ± 114 # | 6.32 ± 1.94 | 12.2 ± 3.11 # | 4.29 ± 0.34 | 8.78 ± 0.14 # |
| 20:4 n-6 (ARA) | 0.037 ± 0.004 | 0.36 ± 0.01 # | 152 ± 37.4 | 330 ± 59.2 # | 4.14 ± 2.37 | 8.62 ± 3.24 ** | 1.30 ± 0.65 | 1.46 ± 0.47 |
| 22:4 n-6 (AdA) | 0.008 ± 0.002 | 0.11 ± 0.02 * | 1.48 ± 0.35 | 2.81 ± 0.60 # | 0.18 ± 0.10 | 0.57 ± 0.27 # | 0.12 ± 0.06 | 0.14 ± 0.03 |
| Other n-6 PUFAs | 0.073 ± 0.008 | 0.70 ± 0.08 ** | 42.0 ± 8.90 | 47.3 ± 14.2 | 1.75 ± 0.98 | 2.19 ± 0.82 | 0.68 ± 0.22 | 0.66 ± 0.09 |
| n-6 PUFA | 2.16 ± 0.03 | 19.7 ± 0.64 # | 552 ± 63.9 | 1295 ± 105 # | 12.4 ± 4.89 | 23.5 ± 7.17 # | 6.39 ± 1.02 | 11.0 ± 0.59 # |
| 18:3 n-3 (ALA) | 0.005 ± 0.002 | 0.049 ± 0.002 # | 5.74 ± 2.18 | 9.97 ± 2.55 * | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.01 |
| 20:5 n-3 (EPA) | 0.008 ± 0.002 | 0.074 ± 0.004 # | 42.9 ± 11.7 | 20.1 ± 2.77 ** | 0.54 ± 0.27 | 0.16 ± 0.05 # | 0.34 ± 0.10 | 0.08 ± 0.01 ** |
| 22:5 n-3 (DPA) | 0.016 ± 0.003 | 0.15 ± 0.007 # | 2.81 ± 0.53 | 7.87 ± 2.28 ** | 0.34 ± 0.23 | 1.87 ± 0.87 # | 0.16 ± 0.08 | 0.36 ± 0.12 * |
| 22:6 n-3 (DHA) | 0.006 ± 0.003 | 0.06 ± 0.02 | 30.9 ± 8.24 | 72.1 ± 8.81 | 2.83 ± 1.93 | 7.91 ± 3.34 # | 0.51 ± 0.29 | 1.10 ± 0.40 * |
| Other n-3 PUFAs | 0.002 ± 0.001 | 0.01 ± 0.01 | 2.87 ± 0.56 | 3.44 ± 0.89 | 0.34 ± 0.23 | 1.87 ± 0.87 # | 0.18 ± 0.09 | 0.38 ± 0.12 * |
| n-3 PUFA | 0.04 ± 0.01 | 0.35 ± 0.007 # | 85.2 ± 21.7 | 114 ± 9.01 * | 3.77 ± 2.36 | 9.99 ± 4.24 # | 1.05 ± 0.47 | 1.59 ± 0.51 |
| PUFA | 2.20 ± 0.03 | 20.1 ± 0.64 # | 637 ± 82.3 | 1408 ± 112 # | 16.16 ± 7.14 | 33.53 ± 11.26 # | 7.44 ± 1.45 | 12.6 ± 1.09 # |
| Free fatty acids | Phospholipids | Acylglycerols | ||||
|---|---|---|---|---|---|---|
| SD | HFD | SD | HFD | SD | HFD | |
| 16:0 | 45.2 ± 4.24 | 41.4 ± 8.53 | 21.0 ± 5.57 | 19.6 ± 4.32 | 23.6 ± 1.66 | 22.5 ± 0.64 |
| 18:0 | 28.4 ± 2.24 | 23.4 ± 3.01 ** | 27.4 ± 3.32 | 27.3 ± 3.95 | 5.30 ± 0.74 | 9.57 ± 1.25 # |
| Other SFAs | 5.68 ± 1.08 | 6.19 ± 1.33 | 2.64 ± 0.83 | 1.92 ± 0.32 | 5.55 ± 0.71 | 6.57 ± 0.86 |
| SFAs | 79.3 ± 3.40 | 71.1 ± 11.4 | 51.1 ± 9.40 | 48.8 ± 8.44 | 34.4 ± 1.92 | 38.7 ± 1.74 ** |
| 16:1 | 3.22 ± 1.42 | 3.00 ± 0.70 | 1.61 ± 0.73 | 0.65 ± 0.13 * | 7.36 ± 1.64 | 3.84 ± 0.98 ** |
| 18:1 | 13.1 ± 2.80 | 16.6 ± 6.23 | 19.8 ± 4.75 | 13.0 ± 2.06 * | 45.7 ± 1.07 | 38.5 ± 3.90 ** |
| Other MUFAs | 0.49 ± 0.19 | 0.79 ± 0.42 | 2.29 ± 0.87 | 1.10 ± 0.12 * | 1.80 ± 0.17 | 1.61 ± 0.29 |
| MUFAs | 16.8 ± 2.53 | 20.4 ± 5.94 | 22.2 ± 5.58 | 14.2 ± 2.17 * | 54.9 ± 2.20 | 43.9 ± 4.89 ** |
| 18:2 n-6 (LA) | 2.48 ± 0.93 | 5.24 ± 3.00 | 4.47 ± 0.73 | 6.24 ± 1.40 * | 9.61 ± 3.08 | 14.3 ± 3.39 |
| 20:4 n-6 (ARA) | 0.53 ± 0.55 | 1.36 ± 1.08 | 9.75 ± 5.82 | 12.7 ± 3.62 | 0.28 ± 0.24 | 1.05 ± 0.65 * |
| 22:4 n-6 (AdA) | 0.11 ± 0.08 | 0.23 ± 0.19 | 0.40 ± 0.25 | 0.71 ± 0.21 | 0.08 ± 0.07 | 0.18 ± 0.12 |
| Other n-6 PUFAs | 0.26 ± 0.16 | 0.52 ± 0.34 | 3.52 ± 2.22 | 2.29 ± 0.61 | 0.37 ± 0.22 | 0.81 ± 0.34 * |
| 18:3 n-3 (ALA) | 0.03 ± 0.02 | 0.05 ± 0.02 | 0.03 ± 0.02 | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.03 ± 0.01 |
| 20:5 n-3 (EPA) | 0.18 ± 0.10 | 0.14 ± 0.07 | 1.09 ± 0.47 | 0.19 ± 0.04 ** | 0.10 ± 0.07 | 0.10 ± 0.02 |
| 22:5 n-3 (DPA) | 0.12 ± 0.09 | 0.44 ± 0.43 | 0.94 ± 0.68 | 2.84 ± 1.13 * | 0.11 ± 0.08 | 0.39 ± 0.24 * |
| 22:6 n-3 (DHA) | 0.17 ± 0.12 | 0.57 ± 0.51 | 6.51 ± 4.89 | 12.0 ± 4.20 | 0.11 ± 0.07 | 0.60 ± 0.36 * |
| Other n-3 PUFAs | 0.001 ± 0.000 | 0.001 ± 0.000 | 0.08 ± 0.01 | 0.06 ± 0.02 | 0.009 ± 0.003 | 0.02 ± 0.01 |
| PUFAs | 3.88 ± 1.61 | 8.56 ± 5.54 | 26.8 ± 14.9 | 37.1 ± 10.5 | 10.7 ± 3.56 | 17.5 ± 3.37 * |
| Neutral lipids | Ceramides | Glycosphingolipids | Sphingomyelins | |||||
|---|---|---|---|---|---|---|---|---|
| SD | HFD | SD | HFD | SD | HFD | SD | HFD | |
| 16:0 | 26.2 ± 2.02 | 24.1 ± 2.08 | 29.6 ± 4.12 | 24.3 ± 0.54 | 43.0 ± 4.14 | 33.7 ± 0.79 * | 24.1 ± 2.13 | 22.3 ± 1.71 |
| 18:0 | 8.51 ± 1.38 | 8.59 ± 0.43 | 16.1 ± 2.74 | 11.9 ± 1.00 * | 34.5 ± 2.80 | 31.2 ± 1.85 | 24.6 ± 1.34 | 25.6 ± 1.29 |
| Other SFAs | 6.38 ± 0.62 | 5.56 ± 0.42 | 12.9 ± 2.43 | 8.61 ± 1.81 | 6.02 ± 2.45 | 4.64 ± 0.25 | 2.11 ± 0.12 | 1.64 ± 0.16 ** |
| SFAs | 41.1 ± 1.26 | 38.3 ± 2.33 | 58.6 ± 8.77 | 44.8 ± 2.91 * | 83.5 ± 4.68 | 69.6 ± 1.43 ** | 50.8 ± 3.13 | 49.6 ± 2.97 |
| 16:1 | 8.28 ± 0.99 | 3.99 ± 0.58 ** | 4.98 ± 0.58 | 4.19 ± 0.69 | 1.55 ± 0.38 | 1.53 ± 0.74 | 1.34 ± 0.09 | 0.54 ± 0.05 # |
| 18:1 | 44.0 ± 1.14 | 42.7 ± 2.02 | 29.8 ± 7.31 | 35.5 ± 2.93 | 9.02 ± 1.70 | 10.6 ± 0.95 | 22.6 ± 1.85 | 14.4 ± 0.52 # |
| Other MUFAs | 2.79 ± 0.63 | 1.59 ± 0.25 | 2.52 ± 0.52 | 1.53 ± 0.23 * | 0.31 ± 0.11 | 0.17 ± 0.01 | 0.47 ± 0.08 | 0.28 ± 0.03 ** |
| MUFAs | 55.1 ± 1.25 | 48.2 ± 2.00 * | 37.3 ± 7.89 | 41.2 ± 2.03 | 10.9 ± 1.32 | 12.3 ± 1.63 | 24.4 ± 1.90 | 15.2 ± 0.57 # |
| 18:2 n-6 (LA) | 3.43 ± 1.26 | 9.70 ± 0.10 ** | 2.71 ± 0.82 | 8.86 ± 1.27 # | 1.74 ± 0.68 | 4.20 ± 0.30 ** | 6.00 ± 0.96 | 8.15 ± 1.55 * |
| 20:4 n-6 (ARA) | 0.30 ± 0.27 | 1.20 ± 0.14 * | 0.75 ± 0.35 | 1.99 ± 0.29 ** | 2.21 ± 1.34 | 6.63 ± 0.21 ** | 9.07 ± 1.66 | 12.4 ± 1.09 * |
| 22:4 n-6 (AdA) | 0.07 ± 0.05 | 0.47 ± 0.06 ** | 0.05 ± 0.03 | 0.37 ± 0.02 # | 0.07 ± 0.04 | 0.36 ± 0.01 # | 0.33 ± 0.07 | 0.70 ± 0.07 # |
| Other n-6 PUFAs | 0.19 ± 0.12 | 0.90 ± 0.17 ** | 0.23 ± 0.09 | 0.59 ± 0.11 ** | 0.50 ± 0.40 | 0.90 ± 0.10 | 2.98 ± 0.71 | 2.34 ± 0.14 |
| 18:3 n-3 (ALA) | 0.02 ± 0.01 | 0.02 ± 0.01 | 0.06 ± 0.02 | 0.04 ± 0.01 | 0.04 ± 0.02 | 0.02 ± 0.01 | 0.01 ± 0.01 | 0.02 ± 0.01 |
| 20:5 n-3 (EPA) | 0.22 ± 0.15 | 0.16 ± 0.04 | 0.4 ± 0.23 | 0.18 ± 0.06 | 0.43 ± 0.17 | 0.09 ± 0.02 * | 1.39 ± 0.24 | 0.18 ± 0.02 # |
| 22:5 n-3 (DPA) | 0.12 ± 0.10 | 1.01 ± 0.14 ** | 0.08 ± 0.04 | 0.88 ± 0.07 # | 0.15 ± 0.12 | 1.35 ± 0.16 # | 0.72 ± 0.20 | 2.57 ± 0.39 |
| 22:6 n-3 (DHA) | 0.12 ± 0.08 | 0.84 ± 0.12 ** | 0.32 ± 0.13 | 1.39 ± 0.07 # | 1.07 ± 0.86 | 4.81 ± 0.23 # | 4.75 ± 1.43 | 9.08 ± 0.64 # |
| Other n-3 PUFAs | 0.013 ± 0.005 | 0.02 ± 0.01 | 0.010 ± 0.007 | 0.03 ± 0.01 * | 0.02 ± 0.01 | 0.04 ± 0.01 | 0.10 ± 0.02 | 0.09 ± 0.03 # |
| PUFA | 4.47 ± 2.01 | 14.3 ± 0.70 ** | 4.61 ± 1.61 | 14.3 ± 1.04 # | 6.24 ± 3.57 | 18.4 ± 0.37 ** | 25.4 ± 5.05 | 35.5 ± 3.43 * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pakiet, A.; Jakubiak, A.; Mierzejewska, P.; Zwara, A.; Liakh, I.; Sledzinski, T.; Mika, A. The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice. Nutrients 2020, 12, 824. https://doi.org/10.3390/nu12030824
Pakiet A, Jakubiak A, Mierzejewska P, Zwara A, Liakh I, Sledzinski T, Mika A. The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice. Nutrients. 2020; 12(3):824. https://doi.org/10.3390/nu12030824
Chicago/Turabian StylePakiet, Alicja, Agnieszka Jakubiak, Paulina Mierzejewska, Agata Zwara, Ivan Liakh, Tomasz Sledzinski, and Adriana Mika. 2020. "The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice" Nutrients 12, no. 3: 824. https://doi.org/10.3390/nu12030824
APA StylePakiet, A., Jakubiak, A., Mierzejewska, P., Zwara, A., Liakh, I., Sledzinski, T., & Mika, A. (2020). The Effect of a High-Fat Diet on the Fatty Acid Composition in the Hearts of Mice. Nutrients, 12(3), 824. https://doi.org/10.3390/nu12030824