Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review

 , , , , , ,
, , , , , ,
Abstract
1. Introduction
2. Materials and Methods
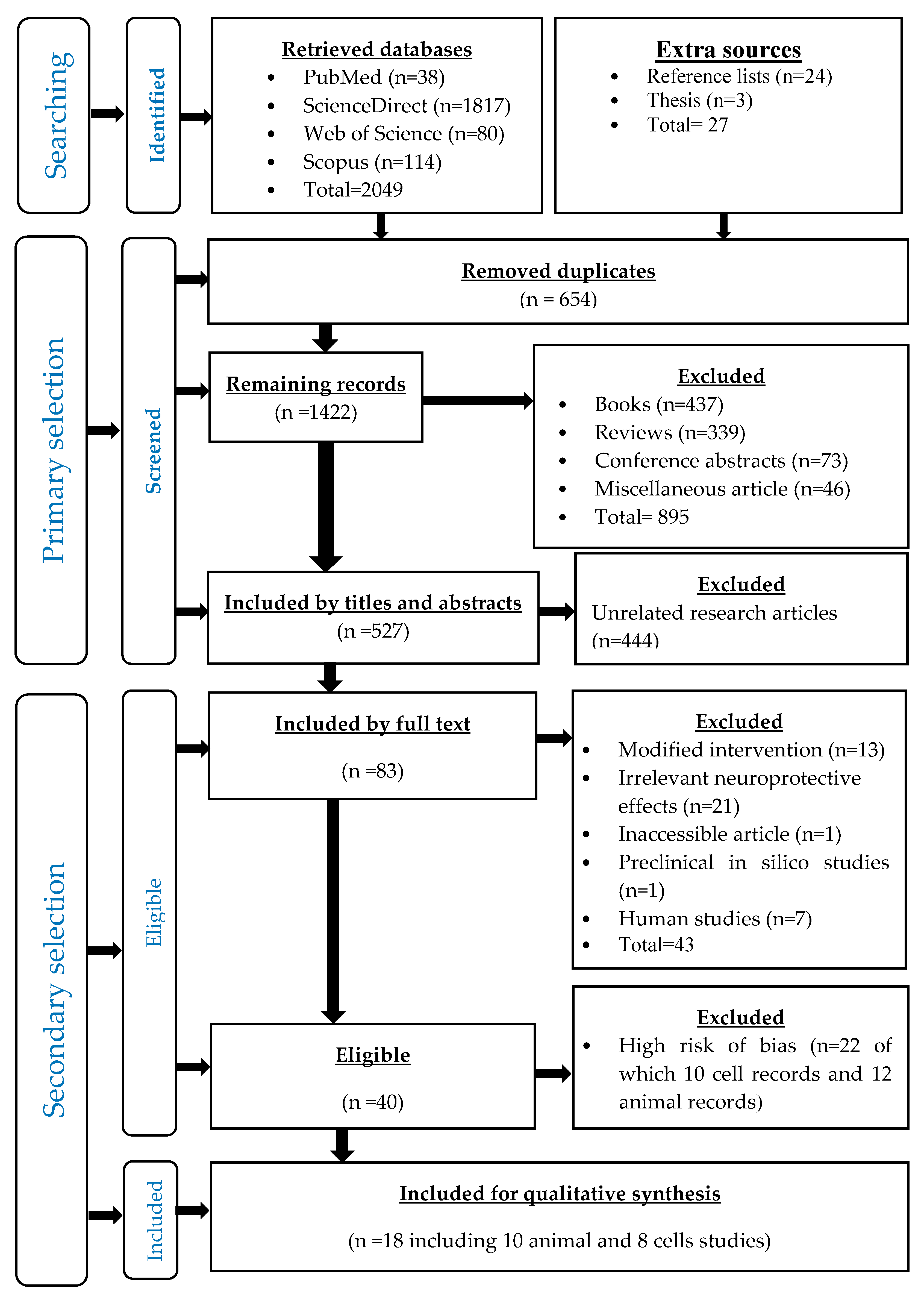
2.1. Sereach Methodology
2.2. Records Identification
2.3. Primary Selection
2.4. Secondary Selection
Eligibility Criteria
2.5. Assessment of the Risks of Bias
2.5.1. Preclinical Cell Studies
2.5.2. Preclinical Animal Studies
2.6. Data Extraction
2.6.1. Preclinical Cell Studies
2.6.2. Preclinical Animal Studies
2.7. Strategy for Data Synthesis
3. Results
3.1. Identified Records
3.2. Primary Selected Records
3.3. Secondary Selected Records
3.4. Asessment of the Risks of Bias
3.5. Outcomes
3.5.1. Preclinical Cell Studies
- Cytoprotective effects of tocotrienol-rich fraction (TRF)
- Simultaneous treatmentTreatment with either 0.1, 1 or 10 µM of TRF at the same time as hydrogen peroxide for 24 h was found to enhance the cellular viability of primary cells of the anterior striatum of foetal Wistar rats (17th–19th day of gestation) [27]. Conversely, treatment with either 0.00003%, 0.0003% or 0.003% of TRF together with Aβ42 aggregates for 24 h did not significantly enhance the cellular viability of a human neuroblastoma cell line (SH-SY5Y) [30], (Table 6).
- Pre-treatmentA five-minute pre-treatment with 250 nm TRF enhanced the cellular viability of mouse hippocampal HT4 neuronal cells line and cerebrocortical neurons of foetuses of Sprague–Dawley rats (17th day of gestation) when subsequently subjected to a 24-h challenge with glutamate neurotoxin, which exerts its effects through the direct inhibition of inducible 12-lipoxignase enzyme and the inhibition of tyrosine phosphorylation of inducible 12-lipoxignase enzyme [29]. Similarly, a five-minute pre-treatment with 200 ng/mL TRF enhanced the cellular viability and survival, and reduced lipid peroxidation in human neuroblastoma cell line (SK-N-SH) through antinecrotic and antiapoptotic effects (early and late apoptosis) against a 24-h challenge with glutamate neurotoxin [28]. However, a five-minute pre-treatment with 100, 200 or 300 ng/mL of TRF neither enhanced cellular viability nor modulated the redox status in a human astrocytes cell line (CRL-2020 cells) against a 24-h challenge with glutamate neurotoxin. Conversely, 200 and 300 ng/mL of TRF attenuated lipid peroxidation and reduced the percentages of apoptotic and necrotic cells [26], (Table 6).
- Post-treatmentA 30-min post treatment with 100, 200 and 300 ng/mL of TRF attenuated cellular apoptosis and necrosis, although only 200 ng/mL TRF could significantly recover the cellular viability of a human neuroblastoma cell line (SK-N-SH) challenged with glutamate neurotoxin for 24 h through the maintenance of the cellular membrane integrity. On the other hand, 300 mg/mL of TRF significantly attenuated lipid peroxidation in the same cells [28]. Similarly, a 30-min post-treatment with 100, 200 or 300 ng/mL of TRF neither recovered the cellular viability nor produced antioxidant activity in human astrocytes cell line (CRL-2020 cells) challenged with glutamate neurotoxin for 24 h, but was able to attenuate lipid peroxidation and exerted antiapoptotic and antinecrotic activities [26].
- Cytoprotective effects of individual tocotrienols isomers
- Simultaneous treatmentA simultaneous treatment with α-tocotrienol (α-TCT) (0.1, 1 and 10 µM), γ-TCT (1 and 10 µM) and δ-TCT (10 µM) for 24 h enhanced the cellular viability of primary cells of the anterior striatum of foetuses of Wistar rats (17th–19th day of gestation) against hydrogen peroxide-induced neurotoxicity [27]. Similarly, a simultaneous treatment with α-, γ- or δ-TCT (0.1, 1 and 10 µM) for 24 h was able to enhance the cellular viability of primary cells of the anterior striatum of fetuses of Wistar rats (17th–19th day of gestation) against parquet-induced neurotoxicity [27]. Likewise, a 24-h simultaneous treatment with α- and γ-TCT (0.1, 1 and 10 µM), as well as δ-TCT (1 and 10 µM), enhanced the cellular viability of primary cells of anterior striatum of fetuses of Wistar rats (17th–19th day of gestation) against S-nitrosocysteine-induced neurotoxicity [27]. Moreover, a similar observation was made when simultaneous treatment of α-TCT, γ-TCT and δ-TCT, and 3-morpholinosydnonimine was performed [27]. When the same cells were challenged for 48 h with L-buthionine (S,R)-sulfoximine and α-TCT, γ-TCT and δ-TCT also enhanced the cellular viability of primary cells of the anterior striatum of fetuses of Wistar rats through preventing DNA fragmentation [27]. Again, the simultaneous treatment with 10 µM of α-TCT rather than γ- or δ-TCT exerted an apoptotic effect in the cells [27], (Table 6).Furthermore, simultaneous treatment with 10 µM of α-TCT reduced the levels of hydrogen peroxide-induced ROS in human neuroblastoma cells [SH-SY5Y wild-type] [23], although the same treatment was observed to increase the levels of β-amyloid proteins in human neuroblastoma cells overexpressing the human APP695 isoform [SH-SY5Y APP] and those expressing C99 [SH-SY5Y cells] through direct stimulation of the β- and γ-secretase proteolytic enzymes. The same treatment was equally found to induce Aβ degradation in a mouse neuroblastoma cell line (N2a) through inhibiting the insulin-degrading enzyme [23]. However, the direct activation of γ-secretase was independent of the transcription of the presenilin 1 (PSEN1), presenilin 2 (PSEN2), nicastrin (NCSTN), presenilin-enhancer 2 (PSENEN) and anterior-pharynx-defective 1A (APH1A) genes [23]. Interestingly, 10 µM of α-TCT significantly reduced the total cholesterol and free cholesterol in a human neuroblastoma cell line [SH-SY5Y wild-type] [23], (Table 6).
- Pre-treatmentA five minute pre-treatment with 250 nM α-TCT enhanced cellular viability of mouse hippocampal cells line (HT4) challenged with either glutamate (for 12, 24 or 36 h) or homocysteic acid (for 2, 6, 12 or 24 h) through a direct inhibiting effect on the inducible 12-lipoxygenase enzyme, thereby maintaining neuronal growth [25], preventing the overexpression of c-Src and 2-lipoxigenase enzymes [13] and exerting an antioxidant activity (increasing the ratio of cellular content of reduced glutathione/oxidized glutathione) [13]. Similar to the mouse hippocampal cell line (HT4), the same pre-treatment enhanced the cellular viability of primary cortical neurons of fetuses of Sprague–Dawley rats (17th day of gestation) challenged with homocysteic acid for 24 h [13]. Similar effects were also observed when lower concentrations (25, 50 and 100 nM) of α-TCT were used on the primary cortical neurons of Sprague–Dawley rats challenged with either glutamate or L-homocysteic acid [25]. The effects of other neurotoxin-like L-buthionine (S,R)-sulfoximine alone or L-buthionine (S,R)-sulfoximine plus arachidonic acid were equally attenuated by 100 nM α-TCT [25]. The viability of the cerebral cortex neurons of mouse foetuses (C57BL/6) and B6.129S2-Alox15tm1Fun mice (14th day of gestation) challenged with glutamate, L-buthionine (S,R)-sulfoximine or L-buthionine (S,R)-sulfoximine + arachidonic acid for 24 h were enhanced following pre-treatment with 100 nM of α-TCT [25], (Table 6).Higher doses (0.25 µM) of α-TCT were, however, found not to be effective in attenuating the neurotoxic effects of L-buthionine (S,R)-sulfoximine plus arachidonic acid on mouse hippocampal HT4 neurons, although it was able to attenuate damage from L-buthionine (S,R)-sulfoximine alone [29]. Similarly, a five minute pre-treatment (0.25 µM of α-TCT) of mouse hippocampal neurons (HT4) challenged with L-arachidonic acid for 24 h protected the cells against toxicity, as evidenced by the inhibition of tyrosine phosphorylation of inducible 12-lipoxignase enzyme and the direct inhibition of the inducible 12-lipoxignase enzyme [29]. Additionally, higher concentrations of α-TCT (2.5 and 10 µM) enhanced cellular viability when the cells were challenged with homocysteic acid for 24 h [13]. Moreover, antioxidant activity was potentiated in these cells for up to 6 h after incubation with homocysteic acid mediated via the increase in the ratio of the reduced/oxidized glutathione ratio [13]. Prolonged incubation with the homocysteic acid (8 h) showed a complete elimination of ROS [13]. When the cells were challenged with linoleic acid under similar conditions, they were found to have been protected against the lipid peroxidation and the build-up of ROS [13]. Furthermore, the viability of murine hippocampal HT4 neuronal cells challenged with glutamate for either 30 min or 24 h was found to have increased when the cells were pretreated with 250 µM α-TCT for 10 min or 2 h. These effects were mediated by decreasing the release of arachidonic and docosahexaenoic acids from the cell membrane through inhibiting the hydrolysing effect of cytosolic phospholipase A2 on the cell membrane, which were thought to have then prevented the translocation of cytosolic phospholipase A2 to the cell membrane, its Ser505 phosphorylation or its direct inhibition [24], (Table 6).
- Post-treatmentWhen hippocampal HT4 neural cells were challenged with homocysteic acid for 24 h and subsequently treated with 250 nM of α-TCT for 8 h, the cellular viability was not improved [13]. Higher concentrations of the α-TCT (0.25, 2.5 and 10 µM), were, however, able to protect the cells from the damaging effects of homocysteic acid [13], (Table 6).
3.5.2. Preclinical Animal Studies
- Neuroprotective effects of palm oilOnly one animal study was eligible, and showed that a long-term (8 months) ad libitum feeding on diet enriched with 5 grams of palm oil could slightly enhance the cognitive performance of young (3 week old) male healthy ICR mice, as evidenced by improved spatial learning and memory abilities [33], (Table 7).
- Neuroprotective effects of tocotrienol-rich fraction (TRF)
- Healthy animal modelsA single oral daily dose of 100 mg/kg of TRF for 10 weeks exerted a slight increase in the cognitive function of healthy male Wistar rats without inducing an inflammatory effect (normal levels of α-TNF, IL-1β, p56 subunit of NFκβ), apoptosis (normal level of caspase-3) or an alteration in the cholinergic function (normal levels of cholinesterase). Moreover, the redox status was also found to be maintained within healthy limits (normal levels of superoxide dismutase enzyme, catalase enzyme, nitrites and malondialdehyde) in the cerebral cortex and hippocampus of these rats [32]. Conversely, the cognitive function of male progeny of Sprague–Dawley rats was improved significantly when given ad libitum diet-admixed TRF during gestation, lactation and post weaning for 8 weeks [34]. The α-tocotrienol isomer of TRF was found to be highest in the plasma and brain of healthy rats as compared to the other isomers of TRF. Similarly, long-term oral single daily doses of 200 mg/kg TRF in young (3 months) male Wistar rats for 8 months significantly enhanced cognitive function and antioxidant activity (increased activity of superoxide dismutase, catalase and glutathione peroxidase) and decreased DNA damage (higher levels of plasma DNA) [38]. However, the oral single daily dose of 200 mg/kg of TRF for three months did not produce similar results, nor did it alter the serum lipid peroxidation of the young (3 months) healthy male Sprague–Dawley rats [39]. When a similarly high dose of TRF (200 mg/kg) was administered for 3 months in elderly (21 months) healthy rats, the cognitive function, plasma lipid peroxidation, and plasma antioxidant activity were significantly improved, while DNA damage was attenuated [39].A five-week single oral daily dose (200 mg/kg) of TRF was also shown to improve morphological features in parts of the brain of rats. Accordingly, it induced significant proliferation of granular cells in the dentate gyrus in the hippocampus of chronic stressed or unstressed healthy male Sprague–Dawley rats [37], (Table 7).
- Disease-induced animal modelsUsing a diabetic model, a single oral daily dose of 50 or 100 mg/kg of α-tocotrienol for 21 days was found to significantly (dose-dependent) enhance cognitive function, attenuate brain lipid peroxidation (decreased level of malondialdehyde (MDA)) and enhance brain antioxidant activity (increased levels of reduced glutathione (GSH), superoxide dismutase (SOD) and catalase (CAT)). Similarly, the brain cholinergic function was slightly enhanced (non-significant dose-dependent reduction in the level of brain acetyl cholinesterase enzyme) [36]. When doses of 25, 50 or 100 mg/kg of TRF were given for 10 weeks, the cognitive function was enhanced in these rats, as well as the cerebrocortical and hippocampal cholinergic function, lipid peroxidation (reduced level of MDA), antioxidant activity (increased SOD and CAT), inflammation (reduced levels of TNF-α, IL-1β and p56 subunit of NFκβ) and antiapoptotic effects (reduced level of cerebrocortical and hippocampal levels of caspase-3) [32], (Table 7).
- Transgenic animal modelsUsing a double transgenic Alzheimer’s disease (AβPP/PS1) animal model, a long-term single oral daily dose of 60 mg/kg of TRF for 10 months in male mice (5 months of age) significantly enhanced recognition abilities and reduced the deposition of β-amyloid proteins in the cortex and hippocampus (soluble and insoluble Aβ isoforms: Aβ 40, Aβ 42 and Aβ oligomer) [30]. Similarly, the same single oral daily dose of TRF (60 mg/kg) for the same duration (10 months) in AβPP/PS1 male mice (5 months of age) slightly enhanced cognitive function, and metabolomics analyses indicated an alteration in 90 putative metabolites that are involved in several metabolic Alzheimer’s disease pathways [31]. In addition, a single oral daily dose of 200 mg/kg of TRF for 6 months in AβPP/PS1 male mice (aged 9 months) upregulated the genes that are responsible for neuroprotective effects, such as Slc24a2 (solute carrier family 24 [sodium/potassium/calcium exchanger]), exo1 (exonuclease 1), and Enox1 (ecto-NOX disulfide-thiol exchanger 1), and downregulated the genes responsible for the pathology of AD, such as Pla2g4a (phospholipase A2, group IVA [cytosolic, calcium-dependent]), Tfap2b (transcription factor AP-2 beta) [35], (Table 7).
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Knopman, D.S.; Mielke, M.M.; Vemuri, P.; Lowe, V.; Senjem, M.L.; Gunter, J.L.; Reyes, D.; et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain 2015, 138, 3747–3759. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nature Med. 2004, 10, S10. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Natteru, P.A.; Selvakumar, G.P.; Saeed, D.; Zahoor, H.; Zaheer, S.; Iyer, S.S.; Zaheer, A. Neuroinflammation Induces Neurodegeneration. J. Neurol. Neurosurg. Spine 2016, 1, 1003. [Google Scholar] [PubMed]
- Chakrabarti, S.; Mohanakumar, K.P. Aging and Neurodegeneration: A Tangle of Models and Mechanisms. Aging Dis. 2016, 7, 111–113. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Zhuang, W.; Zhou, S.; Wang, X. Plant-derived neuroprotective agents in Parkinson’s disease. Am. J. Transl. Res. 2015, 7, 1189–1202. [Google Scholar]
- González-Fuentes, J.; Selva, J.; Moya, C.; Castro-Vázquez, L.; Lozano, M.V.; Marcos, P.; Plaza-Oliver, M.; Rodríguez-Robledo, V.; Santander-Ortega, M.J.; Villaseca-González, N.; et al. Neuroprotective Natural Molecules, From Food to Brain. Front. Neurosci. 2018, 12, 721. [Google Scholar] [CrossRef]
- Miller, D.B.; O’Callaghan, J.P. Biomarkers of Parkinson’s disease: Present and future. Metabolism 2015, 64, S40–S46. [Google Scholar] [CrossRef]
- Essa, M.M.; Vijayan, R.K.; Castellano-Gonzalez, G.; Memon, M.A.; Braidy, N.; Guillemin, G.J. Neuroprotective effect of natural products against Alzheimer’s disease. Neurochem. Res. 2012, 37, 1829–1842. [Google Scholar] [CrossRef]
- Ismail, S.R.; Maarof, S.K.; Ali, S.S.; Ali, A. Systematic review of palm oil consumption and the risk of cardiovascular disease. PLoS ONE 2018, 13, e0193533. [Google Scholar] [CrossRef] [PubMed]
- Sundram, K.; Sambanthamurthi, R.; Tan, Y.-A. Palm fruit chemistry and nutrition. Asia Pac. J. Clin. Nutr. 2003, 12, 355–362. [Google Scholar] [PubMed]
- Khanna, S.; Roy, S.; Parinandi, N.L.; Maurer, M.; Sen, C.K. Characterization of the potent neuroprotective properties of the natural vitamin E α-tocotrienol. J. Neurochem. 2006, 98, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Mitra, A. Health effects of palm oil. J. Hum. Ecol. 2009, 26, 197–203. [Google Scholar] [CrossRef]
- Aggarwal, V.; Kashyap, D.; Sak, K.; Tuli, H.S.; Jain, A.; Chaudhary, A.; Garg, V.K.; Sethi, G.; Yerer, M.B. Molecular mechanisms of action of tocotrienols in cancer: Recent trends and advancements. Int. J. Mol. Sci. 2019, 20, 656. [Google Scholar] [CrossRef]
- Ahsan, H.; Ahad, A.; Iqbal, J.; Siddiqui, W.A. Pharmacological potential of tocotrienols: A review. Nutr. Metab. 2014, 11, 52. [Google Scholar] [CrossRef]
- Obahiagbon, F.I. A review: Aspects of the African oil Palm (Elaeis guineesis jacq.) and the implications of its bioactives in human health. Am. J. Biochem. Mol. Biol. 2012, 2, 106–119. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Sundaram, C.; Prasad, S.; Kannappan, R. Tocotrienols, the vitamin E of the 21st century: Its potential against cancer and other chronic diseases. Biochem. Pharmacol. 2010, 80, 1613–1631. [Google Scholar] [CrossRef]
- Higgins, J.P.; Green, S. Cochrane Handbook for Systematic Reviews of Interventions; John Wiley & Sons: Chichester, UK, 2011; Volume 4. [Google Scholar]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. OHAT Systematic Review. Available online: https://ntp.niehs.nih.gov/pubhealth/hat/review/index-2.html (accessed on 2 December 2019).
- Hooijmans, C.R.; Rovers, M.M.; De Vries, R.B.; Leenaars, M.; Ritskes-Hoitinga, M.; Langendam, M.W. SYRCLE’s risk of bias tool for animal studies. BMC Med. Res. Methodol. 2014, 14, 43. [Google Scholar] [CrossRef]
- Grimm, M.; Regner, L.; Mett, J.; Stahlmann, C.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.; et al. Tocotrienol affects oxidative stress, cholesterol homeostasis and the amyloidogenic pathway in neuroblastoma cells: Consequences for Alzheimer’s disease. Int. J. Mol. Sci. 2016, 17, 1809. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Parinandi, N.L.; Kotha, S.R.; Roy, S.; Rink, C.; Bibus, D.; Sen, C.K. Nanomolar vitamin E α-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection. J. Neurochem. 2010, 112, 1249–1260. [Google Scholar] [CrossRef]
- Khanna, S.; Roy, S.; Ryu, H.; Bahadduri, P.; Swaan, P.W.; Ratan, R.R.; Sen, C.K. Molecular basis of vitamin E action tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. 2003, 278, 43508–43515. [Google Scholar] [CrossRef] [PubMed]
- Musa, I.; Khaza’ai, H.; Abdul Mutalib, M.S.; Yusuf, F.; Sanusi, J.; Chang, S.K. Effects of oil palm tocotrienol rich fraction on the viability and morphology of astrocytes injured with glutamate. Food Biosci. 2017, 20, 168–177. [Google Scholar] [CrossRef]
- Osakada, F.; Hashino, A.; Kume, T.; Katsuki, H.; Kaneko, S.; Akaike, A. alpha-Tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons. Neuropharmacology 2004, 47, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, T.R.; Khazat’ai, H.; Vidyadaran, S.; Abd Mutalib, M.S.; Ramachandran, V.; Hamdan, Y. Cytoprotective Effect of Tocotrienol-Rich Fraction and alpha-Tocopherol Vitamin E Isoforms Against Glutamate-Induced Cell Death in Neuronal Cells. Int. J. Vitam. Nutr. Res. 2014, 84, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Roy, S.; Slivka, A.; Craft, T.K.; Chaki, S.; Rink, C.; Notestine, M.A.; DeVries, A.C.; Parinandi, N.L.; Sen, C.K. Neuroprotective properties of the natural vitamin E α-tocotrienol. Stroke 2005, 36, e144–e152. [Google Scholar] [CrossRef]
- Ibrahim, N.F.; Yanagisawa, D.; Durani, L.W.; Hamezah, H.S.; Damanhuri, H.A.; Wan Ngah, W.Z.; Tsuji, M.; Kiuchi, Y.; Ono, K.; Tooyama, I.; et al. Tocotrienol-Rich Fraction Modulates Amyloid Pathology and Improves Cognitive Function in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2017, 55, 597–612. [Google Scholar] [CrossRef]
- Durani, L.W.; Hamezah, H.S.; Ibrahim, N.F.; Yanagisawa, D.; Nasaruddin, M.L.; Mori, M.; Azizan, K.A.; Damanhuri, H.A.; Makpol, S.; Wan Ngah, W.Z.; et al. Tocotrienol-Rich Fraction of Palm Oil Improves Behavioral Impairments and Regulates Metabolic Pathways in AβPP/PS1 Mice. J. Alzheimer’s Dis. 2018, 64, 249–267. [Google Scholar] [CrossRef]
- Kuhad, A.; Bishnoi, M.; Tiwari, V.; Chopra, K. Suppression of NF-κβ signaling pathway by tocotrienol can prevent diabetes associated cognitive deficits. Pharmacol. Biochem. Behav. 2009, 92, 251–259. [Google Scholar] [CrossRef]
- Lim, S.Y.; Suzuki, H. Effect of dietary docosahexaenoic acid and phosphatidylcholine on maze behavior and fatty acid composition of plasma and brain lipids in mice. Int. J. Vitam. Nutr. Res. 2000, 70, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Nagapan, G.; Goh, Y.M.; Razak, I.S.A.; Nesaretnam, K.; Ebrahimi, M. The effects of prenatal and early postnatal tocotrienol-rich fraction supplementation on cognitive function development in male offspring rats. BMC Neurosci. 2013, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Nasri, W.; Nurzulaikha, W.; Makpol, S.; Mazlan, M.; Tooyama, I.; Ngah, W.Z.W.; Zurinah, W.; Damanhuri, H.A. Tocotrienol Rich Fraction Supplementation Modulate Brain Hippocampal Gene Expression in APPswe/PS1dE9 Alzheimer’s Disease Mouse Model. J. Alzheimer’s Dis. 2018, 70, 1–16. [Google Scholar] [CrossRef]
- Tiwari, V.; Kuhad, A.; Bishnoi, M.; Chopra, K. Chronic treatment with tocotrienol, an isoform of vitamin E, prevents intracerebroventricular streptozotocin-induced cognitive impairment and oxidative–nitrosative stress in rats. Pharmacol. Biochem. Behav. 2009, 93, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Talip, S.B.; Asari, M.A.; Khan, A.A.; Sirajudeen, K.N.S. Evaluation of the protective effects of tocotrienol-rich fraction from palm oil on the dentate gyrus following chronic restraint stress in rats. Braz. J. Pharm. Sci. 2013, 49, 373–380. [Google Scholar] [CrossRef]
- Taridi, N.M.; Yahaya, M.F.; Teoh, S.L.; Latiff, A.A.; Ngah, W.Z.W.; Das, S.; Mazlan, M. Tocotrienol rich fraction (TRF) supplementation protects against oxidative DNA damage and improves cognitive functions in Wistar rats. Clin. Ther. 2011, 162, 93–98. [Google Scholar]
- Taridi, N.M.; Rani, N.A.; Latiff, A.A.; Ngah, W.Z.W.; Mazlan, M. Tocotrienol rich fraction reverses age-related deficits in spatial learning and memory in aged rats. Lipids 2014, 49, 855–869. [Google Scholar] [CrossRef]
- Mancini, A.; Imperlini, E.; Nigro, E.; Montagnese, C.; Daniele, A.; Orrù, S.; Buono, P. Biological and nutritional properties of palm oil and palmitic acid: Effects on health. Molecules 2015, 20, 17339–17361. [Google Scholar] [CrossRef]
- Choudhary, M.; Grover, K. Palm (Elaeis guineensis Jacq.) Oil. In Fruit Oils: Chemistry and Functionality; Springer: Cham, Switzerland, 2019; pp. 789–802. [Google Scholar]
- Ranjan, S.; Passi, S.J.; Singh, S.N. Impact of Crude Palm Oil Fortified Cookies Supplementation on Anthropometry, Vitamin A and Hematological Status of School Children in India. Int. J. Vitam. Nutr. Res. 2019, 89, 321–330. [Google Scholar] [CrossRef]
- Shahidi, F.; De Camargo, A.C. Tocopherols and tocotrienols in common and emerging dietary sources: Occurrence, applications, and health benefits. Int. J. Mol. Sci. 2016, 17, 1745. [Google Scholar] [CrossRef]
- Gibon, V.; De Greyt, W.; Kellens, M. Palm oil refining. Eur. J. Lipid Sci. Technol. 2007, 109, 315–335. [Google Scholar] [CrossRef]
- Selvaraju, T.R.; Khaza’ai, H.; Vidyadaran, S.; Mutalib, M.S.A.; Vasudevan, R. The neuroprotective effects of tocotrienol rich fraction and alpha tocopherol against glutamate injury in astrocytes. Bosn. J. Basic Med. Sci. 2014, 14, 195–204. [Google Scholar] [CrossRef]
- Kamal-Eldin, A.; Appelqvist, L.A. The chemistry and antioxidant properties of tocopherols and tocotrienols. Lipids 1996, 31, 671–701. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Chin, X.W.D.; Schrader, C.; Eckert, G.P.; Rimbach, G. Do tocotrienols have potential as neuroprotective dietary factors? Ageing Res. Rev. 2012, 11, 163–180. [Google Scholar] [CrossRef] [PubMed]
- Haam, J.; Yakel, J.L. Cholinergic modulation of the hippocampal region and memory function. J. Neurochem. 2017, 142, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Bohnen, N.I.; Grothe, M.J.; Ray, N.J.; Müller, M.L.; Teipel, S.J. Recent advances in cholinergic imaging and cognitive decline—Revisiting the cholinergic hypothesis of dementia. Curr. Geriatr. Rep. 2018, 7, 1–11. [Google Scholar] [CrossRef]
- de Wit, N.M.; Vanmol, J.; Kamermans, A.; Hendriks, J.J.A.; de Vries, H.E. Inflammation at the blood-brain barrier: The role of liver X receptors. Neurobiol. Dis. 2017, 107, 57–65. [Google Scholar] [CrossRef]
- Association, A.D. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes—2019. Diabetes Care 2019, 42, S13–S28. [Google Scholar] [CrossRef]
- Glovaci, D.; Fan, W.; Wong, N.D. Epidemiology of diabetes mellitus and cardiovascular disease. Curr. Cardiol. Rep. 2019, 21, 21. [Google Scholar] [CrossRef]
- Morsi, M.; Kobeissy, F.; Magdeldin, S.; Maher, A.; Aboelmagd, O.; Johar, D.; Bernstein, L. A shared comparison of diabetes mellitus and neurodegenerative disorders. J. Cell. Biochem. 2019, 120, 14318–14325. [Google Scholar] [CrossRef]
- Nasrolahi, A.; Mahmoudi, J.; Noori-Zadeh, A.; Haghani, K.; Bakhtiyari, S.; Darabi, S. Shared Pathological Mechanisms Between Diabetes Mellitus and Neurodegenerative Diseases. Curr. Pharmacol. Rep. 2019, 5, 219–231. [Google Scholar] [CrossRef]
- Manocha, G.D.; Ghatak, A.; Puig, K.L.; Kraner, S.D.; Norris, C.M.; Combs, C.K. NFATc2 Modulates Microglial Activation in the AβPP/PS1 Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Jia, L.; Wang, Q.; Zhao, L.; Jin, H.; Li, T.; Quan, M.; Xu, L.; Li, B.; Li, Y.; et al. Identification of a novel PSEN1 Gly111Val missense mutation in a Chinese pedigree with early-onset Alzheimer’s disease. Neurobiol. Aging 2019, 85, e1–e155. [Google Scholar] [CrossRef] [PubMed]
- Ruberg, S.J. Dose response studies II. analysis and interpretation. J. Biopharm. Stat. 1995, 5, 15–42. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Muller, W.E.; Eckert, G.P. Tocotrienols: Constitutional effects in aging and disease. J. Nutr. 2005, 135, 151–154. [Google Scholar] [CrossRef]
- Loera-Valencia, R.; Goikolea, J.; Parrado-Fernandez, C.; Merino-Serrais, P.; Maioli, S. Alterations in cholesterol metabolism as a risk factor for developing Alzheimer’s disease: Potential novel targets for treatment. J. Steroid Biochem. Mol. Biol. 2019, 190, 104–114. [Google Scholar] [CrossRef]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflüg. Arch. Eur. J. Physiol. 2010, 460, 525–542. [Google Scholar] [CrossRef]
- Mikami, Y.; Yamazawa, T. Chlorogenic acid, a polyphenol in coffee, protects neurons against glutamate neurotoxicity. Life Sci. 2015, 139, 69–74. [Google Scholar] [CrossRef]
- Joshi, Y.B.; Giannopoulos, P.F.; Praticò, D. The 12/15-lipoxygenase as an emerging therapeutic target for Alzheimer’s disease. Trends Pharmacol. Sci. 2015, 36, 181–186. [Google Scholar] [CrossRef]


{kind=link}
| Inclusion Criteria | Exclusion Criteria | ||
|---|---|---|---|
| Human disease model | ●Neurotoxicity | ●irrelevant | |
| ●neurodegeneration | |||
| ●neuro-apoptosis | |||
| ●neuro-oxidative stress | |||
| ●neuro-inflammation | |||
| Population | ●normal or transgenic neuronal cell line | ●neuronal or neuroglial cell line derived from an organism with a neurological hereditary disease ●primary neuronal or neuroglial cells derived from an organism with a neurological hereditary disease; ●in silico models | |
| ●normal or transgenic neuroglial cell line | |||
| ●primary neuronal or neuroglial cells | |||
| ●neuronal slices | |||
| Interventions | ●palm oil and palm oil bioactives (tocotrienol-rich fraction, polyphenol-rich fraction, individual tocotrienols or β-carotenes) | ●pure α-, β-, δ- or γ-tocopherols | |
| ●any duration of intervention | |||
| ●any dose of intervention | |||
| ●any timing of intervention (simultaneous treatment: incubation of the intervention and the neurotoxin at the same time) | |||
| ●pre-treatment: cells treated before being challenged with the neurotoxin | |||
| ●post-treatment: cells treated after being challenged with the neurotoxin | |||
| Comparators | ●inert vehicles (water, ethanol, normal saline, phosphate buffer, DMSO or media) | ●comparator with different experimental conditions or exposure compared with the intervention groups | |
| ●comparators subjected to identical experimental conditions and exposure as the intervention groups | ●tocopherols | ||
| ●the same vehicle used to dissolve the intervention | ●a vehicle rather than that used to dissolve the intervention | ||
| Study Design | ●Preclinical in vitro experiments with mono-level or multi-level intervention (pre-, post- and simultaneous exposure) with an appropriate comparator | ●lack of an appropriate comparator | |
| Outcomes | Primary | ●Cellular viability | ●irrelevant |
| ●inflammation | |||
| ●apoptosis; | |||
| ●oxidative stress (lipid peroxidation and antioxidant activity) | |||
| Secondary | ●Cytomorphological and molecular changes | ●irrelevant | |
| Others (article type) | ●published research articles ●full papers in proceedings ●unpublished theses | ●research articles in predatory journals according to Beall’s list | |
| ●Published theses | |||
| ●inaccessible research articles | |||
| ●high-risk biased studies | |||
| Inclusion Criteria | Exclusion Criteria | ||
|---|---|---|---|
| Human disease model | ●neurodegenerative disorders | ●irrelevant | |
| ●neuroinflammation | |||
| ●neurotoxicity | |||
| ●neuro-injury | |||
| ●neuro-oxidative stress | |||
| Population | ●pup, young, young adult, adult or elderly animals | ●none | |
| ●male or female animals | |||
| ●rats or mice | |||
| ●strains of rats or mice | |||
| ●healthy, disease-induced animals or transgenic disease animals | |||
| Interventions | ●palm oil, or its bioactives (e.g., palmitic acid, tocotrienol-rich fraction, polyphenol-rich fraction, individual tocotrienols or β-carotenes) | ●pure α-, β-, δ- and γ-tocopherols ●blended palm oil with other oils ●palm oil combined with other foods ●content-modified palm oil (e.g., vitamin-E-stripped) ●palm oil bioactives extracted from parts of palm tree other than palm fruit | |
| ●any dose of intervention | |||
| ●any timing of intervention | |||
| ●any frequency of intervention (e.g., once or twice… etc. per a day) | |||
| ●any duration of intervention | |||
| ●any technique of intervention administration (admixed with diet, suspended in water, oral via gastric gavage or parenteral) | |||
| Comparators | ●palm oil; inert vehicles (water, normal saline, or tweens) ●comparators subjected to identical experimental conditions and exposure similar to those of the intervention groups ●the same vehicle used to dissolve the intervention | ●blended palm oil with other oils | |
| ●palm oil combined with other foods; content-modified palm oil (e.g., vitamin-E-stripped) | |||
| ●comparator with different experimental conditions or exposure different from the intervention groups | |||
| ●a vehicle rather than that used to dissolve the intervention | |||
| Study Design | ●acute, sub-acute or chronic preclinical animal studies containing at least mono-level or multi-level dosing of oral dietary, oral gavage or parenteral intervention with an appropriate comparator | ●lack an appropriate comparator | |
| Outcomes | Primary | ●cognitive function | ●irrelevant |
| ●locomotor function | |||
| ●healing after neuro-injury | |||
| ●neuroinflammation | |||
| ●apoptosis | |||
| ●oxidative stress (lipid peroxidation and antioxidant activity) | |||
| Secondary | ●structural and molecular changes | ●irrelevant | |
| Others (article type) | ●published research articles ●full papers in proceedings ●Unpublished theses | ●research articles in predator journals according to Beall’s list | |
| ●Published theses | |||
| ●inaccessible research articles | |||
| ● high-risk biased studies | |||
| Keywords | PubMed | Web of Science | Science Direct | Scopus |
|---|---|---|---|---|
| Palm oil and nervous system | 17 | 9 | 499 | 12 |
| Palm oil and brain | 15 | 64 | 1071 | 83 |
| Palm oil and neurodegenerative diseases | 4 | 5 | 135 | 7 |
| Palm oil and cognition | 2 | 2 | 112 | 12 |
| Total | 38 | 80 | 1817 | 114 |
| Studies | Selection Bias | Performance Bias | Detection Bias | Attrition Bias | Reporting Bias | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Randomization and Concurrent Control Group | Allocation Concealment | Identical Vehicle | Blinding | Accuracy of Exposure Characterization | Consistent Exposure Administration | Blinding | Incomplete Outcome Data | Selective Outcome Reporting | Other sources of Bias | |
| [23] | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [24] | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [13] | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [25] | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [26] | PL | NR | PL | NR | DL | DL | NR | NR | PL | PL |
| [27] | PL | PL | DL | DL | DL | DL | PL | NR | DL | PL |
| [28] | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [29] * | PL | NR | DL | NR | PL | DL | NR | NR | PL | PL |
| [30] | PL | NR | PL | NR | PL | DL | NR | NR | PL | PL |
| Studies | Selection Bias | Performance Bias | Detection Bias | Attrition Bias | Reporting Bias | Others | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Random Sequence Generation | Baseline Characteristics | Allocation Concealment | Random Housing | Blinding | Random Outcome Assessment | Blinding | Incomplete Outcome Data | Selective Outcome Reporting | Other sources of Bias | |
| [31] | No | Yes | U | No | No | Yes | No | Yes | Yes | Yes |
| [30] | No | Yes | U | No | No | No | No | Yes | Yes | U |
| [32] | Yes | Yes | U | No | No | Yes | No | No | Yes | U |
| [33] | Yes | Yes | U | No | No | No | No | Yes | Yes | U |
| [34] | Yes | Yes | U | No | No | No | No | Yes | Yes | U |
| [35] | Yes | Yes | U | No | No | Yes | No | No | Yes | Yes |
| [36] | Yes | Yes | U | Yes | No | No | No | No | yes | U |
| [37] | Yes | Yes | U | No | No | No | Yes | No | Yes | U |
| [38] | Yes | Yes | U | No | No | Yes | No | Yes | Yes | U |
| [39] | Yes | Yes | U | No | No | Yes | No | No | Yes | U |
| Reference | Study Design, Human Disease Modelled and Population | Intervention | Comparator | Outcomes | |
|---|---|---|---|---|---|
| Primary | Secondary | ||||
| [25] | ●Glutamate induced-neurotoxicity model for 12, 24 or 36 h ●Mouse hippocampal HT4 cells line | ●5-min pre-treatment with 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant time-dependent enhancement of cellular viability | ●Direct inhibition of inducible 12-lipoxygenase enzyme. ●Morphological changes indicated the prevention of neurodegeneration with the maintenance of neuronal growth. |
| [25] | ●Glutamate or L-homocysteic acid neurotoxicity for 24 h ●Immature primary cortical neurons of Sprague-Dawley rats (17th day of gestation) | ●5-min pre-treatment with 25, 50, 100 and 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement of cellular viability | |
| [25] | ●L-buthionine (S,R)-sulfoximine or L-buthionine (S,R)-sulfoximine +arachidonic acid neurotoxicity for 24 h using immature primary cortical neurons of Sprague-Dawley rats (17th day of gestation) | ●5-min pre-treatment with 100 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement cellular viability | |
| [25] | ●L-buthionine (S,R)-sulfoximine neurotoxicity for 24 h using immature primary cortical neurons of Sprague-Dawley rats (17th day of gestation) | ●5-min pre-treatment with 100 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement of cellular viability, but loss of the cellular reduced glutathione | |
| [25] | ●Glutamate, L-buthionine (S,R)-sulfoximine or L-buthionine (S,R)-sulfoximine + arachidonic acid neurotoxicity for 24 h using cerebral cortex neurons of mouse fetuses (C57BL/6) mice, (14th day of gestation) | ●5-min pre-treatment with 100 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement of cellular viability | |
| [25] | ●Glutamate, L-buthionine (S,R)-sulfoximine or L-buthionine (S,R)-sulfoximine + arachidonic acid for 24 h using cerebral cortex neurons of the fetuses of B6.129S2-Alox15tm1Fun mice | ●5-min pre-treatment with 100 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement of cellular viability | |
| [27] | ●Hydrogen peroxide neurotoxicity for 24 h using primary cells of anterior striatum of fetal Wistar rats (17th–19th day of gestation). | ●Simultaneous treatment with 0.1, 1 or 10 µM of TRF in 0.1% DMSO (TRF: 90% pure contains 14.5. mg α-TCT, 2.5 mg β-TCT, 26 mg γ-TCT and 7.2 δ-TCT) | ●0.1% DMSO | ●A significant enhancement of cellular viability. | |
| [27] | ●Hydrogen peroxide neurotoxicity for 24 h using primary cells of anterior striatum of fetal Wistar rats (17th–19th day of gestation) | ●Simultaneous treatment with 0.1, 1 or 10 µM of either α-, γ- or δ-TCT in 0.1% DMSO | ●0.1% DMSO | ●α-TCT [0.1, 1 and 10 µM], γ-TCT [1 and 10 µM] and δ-TCT [10 µM] significantly enhanced cellular viability | |
| [27] | ●Parquet neurotoxicity with for 24 h using primary cells of anterior striatum of foetal Wistar rats on the 17th–19th day of gestation | ●Simultaneous treatment with 0.1, 1 and 10 µM of either α-, γ- or δ-TCT in 0.1% DMSO | ●0.1% DMSO | ●α-, γ- or δ-TCT [0.1, 1 and 10 µM] significantly enhanced cellular viability | |
| [27] | ●S-nitrosocysteine neurotoxicity for 24 h using primary cells of anterior striatum of foetal Wistar rats on the 17th–19th day of gestation | ●Simultaneous treatment with 0.1, 1 and 10 µM of either α-, γ- or δ-TCT in 0.1% DMSO | ●0.1% DMSO | ●α- and γ-TCT [0.1, 1 and 10 µM] as well as δ-TCT [1 and 10 µM] significantly enhanced cellular viability. | |
| [27] | ●3-morpholinosydnonimine neurotoxicity for 24 h using primary cells of anterior striatum of foetal Wistar rats on the 17th–19th day of gestation | ●Simultaneous treatment with 0.1, 1 and 10 µM of either α-, γ- or δ-TCT in 0.1% DMSO | ●0.1% DMSO | ●α-TCT [0.1, 1 and 10 µM], γ-TCT [1 and 10 µM] and δ-TCT [1 and 10 µM] significantly enhanced cellular viability. | |
| [27] | ●L-buthionine (S,R)-sulfoximine neurotoxicity for 48 h using primary cells of anterior striatum of foetal Wistar rats on the 17th–19th day of gestation | ●Simultaneous treatment with 0.01, 0.1 and 1 µM of either α-, γ- or δ-TCT in 0.1% DMSO | ●0.1% DMSO | ●α-TCT [0.1 and 1 µM], γ-TCT [1 µM] and δ-TCT [1 µM] significantly enhanced cellular viability. α-, γ- and δ-TCT [1 µM] exerted antiapoptotic effects, however, the antiapoptotic effect of α-TCT was superior to that of either γ- or δ-TCT | ●Antiapoptotic effect involved the prevention of DNA fragmentation. |
| [27] | ●Staurosporine neurotoxicity for 24 h using primary cells of anterior striatum of foetal Wistar rats on the 17th–19th day of gestation. | ●Simultaneous treatment with 10 µM of either α-, γ- or δ-TCT 0.1% DMSO | ●0.1% DMSO | ●Only 10 µM of α-TCT exerted a significant antiapoptotic effect, while γ- or δ-TCT field to exert a significant antiapoptotic effect. | ●Antiapoptotic effect involved a significant prevention of DNA fragmentation. |
| [29] | ●Glutamate neurotoxicity for 24 h using mouse Hippocampal HT4 Neurons | ●5-min pre-treatment with 250 nm of TRF in 1% ethanol (TRF: 90% pure contains 14.5. mg α-TCT, 2.5 mg β-TCT, 26 mg γ-TCT and 7.2 δ-TCT) | ●1% ethanol | ●A significant enhancement of cellular viability | ●Inhibiting the tyrosine phosphorylation of inducible 12-lipoxignase enzyme and direct inhibition of inducible 12-lipoxignase enzyme |
| [29] | ●Glutamate neurotoxicity for 24 h using cerebral cortex neurons of foetuses of Sprague-Dawley rats, (17th day of gestation) | ●5-min pre-treatment with 250 nm of TRF in 1% ethanol (TRF: 90% pure contains 14.5. mg α-TCT, 2.5 mg β-TCT, 26 mg γ-TCT and 7.2 δ-TCT) | ●1% ethanol | ●A significant enhancement of cellular viability | ●Inhibiting the tyrosine phosphorylation of inducible 12-lipoxignase enzyme and direct inhibition of inducible 12-lipoxignase enzyme |
| [29] | ●L-buthionine (S,R)-sulfoximine neurotoxicity for 24 h using mouse Hippocampal HT4 Neurons | ●5-min pre-treatment with 0.25 µM of α-TCT in 1% ethanol | ●1% ethanol | ●A relative (nonsignificant) enhancement of cellular viability | |
| [29] | ●L-buthionine (S,R)-sulfoximine + arachidonic acid neurotoxicity for 24 h using mouse Hippocampal HT4 Neurons | ●5-min pre-treatment with 0.25 µM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant loss of cellular viability | |
| [29] | ●L- arachidonic acid neurotoxicity for 24 h using mouse Hippocampal HT4 Neurons | ●5-min pre-treatment with 0.25 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Inhibiting tyrosine phosphorylation of inducible 12-lipoxignase enzyme and direct inhibition of inducible 12-lipoxignase enzyme | |
| [13] | ●Homocysteic acid neurotoxicity for 24 h using mouse hippocampal HT4 neural cells | ●5 min pre- or 8 h post-treatment with 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●Pre-treatment significantly enhanced cellular viability, while post-treatment failed to enhance cellular viability | |
| [13] | ●Homocysteic acid neurotoxicity for 24 h using mouse hippocampal HT4 neural cells | ●5-min pre- or 8 h post-treatment with 0.25, 2.5 and 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Pre- and post-treatment significantly enhanced cellular viability. | |
| [13] | ●Homocysteic acid neurotoxicity for 2 or 6 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●Provided a significant antioxidant activity through enhancing the ratio of cellular levels of reduced glutathione/oxidized glutathione | |
| [13] | ●Homocysteic acid neurotoxicity for 8 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 2.5 and 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Blue fluorescence imaging indicated a completely elimination of ROS | |
| [13] | ●Linoleic acid neurotoxicity for 4 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 0.25, 1, 2.5 and 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●1, 2.5 and 10 µM of α-TCT significantly attenuated lipid peroxidation | ●Fluorescence imaging indicated the attenuation of the build-up of ROS |
| [13] | ●Linoleic acid neurotoxicity for 24 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 0.25, 1, 2.5 and 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Significantly enhanced cellular viability [2.5 and 10 µM] | |
| [13] | ●Homocysteic acid neurotoxicity for 12 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●A significant enhancement of cellular viability | ●Prevented overexpression of c-Src and 2-lipoxigenase |
| [13] | ●Homocysteic acid neurotoxicity for 6 h using mouse hippocampal HT4 neural cells | ●5-min pre-treatment with 0.25, 1, 2.5 and 10 µM of α-TCT 1% ethanol | ●1% ethanol | ●Provided a significant antioxidant activity [2.5 and 10 µM] through enhancing the ratio of cellular levels of reduced glutathione/oxidized glutathione | |
| [13] | ●Homocysteic acid neurotoxicity for 24 h using primary cortical neurons of foetuses of Sprague–Dawley (17th day of gestation) | ●5-min pre-treatment with 250 nM of α-TCT in 1% ethanol | ●1% ethanol | ●Significantly enhanced cellular viability | |
| [13] | ●Homocysteic acid neurotoxicity for 24 h using primary cortical neurons of foetuses of Sprague–Dawley (17th day of gestation) | ●5-min pre-treatment with 0.25, 1, 2.5 and 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Significantly enhanced cellular viability | |
| [24] | ●Glutamate neurotoxicity for 30 min using murine hippocampal HT4 neuronal cells | ●10-min pre-treatment with 250 µM α-TCT in ethanol 1% | ●1% ethanol | ●A significant enhancement of cellular viability | ●Decreasing significantly the release of arachidonic and docosahexaenoic acids from cell membrane through attenuating the hydrolysis activity of cytosolic phospholipase A2 on cell membrane due to inhibiting: ●Translocation of cytosolic phospholipase A2 to cell membrane, ●Ser505 phosphorylation of cytosolic phospholipase A2 ●Phospholipase A2 activity |
| [24] | ●Glutamate neurotoxicity for 24 h using murine hippocampal HT4 neuronal cells | ●2-h pre-treatment with 250 µM α-TCT in ethanol 1% | ●1% ethanol | ●A significant enhancement of cellular viability | ●Direct inhibition of phospholipase A2. |
| [28] | ●Glutamate neurotoxicity for 24 h using human neuroblastoma cells line (SK-N-SH) | ●5-min pre-treatment with 100, 200, or 300 ng/mL of TRF in DMSO (TRF: 25% tocopherol and 75% tocotrienols) | ●DMSO | ●A significant enhancement of cellular viability particularly 200 ng/mL ●A significant dose-dependent attenuation of lipid peroxidation through reducing the levels of MDA | ●Annexin V-FITC/PI staining indicated that 200 mg/kg was significantly the highest against necrosis as well as early and late stage apoptosis |
| [28] | ●Glutamate neurotoxicity for 24 h using human neuroblastoma cells line (SK-N-SH) | ●30-min post-treatment with 100, 200, or 300 ng/mL TRF in DMSO (TRF: 25% tocopherol and 75% tocotrienols) | ●DMSO | ●A significant enhancement of cellular viability particularly 200 mg/kg. ●A significant attenuation of lipid peroxidation through reducing the levels of MDA particularly 300 mg/kg | ●Annexin V-FITC/PI staining indicated slight (nonsignificant) antiapoptotic effect against necrosis as well as early and late stage apoptosis ●Electronic microscope scanning for cellular morphology indicated that only 200 mg/kg could provide a little improvement to the cell membrane integrity. |
| [23] | ●Hydrogen peroxide neurotoxicity for 24 h using human neuroblastoma cells line [SH-SY5Y wild-type] | ●Simultaneous treatment with 10 µM of α-TCT in 1% ethanol | ●1% ethanol | ●Significantly reduced the levels of ROS | ●Significant strong protection of total cholesterol and free cholesterol. |
| [23] | ●Alzheimer’s disease model using human neuroblastoma cells line [SH-SY5Y APP] overexpressing the human APP695 isoform | ●Simultaneous treatment with 10 µM of α-TCT in 1% ethanol for 24 h | ●1% ethanol | ●A nonsignificant increase in the levels of Aβ indicating early onset of AD | ●Direct activation of γ-secretase independent of gene expression |
| [23] | ●Alzheimer’s disease model using human neuroblastoma cells line [SH-SY5Y wild-type] | ●Simultaneous treatment with 10 µM of α-TCT in 1% ethanol for 24 h | ●1% ethanol | ●A significant increase in the levels of Aβ | ●Due to direct increase in β-secretase activity independent of gene transcription of BACE1 |
| [23] | ●Alzheimer’s disease model using human neuroblastoma cells line [SH-SY5Y cells] stably expressing C99 | ●Simultaneous treatment with 10 µM of α-TCT in 1% ethanol for 24 h | ●1% ethanol | ●Significantly increased levels of Aβ | ●Direct activation of γ-secretase independent of gene transcription of PSEN1, PSEN2, NCSTN, PSENEN and APH1A |
| [23] | ●Alzheimer’s disease model using mouse neuroblastoma cell line (N2a) | ●Simultaneous treatment with 10 µM of α-TCT in 1% ethanol for 24 h | ●1% ethanol | ●Significantly decreasing Aβ degradation | ●Inhibiting insulin-degrading enzyme |
| [26] | ●Glutamate neurotoxicity for 24 h using human astrocytes cell line (CRL-2020 cells) derived from glioblastoma with S100B protein | ●5-min pre-treatment with 100, 200 and 300 ng/mL of TRF in absolute ethanol (TRF: 25% tocopherol and 75% tocotrienols) | ●Absolute ethanol | ●TRF could neither promptly (significantly) enhance cellular viability nor modulate the situation of oxidative stress since the level of the reduced glutathione was still low. However, 200 and 300 ng/mL could significantly attenuate lipid peroxidation through reducing the MDA level. | ●Morphological cellular changes indicated a significantly reduction in the percentages of apoptotic and necrotic cells at higher concentrations. |
| [26] | ●Glutamate neurotoxicity for 24 h using human astrocytes cell line (CRL-2020 cells) derived from glioblastoma with S100B protein | ●30-min post-treatment with 100, 200 and 300 ng/mL of TRF in absolute ethanol (TRF: 25% tocopherol and 75% tocotrienols) | ●Absolute ethanol | ●TRF could neither promptly (significantly) enhance cellular viability nor modulate the situation of oxidative stress since the level of the reduced glutathione was still low. However, TRF could significantly attenuate lipid peroxidation through reducing the MDA level. | ●Morphological cellular changes indicated a significant reduction in the percentages of apoptotic and necrotic cells at higher concentrations. |
| [30] * | ●Alzheimer’s disease model with Aβ42 aggregates for 24 h using human neuroblastoma cell line (SH-SY5Y) | ●Simultaneous treatment with 0.00003, 0.0003, 0.003% (v/v) TRF in 0.15% ethanol (TRF: 196 mg/g α-TCT, 24 mg/g β-TCT, 255 mg/g γ-TCT,75mg/gδ-TCT and 168 mg/g α-tocopherol) | ●0.15% ethanol | ●TRF could significantly enhance cellular viability | |
| Reference | Human Modeled Disease, Study Design and Population | Intervention | Comparator | Outcomes | |
|---|---|---|---|---|---|
| Primary | Secondary | ||||
| [33] | ●Nutritionally induced-cognitive dysfunction ●Young healthymale Crj:CD-1 (ICR) mice (3 weeks, n = 5) ●Long-term mono-level oral ad libitum intervention for 8 months. | ●Palm oil (5 g/100 g NRD) | ●100 g NRD | ●Slight (nonsignificant) improvement in cognitive functions as evidenced by the non-significant reduced escape latency. | |
| [32] | ●Diabetes-induced cognitive dysfunction ●Healthy male Wistar rats (age? n = 8). ●IP-injection of 45 mg/kg STZ (pH = 4.4, 0.1 M citrate buffer), while control was IP injected with citrate buffer vehicle. ●Long-term multilevel single oral daily intervention started from the 3rd day of STZ injection for 10 weeks. | ●25, 50 or 100 mg/kg of TRF triturated with 5% tween 80 and dissolved in 5 mL/kg doubled distilled water. (TRF: Purity and composition was not stated) | ●5% tween 80 in 5 mL/kg doubled distilled water | ●Significant dose-dependent improvement in cognitive dysfunctions as evidenced by the deceased transfer latency (the time to reach the platform) and increased the time spent in the target quadrant (improved memory consolidation after learning). ●A significant dose-dependent improve in the cerebrocortical cholinergic activity, while the hippocampal cholinergic function was not significantly improved. ●A significant dose-dependent reversal of the cerebrocortical and hippocampal oxidative stress through attenuating lipid peroxidation and enhancing the activity of the antioxidant enzymes. ●A significant dose-dependent anti-inflammatory effect though reducing the cerebrocortical and hippocampal levels of TNF-α, IL-1β and p56 subunit of NFκβ. ●A significant dose-dependent antiapoptotic effect through reducing cerebrocortical and hippocampal levels of caspase-3. | |
| [32] | ●Normal cognitive function ●Healthy male Wistar rats with (age? n = 8–10). ●Long-term mono-level single oral daily intervention for 10 weeks. | ●100 mg/kg of TRF triturated with 5% tween 80 and dissolved in 5 mL/kg doubled distilled water. (TRF: Purity and composition was not stated) | ●5% tween 80 in 5 mL/kg doubled distilled water. | ●The cognitive performance was slightly (nonsignificant) increased as evidenced by the non-significantly reduced escape latency | |
| [36] | ●Diabetes-induced cognitive dysfunction ●Healthy male Wistar rats, (Age? n = 5–8) were intracerebroventricularly injected with of 2 µL of 3 mg/kg STZ (pH = 4.4 and 0.1 M of citrate buffer) in two divided doses (on day 1 and day 3), while the comparator rats were intraventricular injected with 2 µL of citrate buffer (pH = 4.4, 0.1 M). Post-operative, rats were orally fed on milk and allowed to feed on NRD (ad libitum) for 4 days followed by feeding on NRD up to the end of the treatment. ●Short-term multilevel single oral daily intervention started by the 1st day of injecting STZ to be continued for 21 days. | ●50 and 100 mg/kg α-TCT triturated with 5% tween and dissolved in double distilled water. | ●5% tween and dissolved in double distilled water. | ●A significant dose dependent improvement in cognitive functions as evidenced by the reduced escape latency. ●A significant dose-dependent reversal of neuro-oxidative stress through attenuating lipid peroxidation and enhancing of the activity of the antioxidant enzymes | |
| [38] | ●Healthy cognitive function ●Healthy male Wister rats (age 3 months, n = 10) ● Long-term mono-level single oral daily intervention for 8 months. | ●200 mg/kg TRF in 5 mL/kg of distilled water (TRF: Purity and composition were not stated) | ●5 mL/kg distilled water | ●Significantly enhanced cognitive functions as evidenced by the reduced escape latency ●Significantly reduced plasma DNA damage ●Significant reversal of serum oxidative stress through increasing the activity of antioxidant enzymes | |
| [34] | ●Nutritionally induced cognitive dysfunction. ●Healthy male Sprague-Dawley rats (age? n = 10) ●Long-term mono-level of ad libitum oral feeding on intervention admixed with palm oil base vehicle. ●Rats exposed to the same intervention levels during gestation, 2 weeks during lactation, 8 weeks after weaning. | ●100 mg/kg TRF suspended in 70 g/kg of palm oil base and admixed with 100 g NRD (TRF: Gold-Tri E ™70) | ●70 g/kg of palm oil base admixed with 100 g NRD. | ●Significant improvement in the cognitive functions of rats’ progeny ●Plasm and brain concentrations of tocotrienols indicated that α-TCT was the highest among the other isomers. | |
| [37] | ●Chronic induced-stress condition ●Healthy male Sprague-Dawley rats (5 weeks, n = 9), which were stressed 5 h daily started from the 3rd week of intervention and continued for 21 days. ●Long-term mono-level single oral daily dose intervention for 5 weeks. | ●200 mg/kg of TRF in normal saline (TRF: Tocomin® SuprabioTM 20%) | Normal saline | ●Non-significant enhancement of the cellular proliferation and survival as well as expression of GAP-43 gene of granule cells in dentate gyrus | |
| [37] | ●Unstrained conditions ●Healthy male Sprague-Dawley rats (5 weeks, n = 9) ●Long-term mono-level single oral daily dose intervention for 5 weeks | ●200 mg/kg of TRF in normal saline (TRF: Tocomin® SuprabioTM 20% but compostion was not stated) | Normal saline | ●No significant alteration in the cellular proliferation and survival as well as expression of GAP-43 gene of granule cells in dentate gyrus | |
| [39] | ●Healthy cognitive function ●Healthy young male Wister rats (3 months, n = 9) ●Long-term mono-level oral single daily intervention for 3 months | ●200 mg/kg TRF in 5 mL/kg of olive oil (TRF = 149.2 mg/g α-tocopherol, 164.7 mg/g α-TCT, 48.8 mg/g β-TCT,213.2 mg/g γ-TCT and 171 mg/g δ-TCT). | 5 mL/kg of olive oil | ●No significant alteration in the cognitive functions as evidenced by the non-significant difference in escape latency. ●no significant alteration in plasma lipid peroxidation and the plasma activity of antioxidant enzymes ● A slight (nonsignificant) reduction in plasma DNA damage | |
| [39] | ●cognitive dysfunction ●elderly male Wister rats (21 months, n = 9) ●Long-term mono-level oral single daily intervention for 3 months | ●200 mg/kg TRF in 5 mL/kg of olive oil (TRF = 149.2 mg/g α-tocopherol, 164.7 mg/g α-TCT, 48.8 mg/g β-TCT, 213.2 mg/g γ-TCT and 171 mg/g δ-TCT). | 5 mL/kg of olive oil | ●Significant Improved cognitive functions as evidenced by the significant reduction in escape latency. ●Reversal of the plasma oxidative stress through a significant attenuating lipid peroxidation and enhancing the activity of oxidative enzymes ● Significant attenuation of plasma DNA damage | |
| [30] | ●Transgenic Alzheimer’s disease ●Heterozygous AβPP/PS1 double transgenic male mice (human chimeric amyloid expressing precursor protein and a mutant human presenilin 1 with deletion at exon 9), (5 months) ●Long-term mono-level single oral daily intervention for 10 months. | ●60 mg/kg of TRF in 5 mL/kg 12mg/mL vitamin-E-striped palm oil (n = 11) (TRF = 196.0 mg/g α-TCT, 24 mg/g β-TCT, 255 mg/g γ-TCT, 75 mg/gδ-TCT and 168 mg/g α-tocopherol) | ●5 mL/kg of 12 mg/mL of vitamin-E-striped palm oil (n = 10) | ●Slight (nonsignificant) enhancement of the recognition functions as evidenced by the nonsignificant increase in the recognition index, but the location preference was equivalent as evidenced by the equal spent time to explore the identical objects. ●A non-significant change in the levels of soluble and insoluble cortical or hippocampal Aβ isoforms (Aβ 40, Aβ 42 and Aβ oligomer). | ●A slight (nonsignificant) reduction in the hippocampal Aβ deposition, but significant reduction in the cortical Aβ deposition ●A significant reduction in cortical and hippocampal Aβ plaques |
| [35] | ●Transgenic Alzheimer’s disease ●Heterozygous AβPP/PS1 double transgenic male mice (human chimeric amyloid expressing precursor protein and a mutant human presenilin 1 with deletion at exon 9), (9 months, n = 4) ●Long-term mono-level single oral daily intervention for 6 months. | ●200 mg/kg TRF in 12 mg/mL vitamin E striped palm oil(TRF = f 24% α-tocopherol, 27% α-TCT,4% β-TCT, 32% γ-TCT,and 14% δ-TCT) | 12 mg/mL of vitamin E striped palm oil | Significant upregulation of genes responsible for neuroprotective effects such as Slc24a2, exo1 and Enox1 ●Significant downregulation of genes responsible for the pathology of AD such as Pla2g4a and Tfap2b | |
| [31] | ●Transgenic Alzheimer’s disease ●Heterozygous AβPP/PS1 double transgenic male mice (human chimeric amyloid expressing precursor protein and a mutant human presenilin 1 with deletion at exon 9), (5 months, n = 9). ●Long-term mono-level single oral daily intervention for 10 months. | ●60 mg/kg TRF in 12 mg/mL of vitamin-E-striped palm oil(TRF = 23.40% α-tocopherol (23.40%),27.30% α-TCT; 3.34% β-TCT, 35.51% γ-TCT and 10.45% δ-TCT.) | ●5 mL/kg of vitamin-E-stripped palm oil | ●Slight (nonsignificant) enhancement of cognitive functions as evidenced by non-significantly reduced escape latency. | ●TRF could alter 90 putative metabolites involved in several metabolic AD-related pathways. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ismail, M.; Alsalahi, A.; Imam, M.U.; Ooi, D.J.; Khaza’ai, H.; Aljaberi, M.A.; Shamsudin, M.N.; Idrus, Z. Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review. Nutrients 2020, 12, 521. https://doi.org/10.3390/nu12020521
Ismail M, Alsalahi A, Imam MU, Ooi DJ, Khaza’ai H, Aljaberi MA, Shamsudin MN, Idrus Z. Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review. Nutrients. 2020; 12(2):521. https://doi.org/10.3390/nu12020521
Chicago/Turabian StyleIsmail, Maznah, Abdulsamad Alsalahi, Mustapha Umar Imam, Der Jiun Ooi, Huzwah Khaza’ai, Musheer A. Aljaberi, Mad Nasir Shamsudin, and Zulkifli Idrus. 2020. "Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review" Nutrients 12, no. 2: 521. https://doi.org/10.3390/nu12020521
APA StyleIsmail, M., Alsalahi, A., Imam, M. U., Ooi, D. J., Khaza’ai, H., Aljaberi, M. A., Shamsudin, M. N., & Idrus, Z. (2020). Safety and Neuroprotective Efficacy of Palm Oil and Tocotrienol-Rich Fraction from Palm Oil: A Systematic Review. Nutrients, 12(2), 521. https://doi.org/10.3390/nu12020521