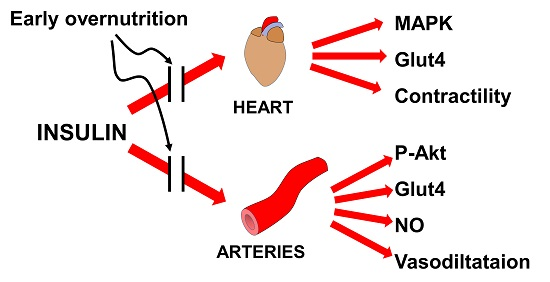
Overfeeding during Lactation in Rats is Associated with Cardiovascular Insulin Resistance in the Short-Term

 ,
,
Abstract

1. Introduction
2. Methods
2.1. Animals
2.2. Plasma Measurements
2.3. Experiments of Heart Perfusion: Langendorff
2.4. Experiments of Vascular Reactivity
2.5. Incubation of Aorta Segments in Presence/Absence of Insulin (10−7 M)
2.6. Nitrite and Nitrate Determination in the Culture Medium
2.7. RNA Extraction and Quantitative RT Real Time PCR
2.8. Protein Quantification by Western Blot
2.9. Detection of Glucose Transporter 4 (GLUT-4) in the Heart by Immunofluorescence
2.10. Statistical Analysis
3. Results
3.1. Body and Organ Weight
3.2. Glycemia, Lipid Profile and Plasma Concentrations of Metabolic Hormones
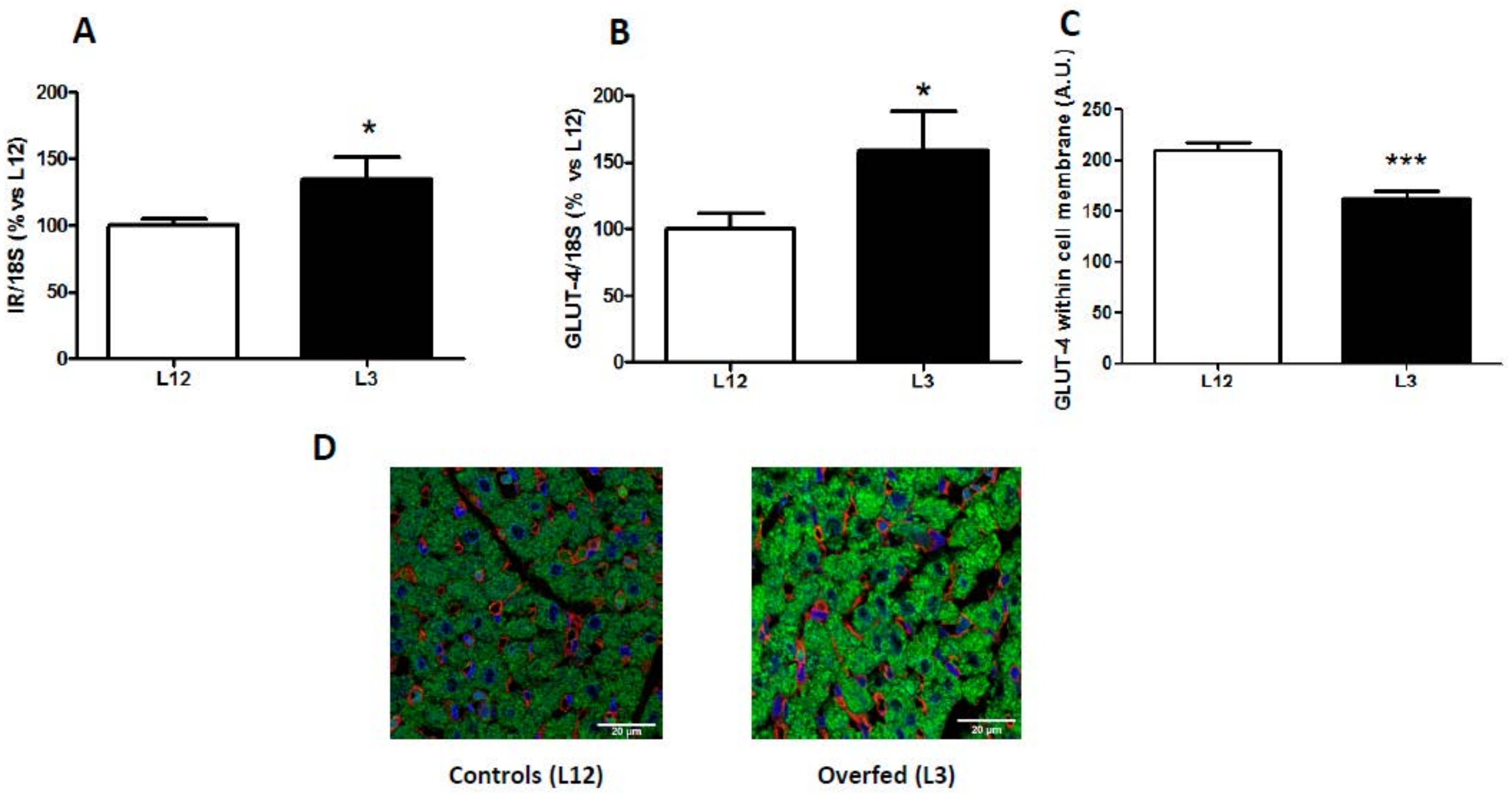
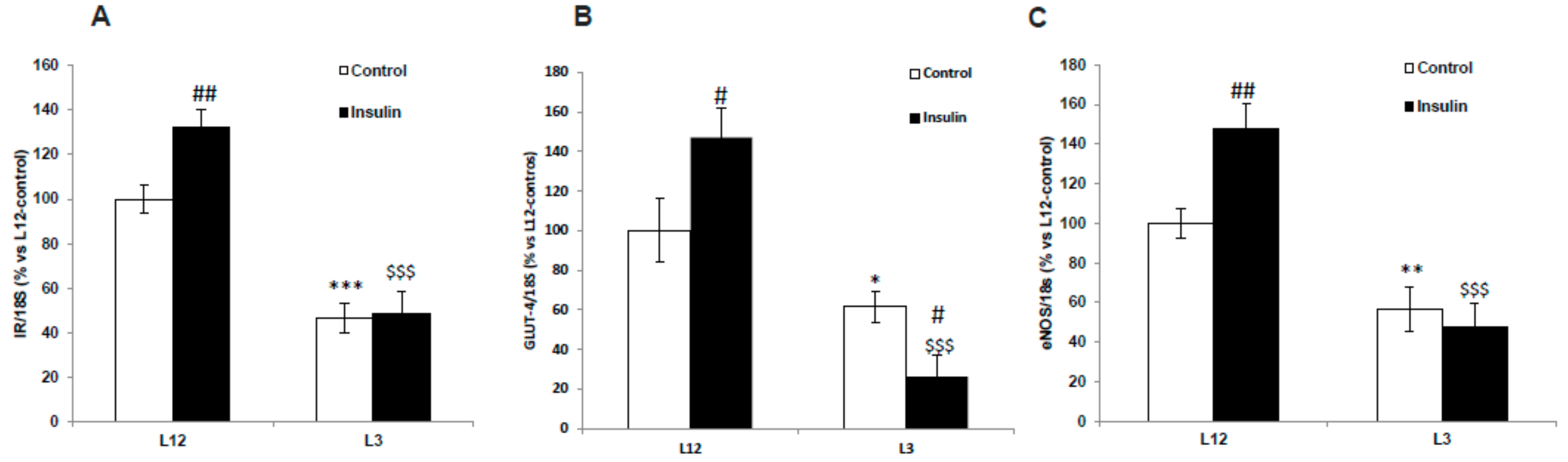
3.3. mRNA Levels of Insulin Receptor and Glucose Transporter 4 in the Myocardium and GLUT-4 Localization
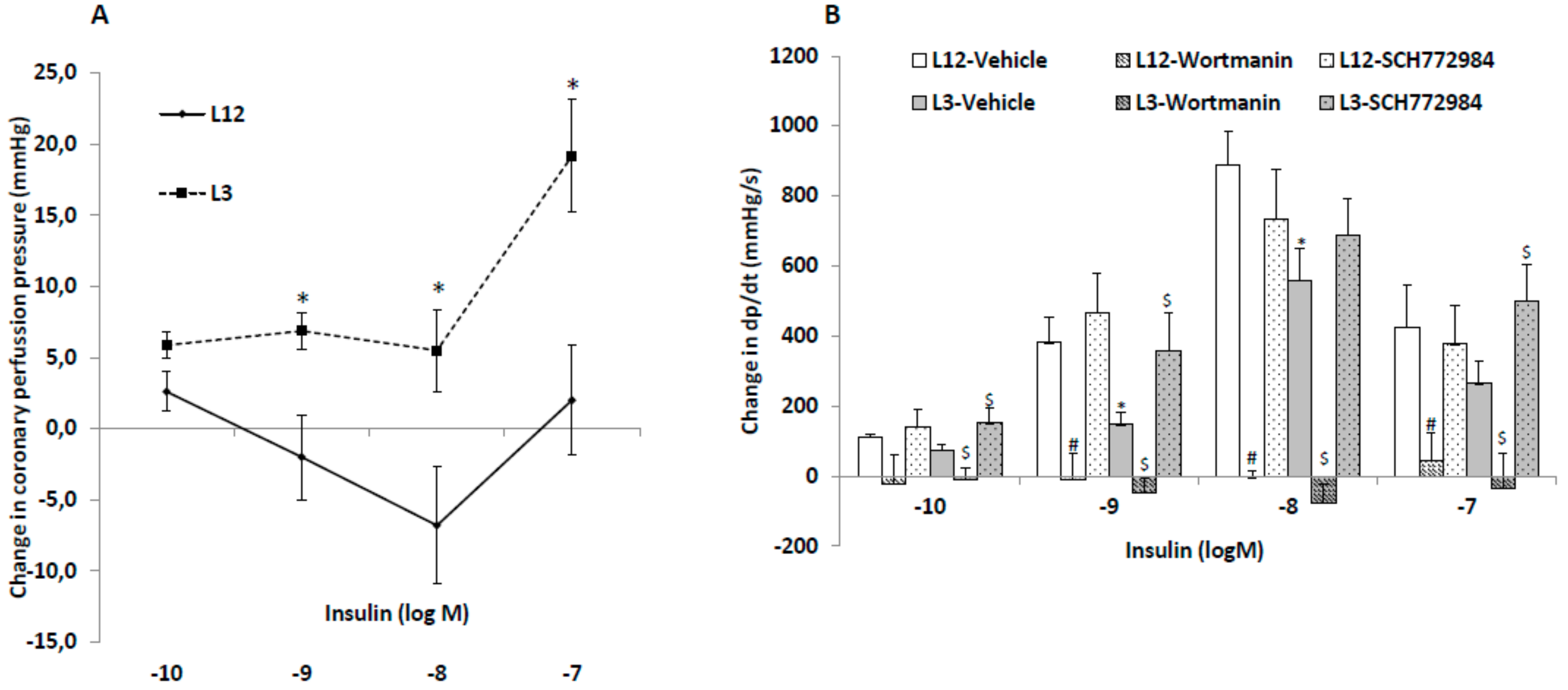
3.4. Changes in Heart Rate, Coronary Perfusion Pressure and Heart Contractility (dp/dt) in Response to Insulin Administration
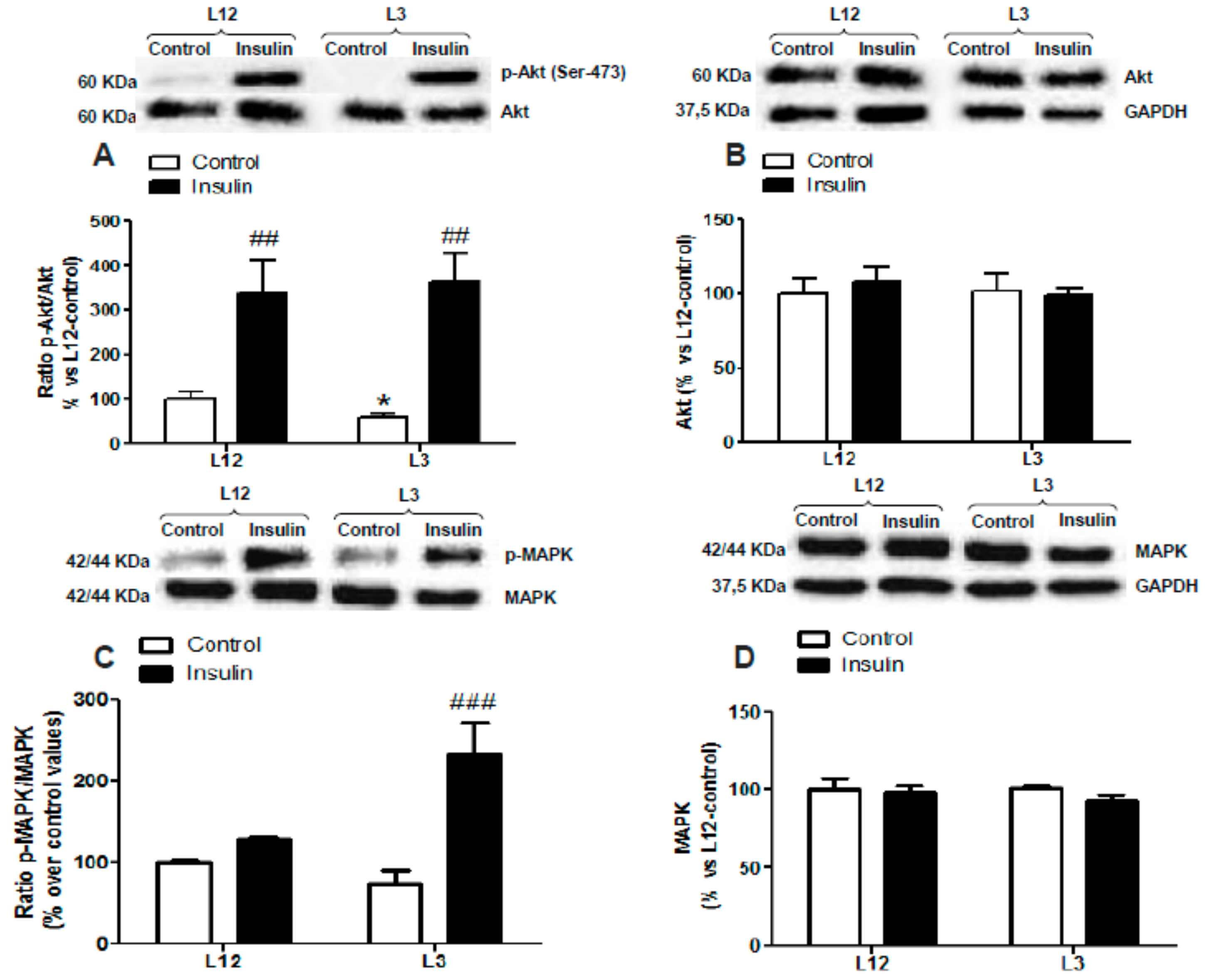
3.5. Myocardial Activation of PI3K/Akt and MAPK Pathways in Response to Insulin Administration
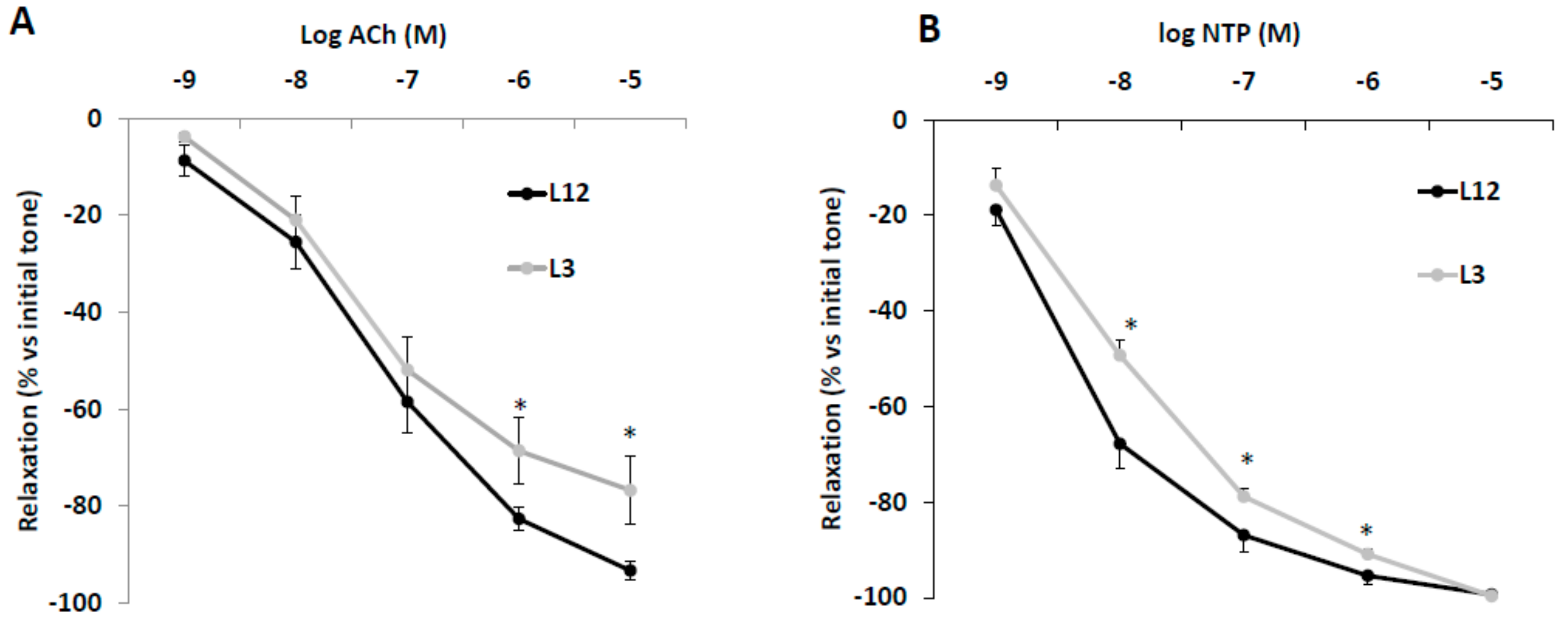
3.6. Vascular Reactivity of Aortic Rings in Response to Acetylcholine (Ach) and Sodium Nitroprusside (NTP)
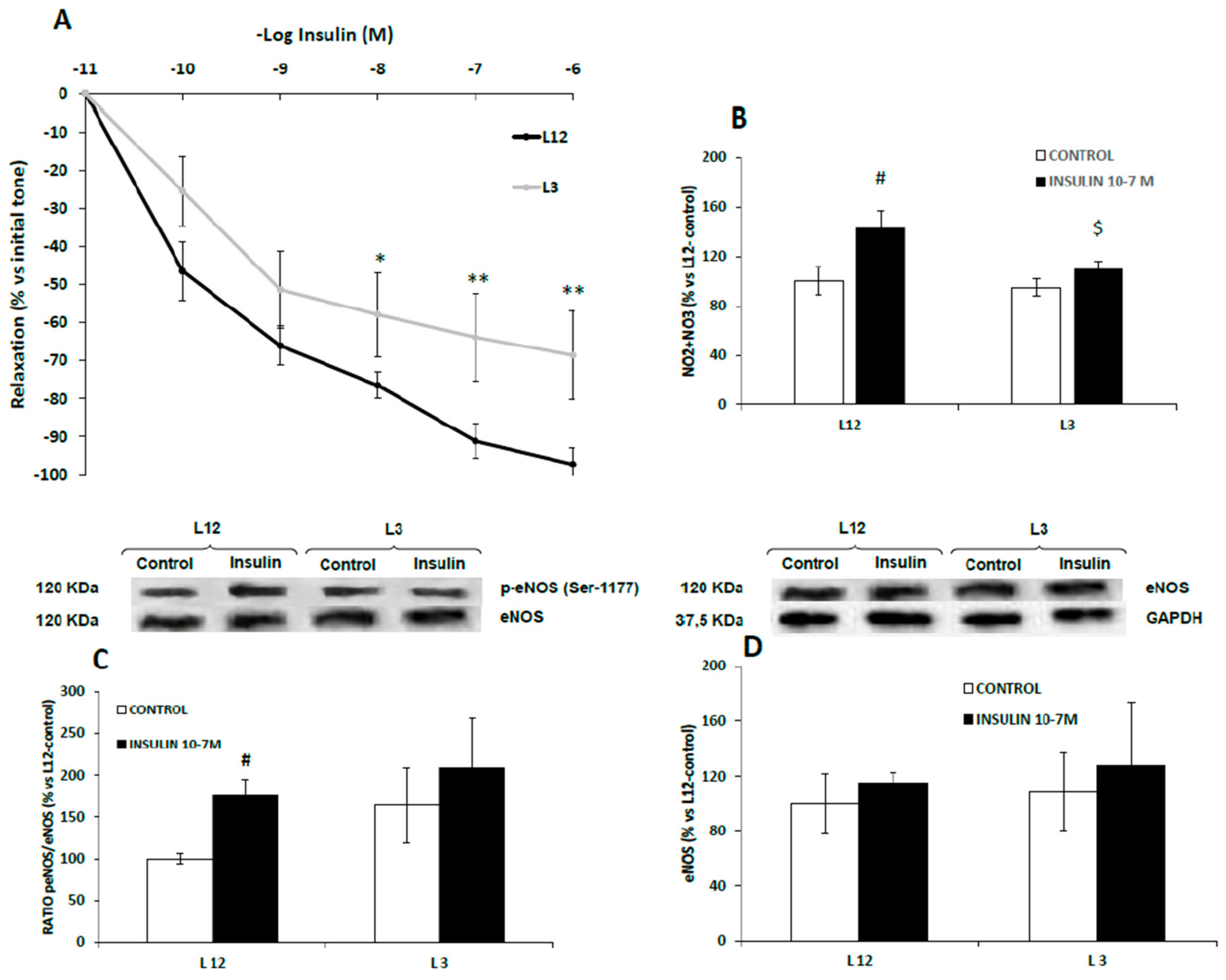
3.7. Vascular Reactivity, Phospho-eNOS Expression and Nitrites Release of Aortic Rings in Response to Insulin
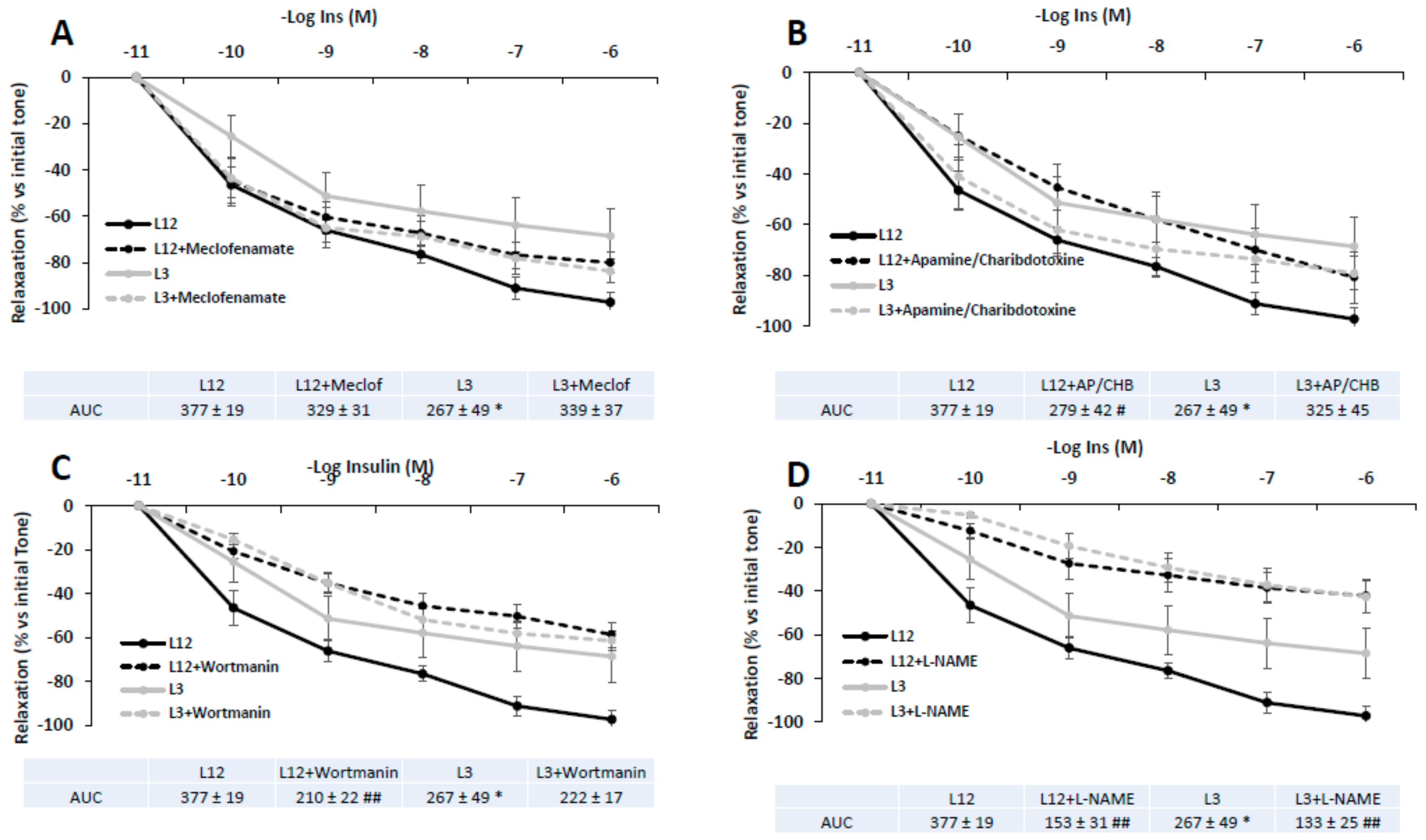
3.8. Vascular Reactivity of Aortic Rings in Response to Insulin in Presence/Absence of Meclofenamate, Apamine/Charibdotoxine, L-NAME, or Wortmanin
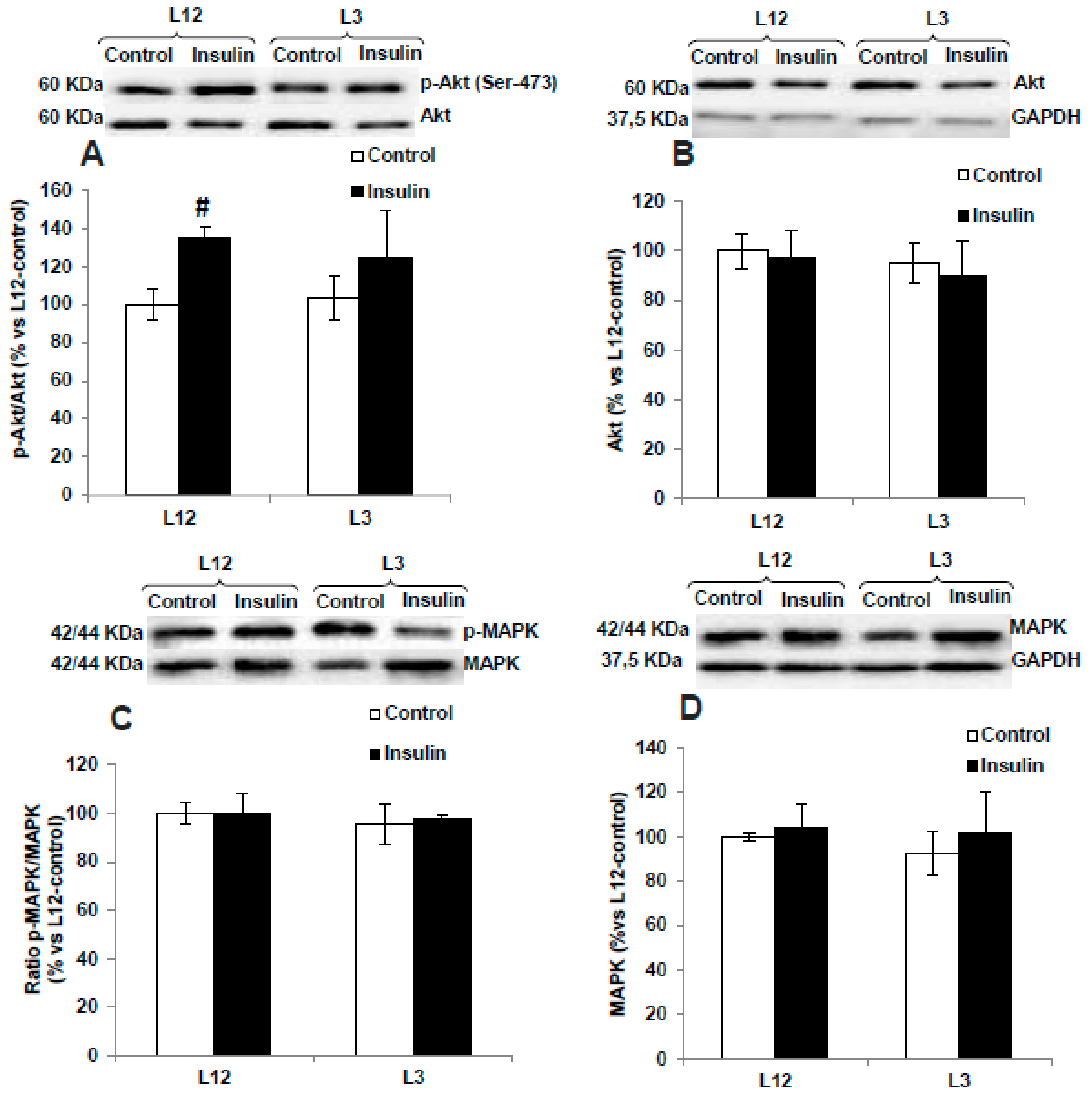
3.9. Activation of PI3K/Akt and MAPK Pathways in Response to Insulin Administration in Arterial Tissue
3.10. Gene Expression of IR, GLUT-4, and eNOS in Arterial Tissue in Response to Insulin
3.11. Gene Expression of Inflammatory and Oxidative Stress-Related Markers in Myocardial and Aortic Tissue
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engin, A. The Definition and Prevalence of Obesity and Metabolic Syndrome. Adv. Exp. Med. Biol. 2017, 960, 1–17. [Google Scholar]
- Graf, C.; Ferrari, N. Metabolic Syndrome in Children and Adolescents. Visc. Med. 2016, 32, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Al-Hamad, D.; Raman, V. Metabolic syndrome in children and adolescents. Transl. Pediatr. 2017, 6, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Caprio, S.; Perry, R.; Kursawe, R. Adolescent Obesity and Insulin Resistance: Roles of Ectopic Fat Accumulation and Adipose Inflammation. Gastroenterology 2017, 152, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, B.W. Cardiovascular consequences of childhood obesity. Can. J. Cardiol. 2015, 31, 124–130. [Google Scholar] [CrossRef]
- Sujana, S.; Gunta, R.H.M. Hypertension in children with obesity. World J. Hyperthens. 2014, 4, 15–24. [Google Scholar]
- Cote, A.T.; Harris, K.C.; Panagiotopoulos, C.; Sandor, G.G.S.; Devlin, A.M. Childhood obesity and cardiovascular dysfunction. J. Am. Coll. Cardiol. 2013, 62, 1309–1319. [Google Scholar] [CrossRef]
- Pires, A.; Martins, P.; Pereira, A.M.; Silva, P.V.; Marinho, J.; Marques, M.; Castela, E.; Sena, C.; Seiça, R. Insulin resistance, dyslipidemia and cardiovascular changes in a group of obese children. Arq. Bras. Cardiol. 2015, 104, 266–273. [Google Scholar] [CrossRef]
- Koopman, L.P.; McCrindle, B.W.; Slorach, C.; Chahal, N.; Hui, W.; Sarkola, T.; Manlhiot, C.; Jaeggi, E.T.; Bradley, T.J.; Mertens, L. Interaction between myocardial and vascular changes in obese children: A pilot study. J. Am. Soc. Echocardiogr. 2012, 25, 401–410. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Baron, A.D. Cardiovascular actions of insulin in humans. Implications for insulin sensitivity and vascular tone. Baillieres. Clin. Endocrinol. Metab. 1993, 7, 961–987. [Google Scholar] [CrossRef]
- Klein, L.J.; Visser, F.C. The effect of insulin on the heart: Part 1: Effects on metabolism and function. Neth. Heart J. 2010, 18, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Montagnani, M.; Chen, H.; Barr, V.A.; Quon, M.J. Insulin-stimulated activation of eNOS is independent of Ca2+ but requires phosphorylation by Akt at Ser(1179). J. Biol. Chem. 2001, 276, 30392–30398. [Google Scholar] [CrossRef]
- Zeng, G.; Quon, M.J. Insulin-stimulated production of nitric oxide is inhibited by wortmannin. Direct measurement in vascular endothelial cells. J. Clin. Invest. 1996, 98, 894–898. [Google Scholar] [CrossRef] [PubMed]
- Cain, A.E.; Tanner, D.M.; Khalil, R.A. Endothelin-1--induced enhancement of coronary smooth muscle contraction via MAPK-dependent and MAPK-independent [Ca(2+)](i) sensitization pathways. Hypertension 2002, 39, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Artunc, F.; Schleicher, E.; Weigert, C.; Fritsche, A.; Stefan, N.; Häring, H.U. The impact of insulin resistance on the kidney and vasculature. Nat. Rev. Nephrol. 2016, 12, 721–737. [Google Scholar] [CrossRef]
- Muniyappa, R.; Sowers, J.R. Role of insulin resistance in endothelial dysfunction. Rev. Endocr. Metab. Disord. 2013, 14, 5–12. [Google Scholar] [CrossRef]
- Plagemann, A.; Heidrich, I.; Götz, F.; Rohde, W.; Dörner, G. Obesity and enhanced diabetes and cardiovascular risk in adult rats due to early postnatal overfeeding. Exp. Clin. Endocrinol. 1992, 99, 154–158. [Google Scholar] [CrossRef]
- Briffa, J.F.; O’Dowd, R.; Romano, T.; Muhlhausler, B.S.; Morits, K.M.; Wlodek, M.E. Reducing Pup Litter Size Alters Early Postnatal Calcium Homeostasis and Programs Adverse Adult Cardiovascular and Bone Health in Male Rats. Nutrients 2019, 11, 118. [Google Scholar] [CrossRef]
- Habbout, A.; Li, N.; Rochette, L.; Vergely, C. Postnatal overfeeding in rodents by litter size reduction induces major short- and long-term pathophysiological consequences. J. Nutr. 2013, 143, 553–562. [Google Scholar] [CrossRef]
- Granado, M.; Fernández, N.; Monge, L.; Figueras, J.C.; Carreño-Tarragona, G.; Amor, S.; García-Villalón, Á.L. Effects of coronary ischemia-reperfusion in a rat model of early overnutrition. Role of angiotensin receptors. PLoS ONE 2013, 8, e54984. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Fernández, N.; Monge, L.; Carreño-Tarragona, G.; Figueras, J.C.; Amor, S.; García-Villalón, Á.L. Long-term effects of early overnutrition in the heart of male adult rats: Role of the renin-angiotensin system. PLoS ONE 2014, 8, e65172. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.; Amor, S.; Fernández, N.; Carreño-Tarragona, G.; Iglesias-Cruz, M.C.; Martín-Carro, B.; Monge, L.; García-Villalón, Á.L. Effects of early overnutrition on the renal response to Ang II and expression of RAAS components in rat renal tissue. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 930–937. [Google Scholar] [CrossRef]
- Rodrigues, A.L.; DE Souza, E.P.G.; Da Silva, S.V.; Rodrigues, D.S.B.; Nascimento, A.B.; Barja-Fidalgo, C.; De Freitas, M.S. Low expression of insulin signaling molecules impairs glucose uptake in adipocytes after early overnutrition. J. Endocrinol. 2007, 195, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Davidowa, H.; Plagemann, A. Inhibition by insulin of hypothalamic VMN neurons in rats overweight due to postnatal overfeeding. Neuroreport 2001, 12, 3201–3204. [Google Scholar] [CrossRef] [PubMed]
- Mangala Priya, V.; Mayilvanan, C.; Akilavalli, N.; Rajesh, P.; Balasubramanian, K. Lactational exposure of phthalate impairs insulin signaling in the cardiac muscle of F1 female albino rats. Cardiovasc. Toxicol. 2014, 14, 10–20. [Google Scholar] [CrossRef]
- Parsanathan, R.; Maria Joseph, A.; Karundevi, B. Postnatal exposure to di-(2-ethylhexyl)phthalate alters cardiac insulin signaling molecules and GLUT4(Ser488) phosphorylation in male rat offspring. J. Cell. Biochem. 2019, 120, 5802–5812. [Google Scholar] [CrossRef]
- Granado, M.; Amor, S.; Martín-Carro, B.; Guerra-Menéndez, L.; Tejera-Muñoz, A.; González-Hedström, D.; Rubio, C.; Carrascosa, J.M.; García-Villalón, Á.L. Caloric restriction attenuates aging-induced cardiac insulin resistance in male Wistar rats through activation of PI3K/Akt pathway. Nutr. Metab. Cardiovasc. Dis. 2019, 29, 97–105. [Google Scholar] [CrossRef]
- Miranda, K.M.; Espey, M.G.; Wink, D.A. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001, 5, 62–71. [Google Scholar] [CrossRef]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 536–537. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Vincent, M.A.; Montagnani, M.; Quon, M.J. Molecular and physiologic actions of insulin related to production of nitric oxide in vascular endothelium. Curr. Diabetes Rep. 2003, 3, 279–288. [Google Scholar] [CrossRef]
- Manrique, C.; Lastra, G.; Sowers, J.R. New insights into insulin action and resistance in the vasculature. Ann. N. Y. Acad. Sci. 2014, 1311, 138–150. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lin, Y.W.; Clemont, A.; Feener, E.P.; Hein, K.D.; Igarashi, M.; Yamauchi, T.; White, M.F.; King, G.L. Characterization of selective resistance to insulin signaling in the vasculature of obese Zucker (fa/fa) rats. J. Clin. Invest. 1999, 104, 447–457. [Google Scholar] [CrossRef]
- Kobayashi, T.; Taguchi, K.; Yasuhiro, T.; Matsumoto, T.; Kamata, K. Impairment of PI3-K/Akt pathway underlies attenuated endothelial function in aorta of type 2 diabetic mouse model. Hypertension 2004, 44, 956–962. [Google Scholar] [CrossRef]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J. Clin. Invest. 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Tomsa, A.; Bartz, S.K.; Krishnamurthy, R.; Krishnamurthy, R.; Bacha, F. Endothelial Function in Youth: A Biomarker Modulated by Adiposity-Related Insulin Resistance. J. Pediatr. 2016, 178, 171–177. [Google Scholar] [CrossRef]
- Haberzettl, P.; McCracken, J.P.; Bhatnagar, A.; Conklin, D.J. Insulin sensitizers prevent fine particulate matter-induced vascular insulin resistance and changes in endothelial progenitor cell homeostasis. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H1423–H1438. [Google Scholar] [CrossRef]
- Paneni, F.; Costantino, S.; Cosentino, F. p66(Shc)-induced redox changes drive endothelial insulin resistance. Atherosclerosis 2014, 236, 426–429. [Google Scholar] [CrossRef]
- Katakam, P.V.; Tulbert, C.D.; Snipes, J.A.; Erdös, B.; Miller, A.W.; Busija, B. Impaired insulin-induced vasodilation in small coronary arteries of Zucker obese rats is mediated by reactive oxygen species. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H854–H860. [Google Scholar] [CrossRef]
- Ren, J.; Walsh, M.F.; Hamaty, M.; Sowers, J.R.; Brown, R.A. Augmentation of the inotropic response to insulin in diabetic rat hearts. Life Sci. 1999, 65, 369–380. [Google Scholar] [CrossRef]
- Cittadini, A.; Ishiguro, Y.; Strömer, H.; Spindler, M.; Moses, A.C.; Clark, R.; Douglas, P.S.; Ingwall, J.S.; Morgan, J.P. Insulin-like growth factor-1 but not growth hormone augments mammalian myocardial contractility by sensitizing the myofilament to Ca2+ through a wortmannin-sensitive pathway: Studies in rat and ferret isolated muscles. Circ. Res. 1998, 83, 50–59. [Google Scholar] [CrossRef]
- Palomeque, J.; Sapia, L.; Hajjar, R.J.; Mattiazzi, A.; Petroff, M.V. Angiotensin II-induced negative inotropy in rat ventricular myocytes: Role of reactive oxygen species and p38 MAPK. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H96–H106. [Google Scholar] [CrossRef]
- Zheng, M.; Hou, R.; Xiao, R.P. Acidosis-induced p38 MAPK activation and its implication in regulation of cardiac contractility. Acta Pharmacol. Sin. 2004, 25, 1299–1305. [Google Scholar]
- Vahebi, S.; Ota, A.; Li, M.; Warren, C.M.; de Tombe, P.P.; Wang, Y.; Solaro, R.J. p38-MAPK induced dephosphorylation of alpha-tropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circ. Res. 2007, 100, 408–415. [Google Scholar] [CrossRef]
- Kumphune, S.; Chattipakorn, S.; Chattipakorn, N. Roles of p38-MAPK in insulin resistant heart: Evidence from bench to future bedside application. Curr. Pharm. Des. 2013, 19, 5742–5754. [Google Scholar] [CrossRef]
- Frazier, W.J.; Xue, J.; Luce, W.A.; Liu, Y. MAPK signaling drives inflammation in LPS-stimulated cardiomyocytes: The route of crosstalk to G-protein-coupled receptors. PLoS ONE 2012, 7, e50071. [Google Scholar] [CrossRef]
- Guo, R.M.; Xiu, W.M.; Lin, J.C.; Mo, L.Q.; Hua, X.X.; Chen, P.X.; Wu, K.; Zheng, D.D.; Feng, J.Q. Activation of the p38 MAPK/NF-kappaB pathway contributes to doxorubicin-induced inflammation and cytotoxicity in H9c2 cardiac cells. Mol. Med. Rep. 2013, 8, 603–608. [Google Scholar] [CrossRef]
- Thandavarayan, R.A.; Giridharan, V.V.; Sari, F.R.; Arumugam, S.; Veeraveedu, P.T.; Pandian, G.N.; Palaniyandi, S.S.; Ma, M.; Suzuki, K.; Gurusamy, N.; et al. Depletion of 14-3-3 protein exacerbates cardiac oxidative stress, inflammation and remodeling process via modulation of MAPK/NF-kB signaling pathways after streptozotocin-induced diabetes mellitus. Cell. Physiol. Biochem. 2011, 28, 911–922. [Google Scholar] [CrossRef]
- Wang, Y.H.; Cai, L. Diabetes/obesity-related inflammation, cardiac cell death and cardiomyopathy. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2006, 31, 814–818. [Google Scholar]
- Matsumoto, K.; Fujita, N.; Ozaki, M.; Tominaga, T.; Ueki, Y.; Miyake, S. Coexistence of insulin resistance and inflammation effectively predicts cardiac disease but not stroke in Japanese patients with type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2006, 74, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.D.; Bassi, R.; Marber, M.S. p38 MAPK in cardioprotection—are we there yet? Br. J. Pharmacol. 2015, 172, 2101–2113. [Google Scholar] [CrossRef] [PubMed]
- Jing, L.; Pulenthiran, A.; Nevius, C.D.; Mejia-Spiegeler, A.; Suever, J.D.; Wehner, G.J.; Kirchner, H.L.; Haggerty, C.M.; Fornwalt, B.K. Impaired right ventricular contractile function in childhood obesity and its association with right and left ventricular changes: A cine DENSE cardiac magnetic resonance study. J. Cardiovasc. Magn. Reson. 2017, 19, 49. [Google Scholar] [CrossRef]
- Mangner, N.; Scheuermann, K.; Winzer, E.; Wagner, I.; Hoellriegel, R.; Sandri, M.; Zimmer, M.; Mende, M.; Linke, A.; Kiess, W.; et al. Childhood obesity: Impact on cardiac geometry and function. JACC Cardiovasc. Imaging 2014, 7, 1198–1205. [Google Scholar] [CrossRef]
- Koopman, L.P.; Mertens, L.L. Impact of childhood obesity on cardiac structure and function. Curr. Treat. Options Cardiovasc. Med. 2014, 16, 345. [Google Scholar] [CrossRef]
- Uner, A.; Doğan, M.; Epcacan, Z.; Epçaçan, S. The effect of childhood obesity on cardiac functions. J. Pediatr. Endocrinol. Metab. 2014, 27, 261–271. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| L12 | L3 | |
|---|---|---|
| Body weight at birth (g) | 6.8 ± 1.1 | 6.9 ± 1.1 |
| Body weight at weaning (g) | 50 ± 1.1 | 65.7 ± 1.1 *** |
| Visceral Epididymal adipose tissue (mg) | 99 ± 9,4 | 185 ± 11 *** |
| Subcutaneous lumbar adipose tissue (mg) | 245 ± 26.7 | 521 ± 40.6 *** |
| Brown adipose tissue (mg) | 259 ± 9 | 344 ± 19.3 *** |
| Periaortic adipose tissue (mg) | 13.6 ± 1.2 | 21.1 ± 1.7 ** |
| Gastrocnemius (mg) | 161 ± 15 | 218 ± 12.2 ** |
| Heart (mg) | 336± 34.5 | 400 ± 21.7 * |
| L12 | L3 | |
|---|---|---|
| Glycemia (mg/dL) | 95 ± 13 | 107 ± 2.5 ** |
| Insulin (ng/mL) | 4.8 ± 0.8 | 14.1 ± 2.9 * |
| Leptin (ng/mL) | 5 ± 0.5 | 17.3 ± 3.4 ** |
| Adiponectin (µg/mL) | 76 ± 7.1 | 117.5 ± 10.5 ** |
| Total Lipids (mg/dL) | 300 ± 16 | 375 ± 21 ** |
| Triglycerides (mg/dL) | 175.8 ± 20.4 | 171.3 ± 24.4 |
| Total Cholesterol (mg/dL) | 255.2 ± 13.2 | 292.1 ± 13.1* |
| LDL-Cholesterol (mg/dL) | 153.4 ± 10.8 | 151.6 ± 9.1 |
| HDL- Cholesterol (mg/dL) | 137.6 ± 10.6 | 112.9 ± 6.6 * |
| L12 | L3 | |
|---|---|---|
| IL-1β | 100 ± 6.8 | 128 ± 12.3 * |
| IL-6 | 101 ± 6.2 | 134 ± 4.6 ** |
| TNF-α | 100 ± 6.1 | 149 ± 31.8 * |
| iNOS | 100 ± 38.3 | 244 ± 141 |
| COX-2 | 100 ± 32.8 | 42 ± 13.7 |
| NOX-1 | 100 ± 24 | 235 ± 50 * |
| NOX-4 | 100 ± 15 | 209 ± 16 ** |
| SOD-1 | 100 ± 5.6 | 139 ± 17.3 * |
| GSR | 100 ± 11 | 143 ± 19 * |
| GPX-3 | 100 ± 8.2 | 115 ± 10 |
| LO | 100 ± 15.8 | 210 ± 68 * |
| L12 | L3 | |
|---|---|---|
| IL-1β | 100 ± 23 | 219 ± 65 |
| IL-6 | 101 ± 6 | 114 ± 2.5 * |
| TNF-α | 100 ± 12 | 111 ± 11 |
| iNOS | 100 ± 19 | 1053 ± 362 * |
| COX-2 | 100 ± 23 | 211 ± 81 |
| NOX-1 | 100 ± 8 | 127 ± 12 * |
| NOX-4 | 100 ± 5 | 99 ± 4 |
| SOD-1 | 100 ± 9 | 72 ± 11 * |
| GSR | 100 ± 5 | 93 ± 3 |
| GPX-3 | 100 ± 6 | 89 ± 6 |
| LO | 100 ± 16 | 92 ± 10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Hedström, D.; Guerra-Menéndez, L.; Tejera-Muñoz, A.; Amor, S.; Fuente-Fernández, M.d.l.; Martín-Carro, B.; Arriazu, R.; García-Villalón, Á.L.; Granado, M. Overfeeding during Lactation in Rats is Associated with Cardiovascular Insulin Resistance in the Short-Term. Nutrients 2020, 12, 549. https://doi.org/10.3390/nu12020549
González-Hedström D, Guerra-Menéndez L, Tejera-Muñoz A, Amor S, Fuente-Fernández Mdl, Martín-Carro B, Arriazu R, García-Villalón ÁL, Granado M. Overfeeding during Lactation in Rats is Associated with Cardiovascular Insulin Resistance in the Short-Term. Nutrients. 2020; 12(2):549. https://doi.org/10.3390/nu12020549
Chicago/Turabian StyleGonzález-Hedström, Daniel, Lucía Guerra-Menéndez, Antonio Tejera-Muñoz, Sara Amor, María de la Fuente-Fernández, Beatriz Martín-Carro, Riansares Arriazu, Ángel Luis García-Villalón, and Miriam Granado. 2020. "Overfeeding during Lactation in Rats is Associated with Cardiovascular Insulin Resistance in the Short-Term" Nutrients 12, no. 2: 549. https://doi.org/10.3390/nu12020549
APA StyleGonzález-Hedström, D., Guerra-Menéndez, L., Tejera-Muñoz, A., Amor, S., Fuente-Fernández, M. d. l., Martín-Carro, B., Arriazu, R., García-Villalón, Á. L., & Granado, M. (2020). Overfeeding during Lactation in Rats is Associated with Cardiovascular Insulin Resistance in the Short-Term. Nutrients, 12(2), 549. https://doi.org/10.3390/nu12020549