Enhanced GIP Secretion in Obesity Is Associated with Biochemical Alteration and miRNA Contribution to the Development of Liver Steatosis

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Anthropometry Measurements
2.3. Sample Collection and Analysis
2.4. Oral Glucose Tolerance Test (OGTT)
2.5. Meal Tolerance Test (MTT)
2.6. Biochemical Tests
2.7. Isolation and Real-Time PCR of miRNA
2.8. Statistical Analyses
3. Results
3.1. Baseline Characteristics of Subjects
3.2. Association of Plasma GIP with Fatty Liver Risk Markers
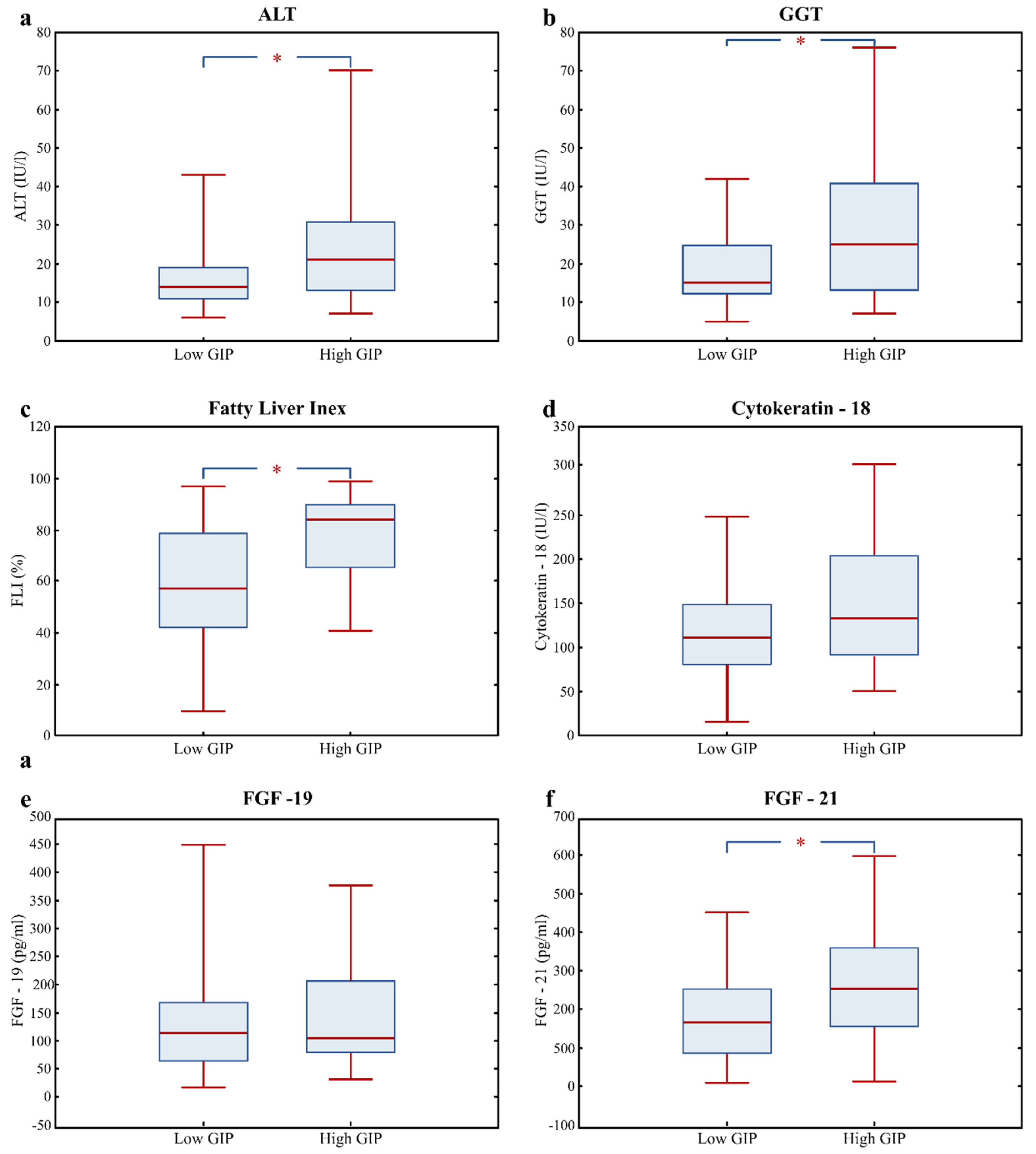
3.3. Fatty Liver Risk Markers in High GIP Subjects
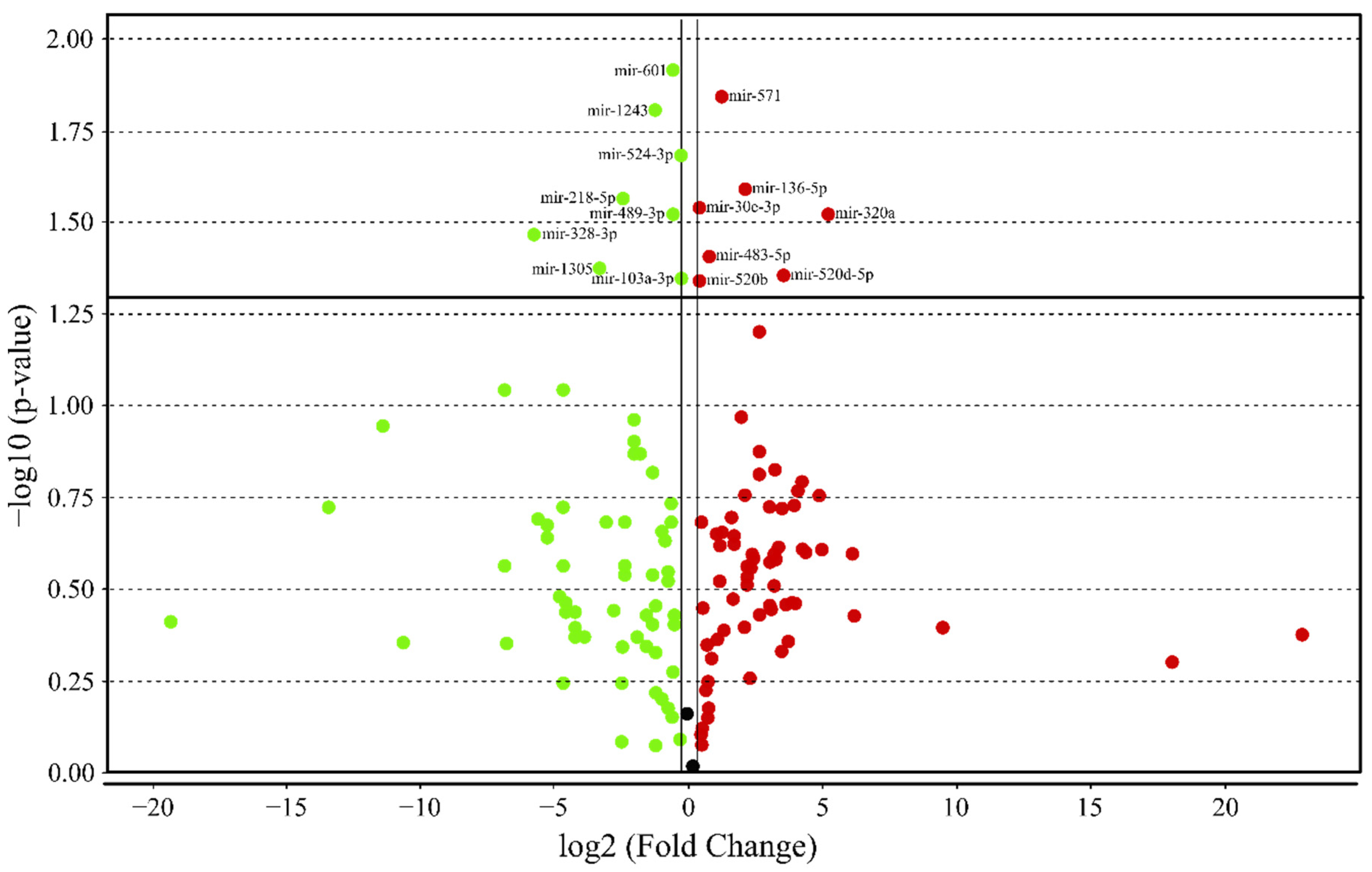
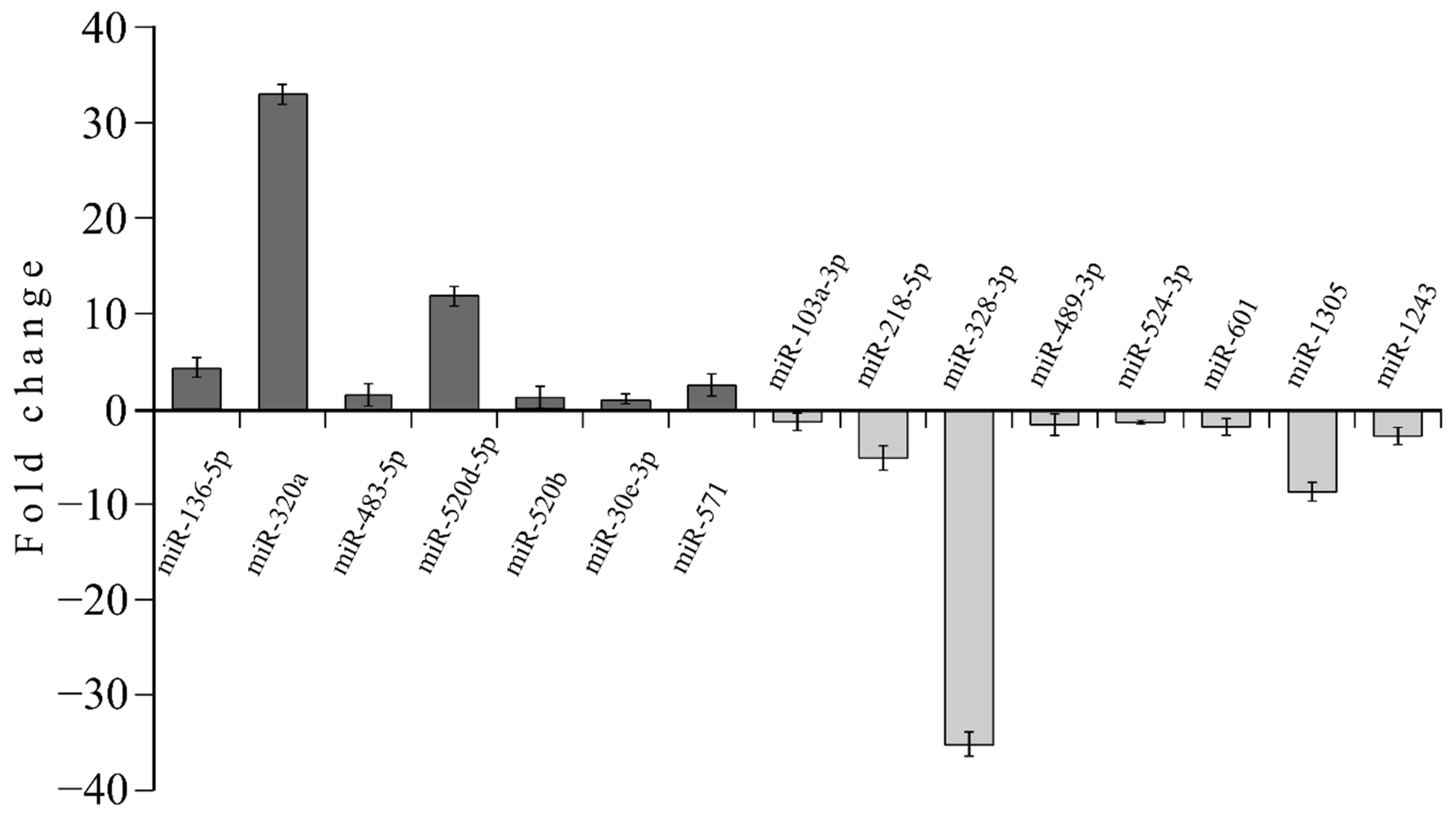
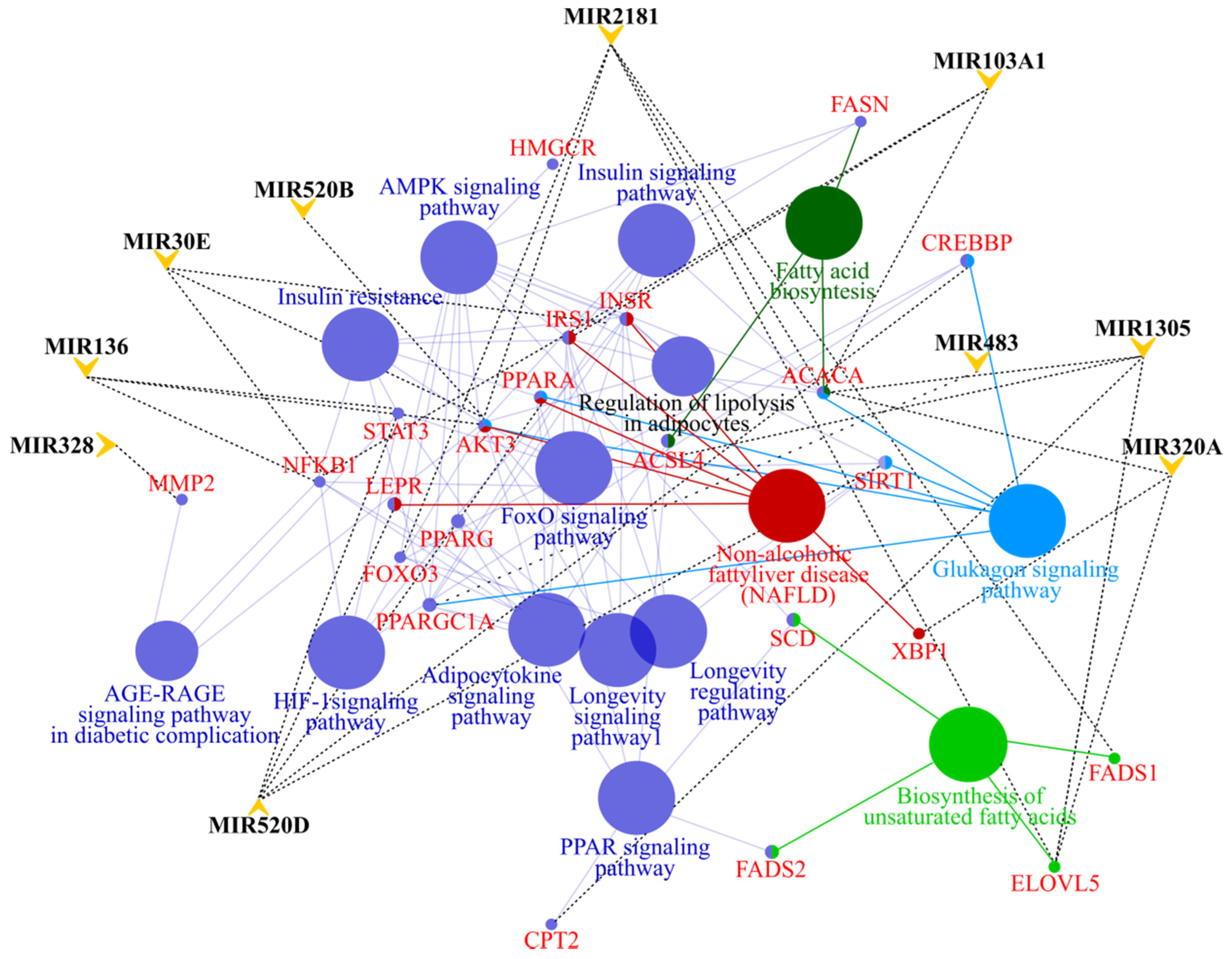
3.4. MicroRNA profile in High GIP Subjects
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pederson, R.A.; Schubert, H.E.; Brown, J.C. The insulinotropic action of gastric inhibitory polypeptide. Can. J. Physiol. Pharmacol. 1975, 53, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, A.F.H.; Keyhani-Nejad, F. High Glycemic Index Metabolic Damage—A Pivotal Role of GIP and GLP-1. Trends Endocrinol. Metab. 2018, 29, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Pacini, G.; De Michieli, F.; Cassader, M. Prolonged saturated fat–induced, glucose-dependent insulinotropic polypeptide elevation is associated with adipokine imbalance and liver injury in nonalcoholic steatohepatitis: Dysregulated enteroadipocyte axis as a novel feature of fatty liver. Am. J. Clin. Nutr. 2009, 89, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.C.; Jarboe, L.A.; Wolfe, M.M. Regulation of glucose-dependent insulinotropic peptide gene expression by a glucose meal. Am. J. Physiol. Liver Physiol. 1994, 266, G887–G891. [Google Scholar] [CrossRef]
- Bailey, C.J.; Flatt, P.R.; Kwasowski, P.; Powell, C.J.; Marks, V. Immunoreactive gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol. 1986, 112, 224–229. [Google Scholar] [CrossRef]
- Sommer, C.A.; Mostoslavsky, G. RNA-Seq Analysis of Enteroendocrine Cells Reveals a Role for FABP5 in the Control of GIP Secretion. Mol. Endocrinol. 2014, 28, 1855–1865. [Google Scholar] [CrossRef]
- Nauck, M.A.; Meier, J.J. Incretin hormones: Their role in health and disease. Diabetes Obes. Metab. 2018, 20, 5–21. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Siepmann, N.; Holst, J.J.; Deacon, C.F.; Schmidt, W.E.; Nauck, M.A. Gastric inhibitory polypeptide (GIP) dose-dependently stimulates glucagon secretion in healthy human subjects at euglycaemia. Diabetologia 2003, 46, 798–801. [Google Scholar] [CrossRef]
- Christensen, M.; Calanna, S.; Sparre-Ulrich, A.H.; Kristensen, P.L.; Rosenkilde, M.M.; Faber, J.; Purrello, F.; Van Hall, G.; Holst, J.J.; Vilsbøll, T.; et al. Glucose-Dependent Insulinotropic Polypeptide Augments Glucagon Responses to Hypoglycemia in Type 1 Diabetes. Diabetes 2014. [Google Scholar] [CrossRef]
- Nauck, M.; Stöckmann, F.; Ebert, R.; Creutzfeldt, W. Reduced incretin effect in type 2 (non-insulin-dependent) diabetes. Diabetologia 1986, 29, 46–52. [Google Scholar] [CrossRef]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.-H.; McIntosh, C.H.S.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef]
- Mohammad, S.; Patel, R.T.; Bruno, J.; Panhwar, M.S.; Wen, J.; McGraw, T.E. A Naturally Occurring GIP Receptor Variant Undergoes Enhanced Agonist-Induced Desensitization, Which Impairs GIP Control of Adipose Insulin Sensitivity. Mol. Cell. Biol. 2014, 34, 3618–3629. [Google Scholar] [CrossRef]
- Sun, E.W.L.; Martin, A.M.; Young, R.L.; Keating, D.J. The Regulation of Peripheral Metabolism by Gut-Derived Hormones. Front. Endocrinol. 2019, 9, 754. [Google Scholar] [CrossRef]
- Campbell, J.E.; Ussher, J.R.; Mulvihill, E.E.; Kolic, J.; Baggio, L.L.; Cao, X.; Liu, Y.; Lamont, B.J.; Morii, T.; Streutker, C.J.; et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016, 22, 84–90. [Google Scholar] [CrossRef]
- Gögebakan, Ö.; Andres, J.; Biedasek, K.; Mai, K.; Kühnen, P.; Krude, H.; Isken, F.; Rudovich, N.; Osterhoff, M.A.; Kintscher, U.; et al. Glucose-Dependent Insulinotropic Polypeptide Reduces Fat-Specific Expression and Activity of 11β-Hydroxysteroid Dehydrogenase Type 1 and Inhibits Release of Free Fatty Acids. Diabetes 2011, 61. [Google Scholar] [CrossRef]
- Mantelmacher, F.D.; Fishman, S.; Cohen, K.; Chor, M.P.; Yamada, Y.; Zvibel, I.; Varol, C. Glucose-Dependent Insulinotropic Polypeptide Receptor Deficiency Leads to Impaired Bone Marrow Hematopoiesis. J. Immunol. 2017, 198, 1601441. [Google Scholar] [CrossRef]
- Yamada, Y.; Tsukiyama, K.; Sato, T.; Shimizu, T.; Fujita, H.; Narita, T. Novel extrapancreatic effects of incretin. J. Diabetes Investig. 2016, 7, 76–79. [Google Scholar] [CrossRef]
- Xie, T.; Leung, P.S. Fibroblast growth factor 21: A regulator of metabolic disease and health span. Am. J. Physiol. Metab. 2017, 313, E292–E302. [Google Scholar] [CrossRef]
- Hadizadeh, F.; Faghihimani, E.; Adibi, P. Nonalcoholic fatty liver disease: Diagnostic biomarkers. World J. Gastrointest. Pathophysiol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Castera, L.; Friedrich-Rust, M.; Loomba, R. Noninvasive Assessment of Liver Disease in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 156, 1264–1281. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Feldstein, A.E.; Wieckowska, A.; Lopez, A.R.; Liu, Y.-C.; Zein, N.N.; McCullough, A.J. Cytokeratin-18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: A multicenter validation study. Hepatology 2009, 50, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- He, L.; Deng, L.; Zhang, Q.; Guo, J.; Zhou, J.; Song, W.; Yuan, F. Diagnostic Value of CK-18, FGF-21, and Related Biomarker Panel in Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Biomed Res. Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rusli, F.; Deelen, J.; Andriyani, E.; Boekschoten, M.V.; Lute, C.; Van den Akker, E.B.; Müller, M.; Beekman, M.; Steegenga, W.T. Fibroblast growth factor 21 reflects liver fat accumulation and dysregulation of signalling pathways in the liver of C57BL/6J mice. Sci. Rep. 2016, 6, 30484. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, J.; Jia, W. Fibroblast growth factor 21: A novel metabolic regulator from pharmacology to physiology. Front. Med. 2013, 7, 25–30. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific Expression of βKlotho and Fibroblast Growth Factor (FGF) Receptor Isoforms Determines Metabolic Activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef]
- Okubo, H.; Kushiyama, A.; Nakatsu, Y.; Yamamotoya, T.; Matsunaga, Y.; Fujishiro, M.; Sakoda, H.; Ohno, H.; Yoneda, M.; Asano, T. Roles of Gut-Derived Secretory Factors in the Pathogenesis of Non-Alcoholic Fatty Liver Disease and Their Possible Clinical Applications. Int. J. Mol. Sci. 2018, 19, 3064. [Google Scholar] [CrossRef]
- Sattar, N.; Ewan, F.; David, P. Non-alcoholic fatty liver disease. Bmj. 2014, 349, g4596. [Google Scholar] [CrossRef]
- Yamane, S.; Harada, N. Gastric inhibitory polypeptide/glucose-dependent insulinotropic polypeptide signaling in adipose tissue. J. Diabetes Investig. 2019, 10, 3–5. [Google Scholar] [CrossRef]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Castro, R.E. Circulating microRNAs as Potential Biomarkers in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma. J. Clin. Med. 2016, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Meroni, M.; Longo, M.; Fargion, S.; Fracanzani, A.L. miRNA Signature in NAFLD: A Turning Point for a Non-Invasive Diagnosis. Int. J. Mol. Sci. 2018, 19, 3966. [Google Scholar] [CrossRef] [PubMed]
- Meroni, M.; Longo, M.; Erconi, V.; Valenti, L.; Gatti, S.; Fracanzani, A.L.; Dongiovanni, P. mir-101-3p Downregulation Promotes Fibrogenesis by Facilitating Hepatic Stellate Cell Transdifferentiation During Insulin Resistance. Nutrients 2019, 11, 2597. [Google Scholar] [CrossRef] [PubMed]
- Radbakhsh, S.; Sathyapalan, T.; Banach, M.; Sahebkar, A. Incretins and microRNAs: Interactions and physiological relevance. Pharmacol. Res. 2020, 104662. [Google Scholar] [CrossRef]
- Razny, U.; Goralska, J.; Zdzienicka, A.; Gruca, A.; Zapala, B.; Micek, A.; Dembinska-Kiec, A.; Solnica, B.; Malczewska-Malec, M. High Fat Mixed Meal Tolerance Test Leads to Suppression of Osteocalcin Decrease in Obese Insulin Resistant Subjects Compared to Healthy Adults. Nutrients 2018, 10, 1611. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Witvrouwen, I.; Vrints, C.J.; Heidbuchel, H.; Van Craenenbroeck, E.M.; Van Laere, S.J.; Van Craenenbroeck, A.H. MicroRNA profiling in plasma samples using qPCR arrays: Recommendations for correct analysis and interpretation. PLoS ONE 2018, 13, e0193173. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.-H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Finan, B.; Müller, T.D.; Clemmensen, C.; Perez-Tilve, D.; DiMarchi, R.D.; Tschöp, M.H. Reappraisal of GIP Pharmacology for Metabolic Diseases. Trends Mol. Med. 2016, 22, 359–376. [Google Scholar] [CrossRef]
- Calanna, S.; Christensen, M.; Holst, J.J.; Laferrère, B.; Gluud, L.L.; Vilsbøll, T.; Knop, F.K. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: Systematic review and meta-analysis of clinical studies. Diabetes Care 2013, 36, 3346–3352. [Google Scholar] [CrossRef]
- Gasbjerg, L.S.; Christensen, M.B.; Hartmann, B.; Lanng, A.R.; Sparre-Ulrich, A.H.; Gabe, M.B.N.; Dela, F.; Vilsbøll, T.; Holst, J.J.; Rosenkilde, M.M.; et al. GIP(3-30)NH2 is an efficacious GIP receptor antagonist in humans: A randomised, double-blinded, placebo-controlled, crossover study. Diabetologia 2018, 61, 413–423. [Google Scholar] [CrossRef]
- Killion, E.A.; Wang, J.; Yie, J.; Shi, S.D.-H.; Bates, D.; Min, X.; Komorowski, R.; Hager, T.; Deng, L.; Atangan, L.; et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Mroz, P.A.; Finan, B.; Gelfanov, V.; Yang, B.; Tschöp, M.H.; DiMarchi, R.D.; Perez-Tilve, D. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol. Metab. 2019, 20, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Frias, J.P.; Nauck, M.A.; Van, J.; Kutner, M.E.; Cui, X.; Benson, C.; Urva, S.; Gimeno, R.E.; Milicevic, Z.; Robins, D.; et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: A randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet 2018, 392, 2180–2193. [Google Scholar] [CrossRef]
- DiMarchi, R.D. “Let’s Stay Together”; GIP and GLP-1 dual agonism in the treatment of metabolic disease. Mol. Metab. 2018, 18, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Sanguineti, R.; Montecucco, F.; Viviani, G.L. Effects of High Glucose Levels and Glycated Serum on GIP Responsiveness in the Pancreatic Beta Cell Line HIT-T15. J. Diabetes Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Góralska, J.; Raźny, U.; Polus, A.; Stancel-Możwiłło, J.; Chojnacka, M.; Gruca, A.; Zdzienicka, A.; Dembińska-Kieć, A.; Kieć-Wilk, B.; Solnica, B.; et al. Pro-inflammatory gene expression profile in obese adults with high plasma GIP levels. Int. J. Obes. 2018, 42, 826–834. [Google Scholar] [CrossRef]
- Martin-Rodriguez, J.L.; Gonzalez-Cantero, J.; Gonzalez-Cantero, A.; Arrebola, J.P.; Gonzalez-Calvin, J.L. Diagnostic accuracy of serum alanine aminotransferase as biomarker for nonalcoholic fatty liver disease and insulin resistance in healthy subjects, using 3T MR spectroscopy. Medicine 2017, 96, e6770. [Google Scholar] [CrossRef]
- Gholam, P.M.; Flancbaum, L.; Machan, J.T.; Charney, D.A.; Kotler, D.P. Nonalcoholic Fatty Liver Disease in Severely Obese Subjects. Am. J. Gastroenterol. 2007, 102, 399–408. [Google Scholar] [CrossRef]
- Cuthbertson, D.J.; Weickert, M.O.; Lythgoe, D.; Sprung, V.S.; Dobson, R.; Shoajee-Moradie, F.; Umpleby, M.; Pfeiffer, A.F.H.; Thomas, E.L.; Bell, J.D.; et al. External validation of the fatty liver index and lipid accumulation product indices, using 1H-magnetic resonance spectroscopy, to identify hepatic steatosis in healthy controls and obese, insulin-resistant individuals. Eur. J. Endocrinol. 2014, 171, 561–569. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M.; Pagano, G. Meta-analysis: Natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann. Med. 2011, 43, 617–649. [Google Scholar] [CrossRef]
- Joka, D.; Wahl, K.; Moeller, S.; Schlue, J.; Vaske, B.; Bahr, M.J.; Manns, M.P.; Schulze-Osthoff, K.; Bantel, H. Prospective biopsy-controlled evaluation of cell death biomarkers for prediction of liver fibrosis and nonalcoholic steatohepatitis. Hepatology 2012, 55, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Alssema, M.; Rijkelijkhuizen, J.M.; Holst, J.J.; Teerlink, T.; Scheffer, P.G.; Eekhoff, E.M.W.; Gastaldelli, A.; Mari, A.; Hart, L.M.; Nijpels, G.; et al. Preserved GLP-1 and exaggerated GIP secretion in type 2 diabetes and relationships with triglycerides and ALT. Eur. J. Endocrinol. 2013, 169, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Seghieri, M.; Christensen, A.S.; Andersen, A.; Solini, A.; Knop, F.K.; Vilsbøll, T. Future Perspectives on GLP-1 Receptor Agonists and GLP-1/glucagon Receptor Co-agonists in the Treatment of NAFLD. Front. Endocrinol. 2018, 9, 649. [Google Scholar] [CrossRef] [PubMed]
- Junker, A.E.; Gluud, L.L.; Holst, J.J.; Knop, F.K.; Vilsbøll, T. Influence of gastrointestinal factors on glucose metabolism in patients with cirrhosis. J. Gastroenterol. Hepatol. 2015, 30, 1522–1528. [Google Scholar] [CrossRef]
- Junker, A.E. The role of incretin hormones and glucagon in patients with liver disease. Dan. Med. J. 2017, 64. [Google Scholar] [CrossRef]
- Joo, E.; Harada, N.; Yamane, S.; Fukushima, T.; Taura, D.; Iwasaki, K.; Sankoda, A.; Shibue, K.; Harada, T.; Suzuki, K.; et al. Inhibition of Gastric Inhibitory Polypeptide Receptor Signaling in Adipose Tissue Reduces Insulin Resistance and Hepatic Steatosis in High-Fat Diet-Fed Mice. Diabetes 2017, 66, 868–879. [Google Scholar] [CrossRef]
- Keyhani-Nejad, F.; Irmler, M.; Isken, F.; Wirth, E.K.; Beckers, J.; Birkenfeld, A.L.; Pfeiffer, A.F.H. Nutritional strategy to prevent fatty liver and insulin resistance independent of obesity by reducing glucose-dependent insulinotropic polypeptide responses in mice. Diabetologia 2015, 58, 374–383. [Google Scholar] [CrossRef]
- Kruse, M.; Keyhani-Nejad, F.; Isken, F.; Nitz, B.; Kretschmer, A.; Reischl, E.; De Las Heras Gala, T.; Osterhoff, M.A.; Grallert, H.; Pfeiffer, A.F.H. High-Fat Diet During Mouse Pregnancy and Lactation Targets GIP-Regulated Metabolic Pathways in Adult Male Offspring. Diabetes 2016, 65, 574–584. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Mangelsdorf, D.J. Fibroblast growth factor 21: From pharmacology to physiology. Am. J. Clin. Nutr. 2010, 91, 254S–257S. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Inagaki, T.; Satapati, S.; Ding, X.; He, T.; Goetz, R.; Mohammadi, M.; Finck, B.N.; Mangelsdorf, D.J.; Kliewer, S.A.; et al. FGF21 induces PGC-1 and regulates carbohydrate and fatty acid metabolism during the adaptive starvation response. Proc. Natl. Acad. Sci. USA 2009, 106, 10853–10858. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Linden, M.A.; Sheldon, R.D.; Meers, G.M.; Morris, E.M.; Butterfield, A.; Perfield, J.W.; Rector, R.S.; Thyfault, J.P. Fibroblast growth factor 21 increases hepatic oxidative capacity but not physical activity or energy expenditure in hepatic peroxisome proliferator-activated receptor γ coactivator-1α-deficient mice. Exp. Physiol. 2018, 103, 408–418. [Google Scholar] [CrossRef]
- Chu, A.Y.; Workalemahu, T.; Paynter, N.P.; Rose, L.M.; Giulianini, F.; Tanaka, T.; Ngwa, J.S.; Qi, Q.; Curhan, G.C.; Rimm, E.B.; et al. Novel locus including FGF21 is associated with dietary macronutrient intake. Hum. Mol. Genet. 2013, 22, 1895–1902. [Google Scholar] [CrossRef]
- Berti, L.; Hartwig, S.; Irmler, M.; Rädle, B.; Siegel-Axel, D.; Beckers, J.; Lehr, S.; Al-Hasani, H.; Häring, H.-U.; Hrabě de Angelis, M.; et al. Impact of fibroblast growth factor 21 on the secretome of human perivascular preadipocytes and adipocytes: A targeted proteomics approach. Arch. Physiol. Biochem. 2016, 122, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G893–G902. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1α Pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic Mice Expressing Human Fibroblast Growth Factor-19 Display Increased Metabolic Rate and Decreased Adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast Growth Factor 19 Increases Metabolic Rate and Reverses Dietary and Leptin-Deficient Diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, M.; Janus, D.; Dolezal-Oltarzewska, K.; Kalicka-Kasperczyk, A.; Poplawska, K.; Drozdz, D.; Sztefko, K.; Starzyk, J.B. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. J. Pediatr. Endocrinol. Metab. 2012, 25. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- Shima, K.R.; Ota, T.; Kato, K.-I.; Takeshita, Y.; Misu, H.; Kaneko, S.; Takamura, T. Ursodeoxycholic acid potentiates dipeptidyl peptidase-4 inhibitor sitagliptin by enhancing glucagon-like peptide-1 secretion in patients with type 2 diabetes and chronic liver disease: A pilot randomized controlled and add-on study. BMJ Open Diabetes Res. care 2018, 6, e000469. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; Seok, S.; Choi, S.; Huang, Z.; Suino-Powell, K.; Xu, H.E.; Kemper, B.; Kemper, J.K. MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyte fibroblast growth factor 21 signaling and SIRT1 function. Mol. Cell. Biol. 2014, 34, 4130–4142. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xie, X.; Yang, X.; Li, Y.; Wang, Y.; Xu, D. Enhanced inflammatory damage by microRNA-136 targeting Klotho expression in HK-2 cells by modulating JAK/STAT pathway. Pharmazie 2017, 72, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kuang, Y.; Zhou, J. Klotho protects against LPS-induced inflammation injury by inhibiting Wnt and NF-κB pathways in HK-2 cells. Pharmazie 2017, 72, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.-J.; Jin, P.-Y.; Tang, Y.; Fan, S.-H.; Zhang, Z.-F.; Wang, F.; Wu, D.-M.; Lu, J.; Zheng, Y.-L. microRNA-136 inhibits proliferation and promotes apoptosis and radiosensitivity of cervical carcinoma through the NF-κB pathway by targeting E2F1. Life Sci. 2018, 199, 167–178. [Google Scholar] [CrossRef]
- Delerive, P.; Galardi, C.M.; Bisi, J.E.; Nicodeme, E.; Goodwin, B. Identification of Liver Receptor Homolog-1 as a Novel Regulator of Apolipoprotein AI Gene Transcription. Mol. Endocrinol. 2004, 18, 2378–2387. [Google Scholar] [CrossRef]
- Lee, Y.-K.; Schmidt, D.R.; Cummins, C.L.; Choi, M.; Peng, L.; Zhang, Y.; Goodwin, B.; Hammer, R.E.; Mangelsdorf, D.J.; Kliewer, S.A. Liver receptor homolog-1 regulates bile acid homeostasis but is not essential for feedback regulation of bile acid synthesis. Mol. Endocrinol. 2008, 22, 1345–1356. [Google Scholar] [CrossRef]
- Mo, F.-F.; An, T.; Zhang, Z.-J.; Liu, Y.-F.; Liu, H.-X.; Pan, Y.-Y.; Miao, J.-N.; Zhao, D.-D.; Yang, X.-Y.; Zhang, D.-W.; et al. Jiang Tang Xiao Ke Granule Play an Anti-diabetic Role in Diabetic Mice Pancreatic Tissue by Regulating the mRNAs and MicroRNAs Associated with PI3K-Akt Signaling Pathway. Front. Pharmacol. 2017, 8, 795. [Google Scholar] [CrossRef]
- Li, T.; Ma, J.; Han, X.; Jia, Y.; Yuan, H.; Shui, S.; Guo, D. MicroRNA-320 Enhances Radiosensitivity of Glioma Through Down-Regulation of Sirtuin Type 1 by Directly Targeting Forkhead Box Protein M1. Transl. Oncol. 2018, 11, 205–212. [Google Scholar] [CrossRef]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-Specific Deletion of SIRT1 Alters Fatty Acid Metabolism and Results in Hepatic Steatosis and Inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef]
- Colak, Y.; Ozturk, O.; Senates, E.; Tuncer, I.; Yorulmaz, E.; Adali, G.; Doganay, L.; Enc, F.Y. SIRT1 as a potential therapeutic target for treatment of nonalcoholic fatty liver disease. Med. Sci. Monit. 2011, 17. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Chen, X.; Liang, Z. MicroRNA-320 regulates autophagy in retinoblastoma by targeting hypoxia inducible factor-1α. Exp. Ther. Med. 2017, 14, 2367–2372. [Google Scholar] [CrossRef] [PubMed]
- Lamming, D.W.; Sabatini, D.M. A Central Role for mTOR in Lipid Homeostasis. Cell Metab. 2013, 18, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wan, S.; Yang, T.; Niu, D.; Zhang, A.; Yang, C.; Cai, J.; Wu, J.; Song, J.; Zhang, C.-Y.; et al. Increased serum microRNAs are closely associated with the presence of microvascular complications in type 2 diabetes mellitus. Sci. Rep. 2016, 6, 20032. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Mollnow, T.; Bongaerts, B.; Elfimova, N.; Vargas Cardenas, D.; Berger, K.; Zimmermann, H.; Koch, A.; Vucur, M.; Luedde, M.; et al. Micro-RNA Profiling in Human Serum Reveals Compartment-Specific Roles of miR-571 and miR-652 in Liver Cirrhosis. PLoS ONE 2012, 7, e32999. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, Y.; Tsuno, S.; Kuwamoto, S.; Yamashita, T.; Endo, Y.; Hasegawa, J.; Miura, N. Hsa-miR-520d Converts Fibroblasts into CD105+ Populations. Drugs RD 2014, 14, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, A.; Muyleart, M.; Kadlec, A.; Gutterman, D.; Mammoto, T. YAP1-TEAD1 signaling controls angiogenesis and mitochondrial biogenesis through PGC1α. Microvasc. Res. 2018, 119, 73–83. [Google Scholar] [CrossRef]
- Yang, S.-F.; Lee, W.-J.; Tan, P.; Tang, C.-H.; Hsiao, M.; Hsieh, F.-K.; Chien, M.-H. Upregulation of miR-328 and inhibition of CREB-DNA-binding activity are critical for resveratrol-mediated suppression of matrix metalloproteinase-2 and subsequent metastatic ability in human osteosarcomas. Oncotarget 2015, 6, 2736–2753. [Google Scholar] [CrossRef]
- He, W.; Zhuang, J.; Zhao, Z.G.; Luo, H.; Zhang, J. miR-328 prevents renal fibrogenesis by directly targeting TGF-β2. Bratisl. Lek. Listy 2018, 119, 434–440. [Google Scholar] [CrossRef]
- Yi, W.; Tu, M.J.; Liu, Z.; Zhang, C.; Batra, N.; Yu, A.X.; Yu, A.M. Bioengineered miR-328-3p modulates GLUT1-mediated glucose uptake and metabolism to exert synergistic antiproliferative effects with chemotherapeutics. Acta Pharm. Sin. B 2020, 10, 159–170. [Google Scholar] [CrossRef]
- Zhang, M.; Sun, W.; Zhou, M.; Tang, Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci. Rep. 2017, 7, 14493. [Google Scholar] [CrossRef]
- Kurtz, C.L.; Fannin, E.E.; Toth, C.L.; Pearson, D.S.; Vickers, K.C.; Sethupathy, P. Inhibition of miR-29 has a significant lipid-lowering benefit through suppression of lipogenic programs in liver. Sci. Rep. 2015, 5, 12911. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Hsu, S.-D.; Hsu, C.-S.; Lai, T.-C.; Chen, S.-J.; Shen, R.; Huang, Y.; Chen, H.-C.; Lee, C.-H.; Tsai, T.-F.; et al. MicroRNA-122 plays a critical role in liver homeostasis and hepatocarcinogenesis. J. Clin. Invest. 2012, 122, 2884–2897. [Google Scholar] [CrossRef]
- Hanin, G.; Yayon, N.; Tzur, Y.; Haviv, R.; Bennett, E.R.; Udi, S.; Krishnamoorthy, Y.R.; Kotsiliti, E.; Zangen, R.; Efron, B.; et al. miRNA-132 induces hepatic steatosis and hyperlipidaemia by synergistic multitarget suppression. Gut 2018, 67, 1124–1134. [Google Scholar] [CrossRef]
- Rottiers, V.; Näär, A.M. MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol. 2012, 13, 239–250. [Google Scholar] [CrossRef]
- Yan, X.; Wang, Z.; Westberg-Rasmussen, S.; Tarbier, M.; Rathjen, T.; Tattikota, S.G.; Peck, B.C.E.; Kanke, M.; Oxvig, C.; Frystyk, J.; et al. Differential Impact of Glucose Administered Intravenously and Orally on Circulating miR-375 Levels in Human Subjects. J. Clin. Endocrinol. Metab. 2017, 102, 3749–3755. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All (n = 128) | Low GIP (n = 85) | High GIP (n = 43) | ||
|---|---|---|---|---|
| Median (Q1; Q3) | Median (Q1; Q3) | Median (Q1; Q3) | p * | |
| Sex (% of female) | 74% | 81% | 61% | 0.021 § |
| Age (yrs) | 47 (39; 57) | 46 (39; 57) | 54 (41.5; 58) | 0.134 |
| BMI (kg/m2) | 32.28 (29.84; 34.77) | 32.03 (28.87; 34.55) | 32.97 (31.13; 36.02) | 0.065 |
| Waist circumference (cm) | 100 (94; 110) | 99 (93; 108) | 110 (97; 117) | 0.009 |
| Women | 98 (93; 106) | 96 (91; 103) | 100 (95; 110) | 0.052 |
| Men | 114 (106; 118) | 114 (107; 116) | 116 (105; 121) | 0.505 |
| WHR | 0.88 (0.82; 0.96) | 0.85 (0.81; 0.93) | 0.91 (0.84; 0.98) | 0.016 |
| Women | 0.84 (0.81; 0.90) | 0.83 (0.8; 0.89) | 0.87 (0.84; 0.91) | 0.051 |
| Men | 0.99 (0.89; 1.51) | 1.01 (0.98; 1.04) | 0.98 (0.96; 1.03) | 0.263 |
| Body fat (%) | 38.30 (33.50; 42.00) | 38.1 (33.58; 42.05) | 38.8 (32.6; 42) | 0.959 |
| Systolic blood pressure (mmHg) | 130 (120; 140) | 125 (120; 134.5) | 130 (120; 140) | 0.184 |
| Diastolic blood pressure (mmHg) | 84 (80; 90) | 82 (80; 90) | 90 (80; 90) | 0.111 |
| Fasting glucose (mmol/l) | 5.1 (4.8; 5.6) | 5.0 (4.8; 5.4) | 5.6 (5.0; 6.0) | <0.001 |
| Fasting insulin (µIU/mL) | 13.05 (9.68; 19.03) | 12.4 (9.10; 18.00) | 15.00 (11.40; 20.55) | 0.075 |
| HOMA-IR | 2.93 (2.17; 4,56) | 2.7 (2.13; 4.33) | 3.68 (2.72; 5.42) | 0.021 |
| GIP (pg/mL) | 26.2 (18.3; 38.5) | 19.44 (15.27; 26.12) | 44.86 (38.72; 53.43) | <0.001 |
| GIP AUC MTT (ng/mL*min) | 347.7 (279.4; 436.9) | 320.3 (248.8; 389.7) | 401.0 (324.7; 573.6) | <0.001 |
| GIP AUC OGTT (ng/mL*min) | 63.8 (47.6; 86.1) | 60.97 (46.91; 79.73) | 74.42 (58.18; 94.43) | <0.013 |
| FFA (mmol/l) | 0.67 (0.52; 0.86) | 0.67 (0.52; 0.83) | 0.66 (0.53; 0.93) | 0.714 |
| FFA AUC MTT (mol/l*min) | 1.45 (1.22; 1.78) | 1.41 (1.22; 1.68) | 1.56 (1.17; 2.03) | 0.338 |
| TG (mmol/l) | 1.24 (0.92; 1.80) | 1.16 (0.90; 1.49) | 1.57 (1.17; 2.28) | 0.002 |
| TG AUC MTT (mol/l*min) | 3.47 (2.43; 4.89) | 3.22 (2.30; 4.34) | 4.19 (2.83; 5.84) | 0.017 |
| Cholesterol total (mmol/l) | 5.43 (4.82; 6.07) | 5.39 (4.8; 6.03) | 5.46 (4.94; 6.3) | 0.311 |
| HDL-cholesterol (mmol/l) | 1.28 (1.13; 1.48) | 1.32 (1.16; 1.5) | 1.20 (1.11; 1.32) | 0.004 |
| LDL-cholesterol (mmol/l) | 3.45 (2.87; 4.13) | 3.41 (2.90; 4.08) | 3.54 (2.78; 4.39) | 0.527 |
| ALT (IU/l) | 16.0 (12.0; 22.0) | 14.0 (11.0; 19.0) | 21.0 (13.5; 30.5) | 0.004 |
| GGT (IU/l) | 18.0 (12.0; 31.0) | 15.0 (12.0; 25.0) | 24.0 (15.3; 40.5) | 0.007 |
| Fatty Liver Index (%) | 70.5 (45.1; 86.1) | 57.4 (42.0; 79.1) | 84.2 (66.4; 90.3) | 0.000 |
| Cytokeratin; 18 (U/l) | 115.4 (86.1; 159.5) | 110.8 (80.5; 148.3) | 132.5 (91.1; 193.2) | 0.090 |
| FGF-19 (pg/mL) | 108.8 (65.5; 178.4) | 112.6 (62.8; 168.4) | 104.2 (81.0; 206.2) | 0.567 |
| FGF-21 (pg/mL) | 187.4 (120.2; 285.5) | 165.3 (85.3; 255.4) | 253.3 (154.3; 357.0) | 0.007 |
| Model 1 β (95% CI) | Model 2 β (95% CI) | |
|---|---|---|
| Fasting triglycerides | ||
| Fasting GIP | 0.21 (0.06–0.36) | 0.20 (0.04–0.35) |
| R2 | 18% | 19% |
| VIFs | <1.0 | <1.3 |
| GIP AUC MTT | 0.21 (0.05–0.37) | 0.20 (0.04–0.36) |
| R2 | 21% | 24% |
| VIFs | <1.0 | <1.2 |
| GIP AUC OGTT | 0.16 (0.01–0.31) | 0.17 (0.02–0.33) |
| R2 | 16% | 18% |
| VIFs | <1.0 | <1.2 |
| TG AUC MTT | ||
| GIP AUC MTT | 0.24 (0.09–0.40) | 0.23 (0.07–0.39) |
| R2 | 24% | 25% |
| VIFs | <1.0 | <1.2 |
| ALT | ||
| Fasting GIP | 0.16 (0.01–0.32) | 0.14 (−0.02–0.3) |
| R2 | 21% | 24% |
| VIFs | <1.0 | <1.3 |
| GGT | ||
| GIP AUC MTT | 0.17 (0.03–0.32) | 0.15 (0.01–0.30) |
| R2 | 35% | 36% |
| VIFs | <1.0 | <1.2 |
| FGF-21 | ||
| Fasting GIP | 0.20 (0.03–0.37) | 0.22 (0.05–0.4) |
| R2 | 7% | 10% |
| VIFs | <1.1 | <1.3 |
| GIP AUC MTT | 1.14 (−0.04–0.31) | 1.17 (−0.01–0.34) |
| R2 | 5% | 7% |
| VIFs | <1.1 | <1.3 |
| High GIP | OR (95% CI) for ALT > 23 IU/l | OR (95% CI) for ALT > 31 IU/l | OR (95% CI) for FLI > 73% | OR (95% CI) for FGF-21 > Median | OR (95% CI) for FGF-21 > 3rd Tertile |
|---|---|---|---|---|---|
| Model 1 (adjusted for age and sex) | 3.16 (1.23–8.13) * | 4.82 (1.46–15.91) * | 2.42 (1.02–5.72) * | 2.43 (1.12–5.40) * | 2.53 (1.14–5.67) * |
| Model 2 (adjusted for age, sex, BMI, fasting glucose) | 3.11 (1.13–8.57) * | 4.31 (1.20–15.43) * | 1.85 (0.57–6.06) | 2.04 (0.89–4.75) | 2.46 (1.05–5.84) * |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Góralska, J.; Raźny, U.; Polus, A.; Dziewońska, A.; Gruca, A.; Zdzienicka, A.; Dembińska-Kieć, A.; Solnica, B.; Micek, A.; Kapusta, M.; et al. Enhanced GIP Secretion in Obesity Is Associated with Biochemical Alteration and miRNA Contribution to the Development of Liver Steatosis. Nutrients 2020, 12, 476. https://doi.org/10.3390/nu12020476
Góralska J, Raźny U, Polus A, Dziewońska A, Gruca A, Zdzienicka A, Dembińska-Kieć A, Solnica B, Micek A, Kapusta M, et al. Enhanced GIP Secretion in Obesity Is Associated with Biochemical Alteration and miRNA Contribution to the Development of Liver Steatosis. Nutrients. 2020; 12(2):476. https://doi.org/10.3390/nu12020476
Chicago/Turabian StyleGóralska, Joanna, Urszula Raźny, Anna Polus, Agnieszka Dziewońska, Anna Gruca, Anna Zdzienicka, Aldona Dembińska-Kieć, Bogdan Solnica, Agnieszka Micek, Maria Kapusta, and et al. 2020. "Enhanced GIP Secretion in Obesity Is Associated with Biochemical Alteration and miRNA Contribution to the Development of Liver Steatosis" Nutrients 12, no. 2: 476. https://doi.org/10.3390/nu12020476
APA StyleGóralska, J., Raźny, U., Polus, A., Dziewońska, A., Gruca, A., Zdzienicka, A., Dembińska-Kieć, A., Solnica, B., Micek, A., Kapusta, M., Słowińska-Solnica, K., & Malczewska-Malec, M. (2020). Enhanced GIP Secretion in Obesity Is Associated with Biochemical Alteration and miRNA Contribution to the Development of Liver Steatosis. Nutrients, 12(2), 476. https://doi.org/10.3390/nu12020476