Endocrine Disruptor Bisphenol A (BPA) Triggers Systemic Para-Inflammation and is Sufficient to Induce Airway Allergic Sensitization in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Experiments
2.2. Bronchoalveolar Lavage, Lung Digestion, and Flow Cytometry
2.3. Cell Culture
2.4. Quantitative PCR
2.5. Cytotoxicity Assay
2.6. Measurements of Serum Proteins and Antibodies
2.7. Statistical Analysis
3. Results
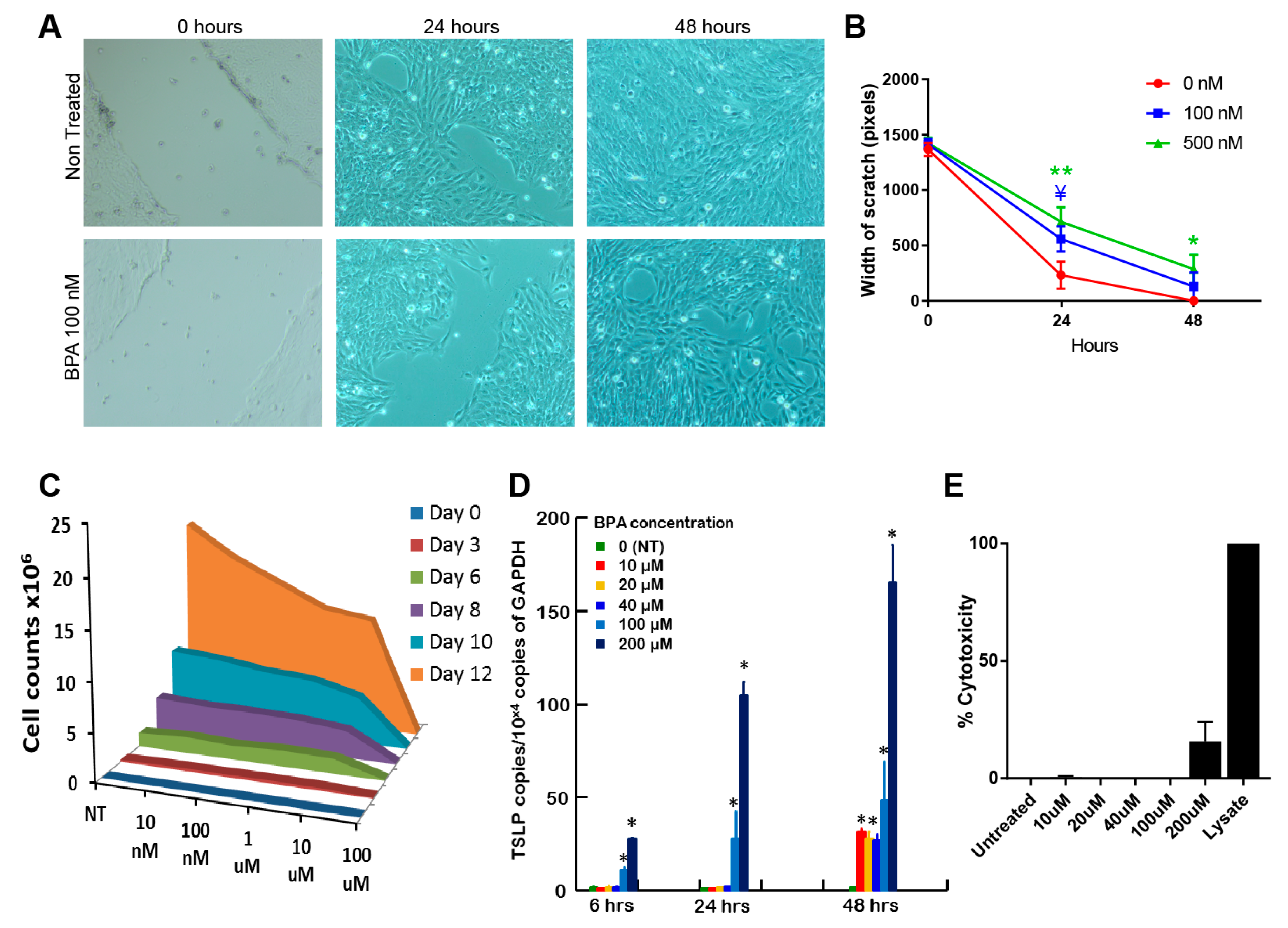
3.1. Bisphenol A Has An Inhibitory Effect on BEAS-2B Epithelial Cell Proliferation and Wound Healing
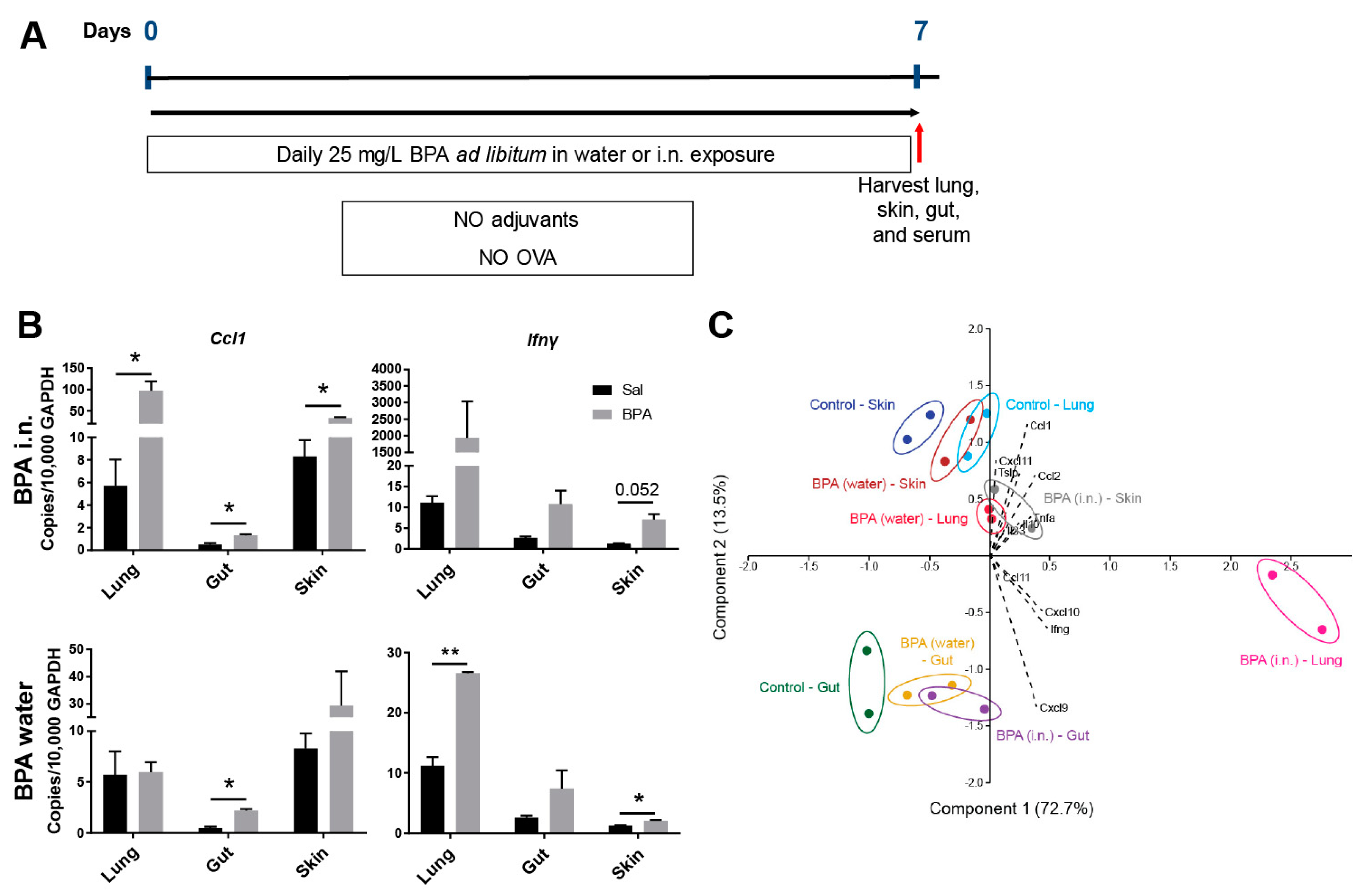
3.2. Ingestion of BPA Promotes Systemic Para-Inflammation in Mice
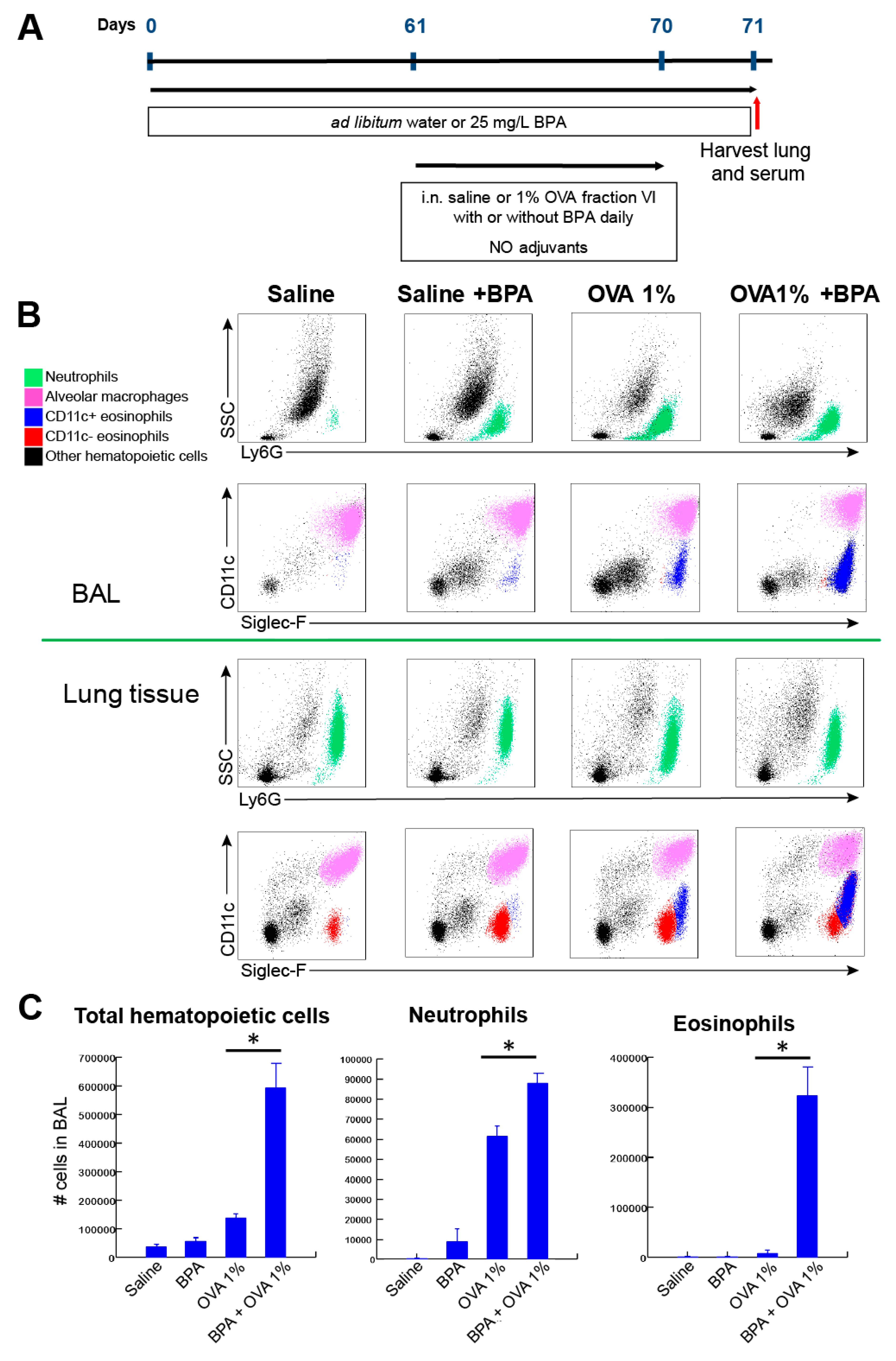
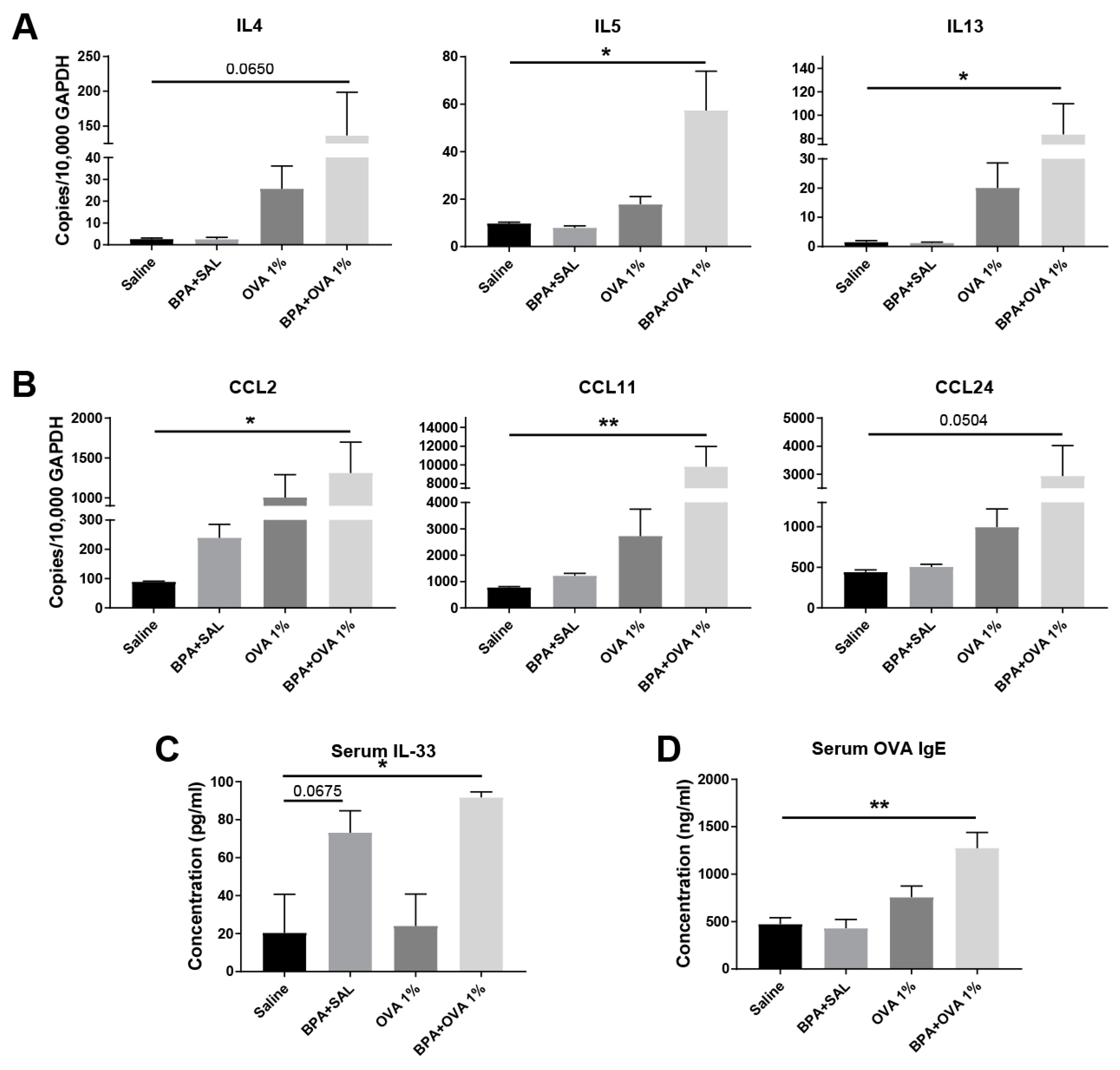
3.3. Chronic Systemic BPA Exposure Induces Allergic Sensitization to Innocuous OVA Antigen Exposure and Facilitates the Development of Allergic Inflammation
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Van Ree, R.; Hummelshoj, L.; Plantinga, M.; Poulsen, L.K.; Swindle, E. Allergic sensitization: Host-immune factors. Clin. Transl. Allergy 2014, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, K.M.; Konstantinou, G.N.; Pilapil, M.; Arrieta, M.C.; Noone, S.; Sampson, H.A.; Meddings, J.; Nowak-Wegrzyn, A. Intestinal permeability in children with food allergy on specific elimination diets. Pediatr. Allergy Immunol. 2013, 24, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.; Wolf, D. Abnormal epidermal barrier in the pathogenesis of atopic dermatitis. Clin. Dermatol. 2012, 30, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Puddicombe, S.M.; Field, S.; Haywood, J.; Broughton-Head, V.; Puxeddu, I.; Haitchi, H.M.; Vernon-Wilson, E.; Sammut, D.; Bedke, N.; et al. Defective epithelial barrier function in asthma. J. Allergy Clin. Immunol. 2011, 128, 549–556 e541-512. [Google Scholar] [CrossRef]
- Perrier, C.; Corthesy, B. Gut permeability and food allergies. Clin. Exp. Allergy 2011, 41, 20–28. [Google Scholar] [CrossRef]
- Fallon, P.G.; Sasaki, T.; Sandilands, A.; Campbell, L.E.; Saunders, S.P.; Mangan, N.E.; Callanan, J.J.; Kawasaki, H.; Shiohama, A.; Kubo, A.; et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet. 2009, 41, 602–608. [Google Scholar] [CrossRef]
- Chan, L.S.; Robinson, N.; Xu, L. Expression of interleukin-4 in the epidermis of transgenic mice results in a pruritic inflammatory skin disease: An experimental animal model to study atopic dermatitis. J. Invest. Dermatol. 2001, 117, 977–983. [Google Scholar] [CrossRef]
- Li, B.; Zou, Z.; Meng, F.; Raz, E.; Huang, Y.; Tao, A.; Ai, Y. Dust mite-derived Der f 3 activates a pro-inflammatory program in airway epithelial cells via PAR-1 and PAR-2. Mol. Immunol. 2019, 109, 1–11. [Google Scholar] [CrossRef]
- Hiraishi, Y.; Yamaguchi, S.; Yoshizaki, T.; Nambu, A.; Shimura, E.; Takamori, A.; Narushima, S.; Nakanishi, W.; Asada, Y.; Numata, T.; et al. IL-33, IL-25 and TSLP contribute to development of fungal-associated protease-induced innate-type airway inflammation. Sci. Rep. 2018, 8, 18052. [Google Scholar] [CrossRef]
- Schleimer, R.P.; Berdnikovs, S. Etiology of epithelial barrier dysfunction in patients with type 2 inflammatory diseases. J. Allergy Clin. Immunol. 2017, 139, 1752–1761. [Google Scholar] [CrossRef]
- Bonds, R.S.; Midoro-Horiuti, T. Estrogen effects in allergy and asthma. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.; Alqahtani, S.S.; Alshahrani, S. Diet: A Source of Endocrine Disruptors. Endocr. Metab. Immune Disord. Drug Targets 2019. [Google Scholar] [CrossRef] [PubMed]
- Paterni, I.; Granchi, C.; Minutolo, F. Risks and benefits related to alimentary exposure to xenoestrogens. Crit. Rev. Food Sci. Nutr. 2017, 57, 3384–3404. [Google Scholar] [CrossRef] [PubMed]
- Welshons, W.V.; Nagel, S.C.; vom Saal, F.S. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol A at levels of human exposure. Endocrinology 2006, 147, S56–S69. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Goldblum, R.M.; Midoro-Horiuti, T. Fetal exposure to bisphenol A as a risk factor for the development of childhood asthma: An animal model study. Environ. Health 2012, 11, 8. [Google Scholar] [CrossRef]
- Donohue, K.M.; Miller, R.L.; Perzanowski, M.S.; Just, A.C.; Hoepner, L.A.; Arunajadai, S.; Canfield, S.; Resnick, D.; Calafat, A.M.; Perera, F.P.; et al. Prenatal and postnatal bisphenol A exposure and asthma development among inner-city children. J. Allergy Clin. Immunol. 2013, 131, 736–742. [Google Scholar] [CrossRef]
- Houston, T.J.; Ghosh, R. Untangling the association between environmental endocrine disruptive chemicals and the etiology of male genitourinary cancers. Biochem. Pharmacol. 2019, 113743. [Google Scholar] [CrossRef]
- Verdier-Sevrain, S.; Bonte, F.; Gilchrest, B. Biology of estrogens in skin: Implications for skin aging. Exp. Dermatol. 2006, 15, 83–94. [Google Scholar] [CrossRef]
- Morani, A.; Warner, M.; Gustafsson, J.A. Biological functions and clinical implications of oestrogen receptors alfa and beta in epithelial tissues. J. Intern. Med. 2008, 264, 128–142. [Google Scholar] [CrossRef]
- Fu, X.D.; Simoncini, T. Extra-nuclear signaling of estrogen receptors. IUBMB Life 2008, 60, 502–510. [Google Scholar] [CrossRef]
- Roarty, K.; Rosen, J.M. Wnt and mammary stem cells: Hormones cannot fly wingless. Curr. Opin. Pharmacol. 2010, 10, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Skandalis, S.S.; Afratis, N.; Smirlaki, G.; Nikitovic, D.; Theocharis, A.D.; Tzanakakis, G.N.; Karamanos, N.K. Cross-talk between estradiol receptor and EGFR/IGF-IR signaling pathways in estrogen-responsive breast cancers: Focus on the role and impact of proteoglycans. Matrix Biol. 2014, 35, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yi, B.; Zhou, X.; Wu, Y.; Wang, L. Overexpression Of ERbeta Participates In The Progression Of Liver Cancer Via Inhibiting The Notch Signaling Pathway. Onco Targets Ther. 2019, 12, 8715–8724. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, E.M.; Maggiolini, M.; Musti, A.M. Crosstalk between Notch, HIF-1alpha and GPER in Breast Cancer EMT. Int. J. Mol. Sci. 2018, 19, 2011. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Tan, Y.; Li, M.; Dey, S.K.; Das, S.K. Canonical Wnt signaling is critical to estrogen-mediated uterine growth. Mol. Endocrinol. 2004, 18, 3035–3049. [Google Scholar] [CrossRef] [PubMed]
- Abdala Valencia, H.; Loffredo, L.F.; Misharin, A.V.; Berdnikovs, S. Phenotypic plasticity and targeting of Siglec-F(high) CD11c(low) eosinophils to the airway in a murine model of asthma. Allergy 2016, 71, 267–271. [Google Scholar] [CrossRef]
- Bryce, P.J.; Geha, R.; Oettgen, H.C. Desloratadine inhibits allergen-induced airway inflammation and bronchial hyperresponsiveness and alters T-cell responses in murine models of asthma. J. Allergy Clin. Immunol. 2003, 112, 149–158. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Kintscher, U. Metabolic impact of estrogen signalling through ERalpha and ERbeta. J. Steroid Biochem. Mol. Biol. 2010, 122, 74–81. [Google Scholar] [CrossRef]
- Honeth, G.; Lombardi, S.; Ginestier, C.; Hur, M.; Marlow, R.; Buchupalli, B.; Shinomiya, I.; Gazinska, P.; Bombelli, S.; Ramalingam, V.; et al. Aldehyde dehydrogenase and estrogen receptor define a hierarchy of cellular differentiation in the normal human mammary epithelium. Breast Cancer Res. 2014, 16, R52. [Google Scholar] [CrossRef]
- Wang, M.H.; Baskin, L.S. Endocrine disruptors, genital development, and hypospadias. J. Androl. 2008, 29, 499–505. [Google Scholar] [CrossRef]
- Barros, R.P.; Gustafsson, J.A. Estrogen receptors and the metabolic network. Cell Metab. 2011, 14, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Faulds, M.H.; Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.A. The diversity of sex steroid action: Regulation of metabolism by estrogen signaling. J. Endocrinol. 2012, 212, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Cruz, S.; Togno-Pierce, C.; Morales-Montor, J. Non-reproductive effects of sex steroids: Their immunoregulatory role. Curr. Top. Med. Chem. 2011, 11, 1714–1727. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Janesick, A.; Blumberg, B.; Heindel, J.J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 2011, 127, 204–215. [Google Scholar] [CrossRef]
- Rochester, J.R. Bisphenol A and human health: A review of the literature. Reprod. Toxicol. 2013, 42, 132–155. [Google Scholar] [CrossRef]
- Crews, D.; McLachlan, J.A. Epigenetics, evolution, endocrine disruption, health, and disease. Endocrinology 2006, 147, S4–S10. [Google Scholar] [CrossRef]
- Tajiki-Nishino, R.; Makino, E.; Watanabe, Y.; Tajima, H.; Ishimota, M.; Fukuyama, T. Oral Administration of Bisphenol A Directly Exacerbates Allergic Airway Inflammation but Not Allergic Skin Inflammation in Mice. Toxicol. Sci. 2018, 165, 314–321. [Google Scholar] [CrossRef]
- Koike, E.; Yanagisawa, R.; Win-Shwe, T.T.; Takano, H. Exposure to low-dose bisphenol A during the juvenile period of development disrupts the immune system and aggravates allergic airway inflammation in mice. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418774897. [Google Scholar] [CrossRef]
- He, M.; Ichinose, T.; Yoshida, S.; Takano, H.; Nishikawa, M.; Shibamoto, T.; Sun, G. Exposure to bisphenol A enhanced lung eosinophilia in adult male mice. Allergy Asthma Clin. Immunol. 2016, 12, 16. [Google Scholar] [CrossRef]
- Yanagisawa, R.; Koike, E.; Win-Shwe, T.T.; Takano, H. Oral exposure to low dose bisphenol A aggravates allergic airway inflammation in mice. Toxicol. Rep. 2019, 6, 1253–1262. [Google Scholar] [CrossRef]
- Mendy, A.; Salo, P.M.; Wilkerson, J.; Feinstein, L.; Ferguson, K.K.; Fessler, M.B.; Thorne, P.S.; Zeldin, D.C. Association of urinary levels of bisphenols F and S used as bisphenol A substitutes with asthma and hay fever outcomes. Environ. Res. 2019, 108944. [Google Scholar] [CrossRef]
- Youssef, M.M.; El-Din, E.; AbuShady, M.M.; El-Baroudy, N.R.; Abd El Hamid, T.A.; Armaneus, A.F.; El Refay, A.S.; Hussein, J.; Medhat, D.; Latif, Y.A. Urinary bisphenol A concentrations in relation to asthma in a sample of Egyptian children. Hum. Exp. Toxicol. 2018, 37, 1180–1186. [Google Scholar] [CrossRef]
- Zhou, A.; Chang, H.; Huo, W.; Zhang, B.; Hu, J.; Xia, W.; Chen, Z.; Xiong, C.; Zhang, Y.; Wang, Y.; et al. Prenatal exposure to bisphenol A and risk of allergic diseases in early life. Pediatr. Res. 2017, 81, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Hauser, R.; Marcus, M.; Olea, N.; Welshons, W.V. Human exposure to bisphenol A (BPA). Reprod. Toxicol. 2007, 24, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Ordonez-Moran, P.; Munoz, A. Nuclear receptors: Genomic and non-genomic effects converge. Cell Cycle 2009, 8, 1675–1680. [Google Scholar] [CrossRef] [PubMed]
- Schmuth, M.; Watson, R.E.; Deplewski, D.; Dubrac, S.; Zouboulis, C.C.; Griffiths, C.E. Nuclear hormone receptors in human skin. Horm. Metab. Res. 2007, 39, 96–105. [Google Scholar] [CrossRef]
- McPherson, S.J.; Ellem, S.J.; Risbridger, G.P. Estrogen-regulated development and differentiation of the prostate. Differentiation 2008, 76, 660–670. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, V.; Edwards, D.P. Receptor mechanisms mediating non-genomic actions of sex steroids. Semin. Reprod. Med. 2007, 25, 139–153. [Google Scholar] [CrossRef]
- Demehri, S.; Liu, Z.; Lee, J.; Lin, M.H.; Crosby, S.D.; Roberts, C.J.; Grigsby, P.W.; Miner, J.H.; Farr, A.G.; Kopan, R. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008, 6, e123. [Google Scholar] [CrossRef]
- Lobaccaro, J.M.; Trousson, A. Environmental estrogen exposure during fetal life: A time bomb for prostate cancer. Endocrinology 2014, 155, 656–658. [Google Scholar] [CrossRef][Green Version]
- Takai, T. TSLP expression: Cellular sources, triggers, and regulatory mechanisms. Allergol. Int. 2012, 61, 3–17. [Google Scholar] [CrossRef]
- Heijink, I.H.; Kies, P.M.; Kauffman, H.F.; Postma, D.S.; van Oosterhout, A.J.; Vellenga, E. Down-regulation of E-cadherin in human bronchial epithelial cells leads to epidermal growth factor receptor-dependent Th2 cell-promoting activity. J. Immunol. 2007, 178, 7678–7685. [Google Scholar] [CrossRef]
- Lee, K.H.; Cho, K.A.; Kim, J.Y.; Kim, J.Y.; Baek, J.H.; Woo, S.Y.; Kim, J.W. Filaggrin knockdown and Toll-like receptor 3 (TLR3) stimulation enhanced the production of thymic stromal lymphopoietin (TSLP) from epidermal layers. Exp. Dermatol. 2011, 20, 149–151. [Google Scholar] [CrossRef]
- Chang, K.K.; Liu, L.B.; Li, H.; Mei, J.; Shao, J.; Xie, F.; Li, M.Q.; Li, D.J. TSLP induced by estrogen stimulates secretion of MCP-1 and IL-8 and growth of human endometrial stromal cells through JNK and NF-kappaB signal pathways. Int. J. Clin. Exp. Pathol. 2014, 7, 1889–1899. [Google Scholar]
- Wang, S.B.; Hu, K.M.; Seamon, K.J.; Mani, V.; Chen, Y.; Gronert, K. Estrogen negatively regulates epithelial wound healing and protective lipid mediator circuits in the cornea. FASEB J. 2012, 26, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Nakajima, Y.; Asano, K.; Nakatani, T. Topical estrogen application to wounds promotes delayed cutaneous wound healing in 80-week-old female mice. PLoS ONE 2019, 14, e0225880. [Google Scholar] [CrossRef] [PubMed]
- Hardman, M.J.; Ashcroft, G.S. Estrogen, not intrinsic aging, is the major regulator of delayed human wound healing in the elderly. Genome Biol. 2008, 9, R80. [Google Scholar] [CrossRef] [PubMed]
- Mukai, K.; Nakajima, Y.; Urai, T.; Komatsu, E.; Nasruddin; Sugama, J.; Nakatani, T. 17beta-Estradiol administration promotes delayed cutaneous wound healing in 40-week ovariectomised female mice. Int. Wound J. 2016, 13, 636–644. [Google Scholar] [CrossRef]
- Bishop, B.; Lloyd, C.M. CC chemokine ligand 1 promotes recruitment of eosinophils but not Th2 cells during the development of allergic airways disease. J. Immunol. 2003, 170, 4810–4817. [Google Scholar] [CrossRef]
- Rowe, J.; Heaton, T.; Kusel, M.; Suriyaarachchi, D.; Serralha, M.; Holt, B.J.; de Klerk, N.; Sly, P.D.; Holt, P.G. High IFN-gamma production by CD8+ T cells and early sensitization among infants at high risk of atopy. J. Allergy Clin. Immunol. 2004, 113, 710–716. [Google Scholar] [CrossRef]
- Chovatiya, R.; Medzhitov, R. Stress, inflammation, and defense of homeostasis. Mol. Cell 2014, 54, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loffredo, L.F.; Coden, M.E.; Berdnikovs, S. Endocrine Disruptor Bisphenol A (BPA) Triggers Systemic Para-Inflammation and is Sufficient to Induce Airway Allergic Sensitization in Mice. Nutrients 2020, 12, 343. https://doi.org/10.3390/nu12020343
Loffredo LF, Coden ME, Berdnikovs S. Endocrine Disruptor Bisphenol A (BPA) Triggers Systemic Para-Inflammation and is Sufficient to Induce Airway Allergic Sensitization in Mice. Nutrients. 2020; 12(2):343. https://doi.org/10.3390/nu12020343
Chicago/Turabian StyleLoffredo, Lucas Fedele, Mackenzie Elyse Coden, and Sergejs Berdnikovs. 2020. "Endocrine Disruptor Bisphenol A (BPA) Triggers Systemic Para-Inflammation and is Sufficient to Induce Airway Allergic Sensitization in Mice" Nutrients 12, no. 2: 343. https://doi.org/10.3390/nu12020343
APA StyleLoffredo, L. F., Coden, M. E., & Berdnikovs, S. (2020). Endocrine Disruptor Bisphenol A (BPA) Triggers Systemic Para-Inflammation and is Sufficient to Induce Airway Allergic Sensitization in Mice. Nutrients, 12(2), 343. https://doi.org/10.3390/nu12020343