Genomics in Personalized Nutrition: Can You “Eat for Your Genes”?


 ,
,
Abstract
1. Introduction
2. Genomic Architecture
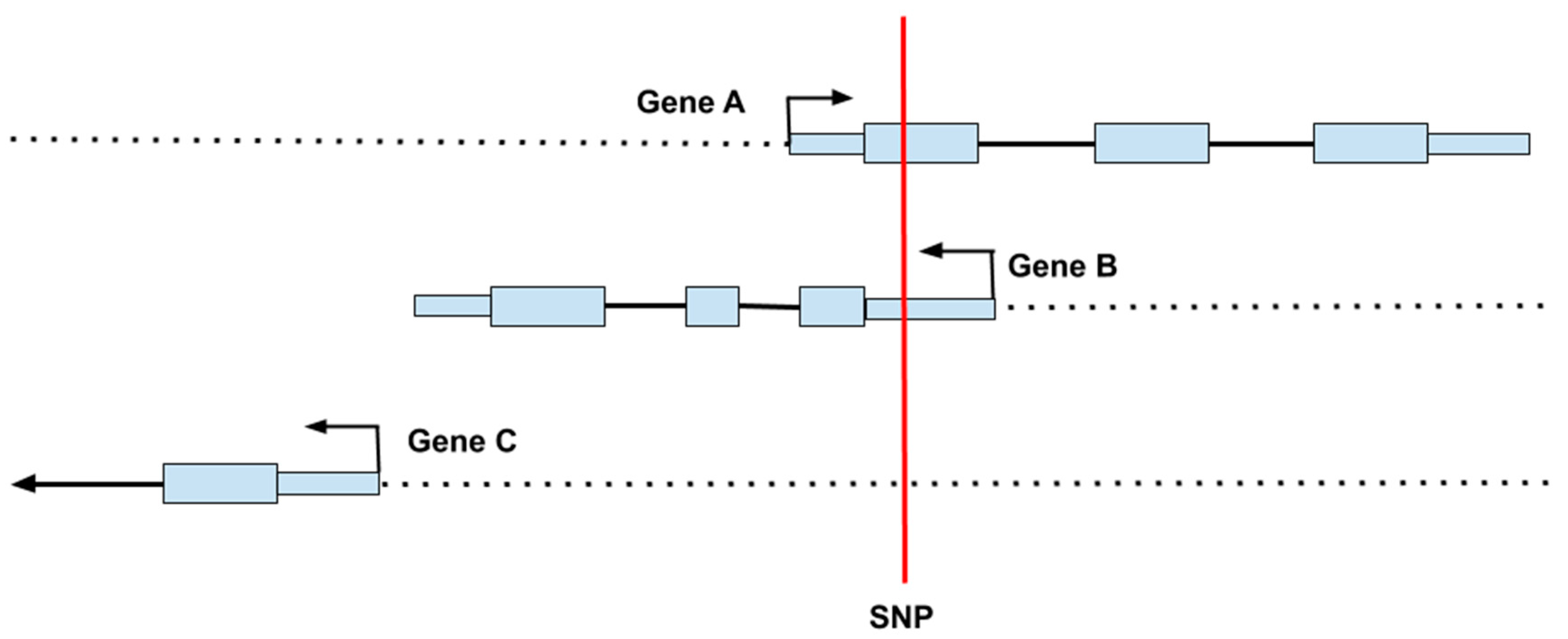
2.1. Genetic Variation
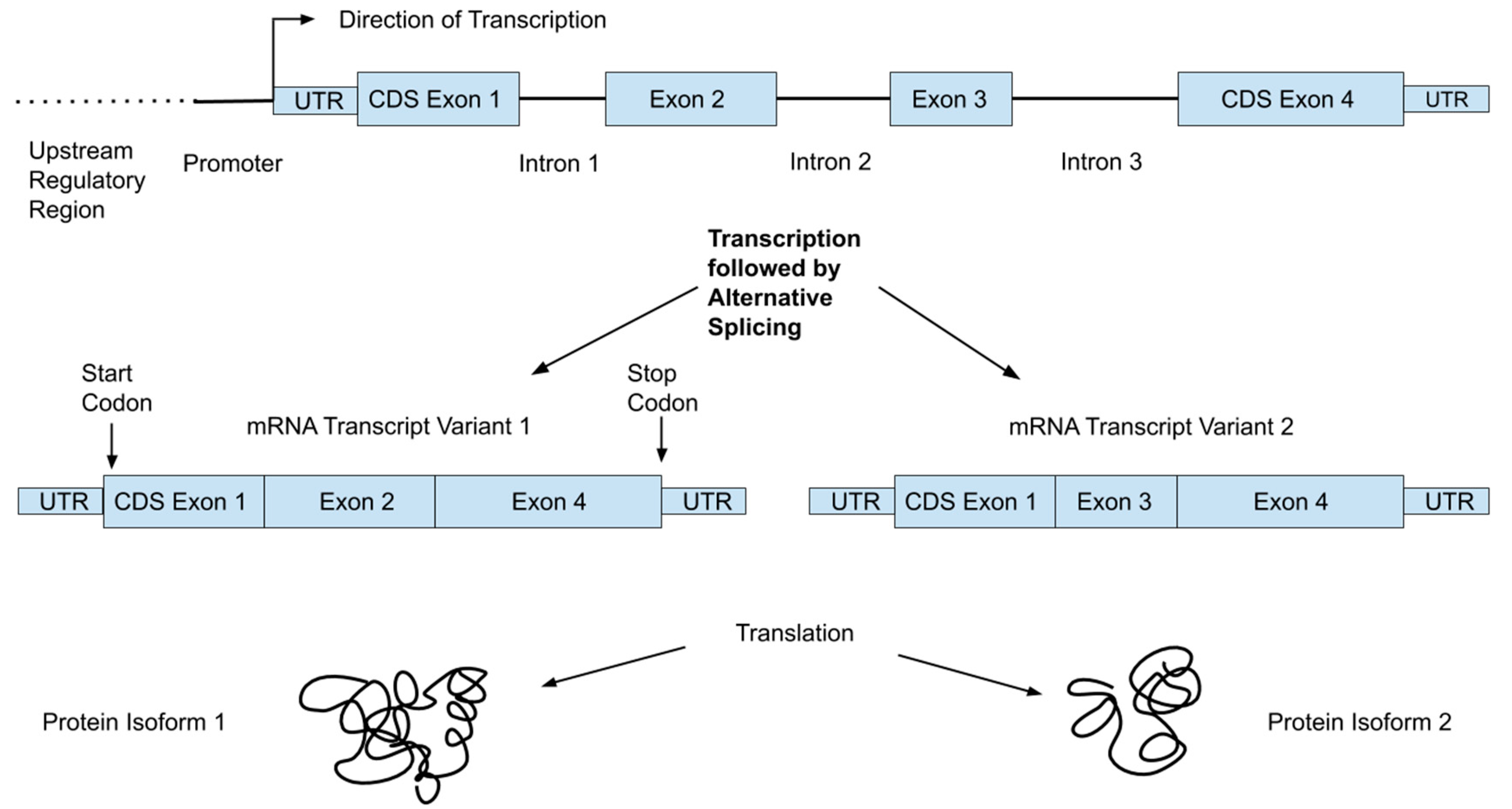
2.2. From Genotype to Phenotype
2.3. Penetrance, Pleiotropy, Epistasis, and Polygenicity
2.4. Genome-Wide Genetic Association Studies (GWAS)
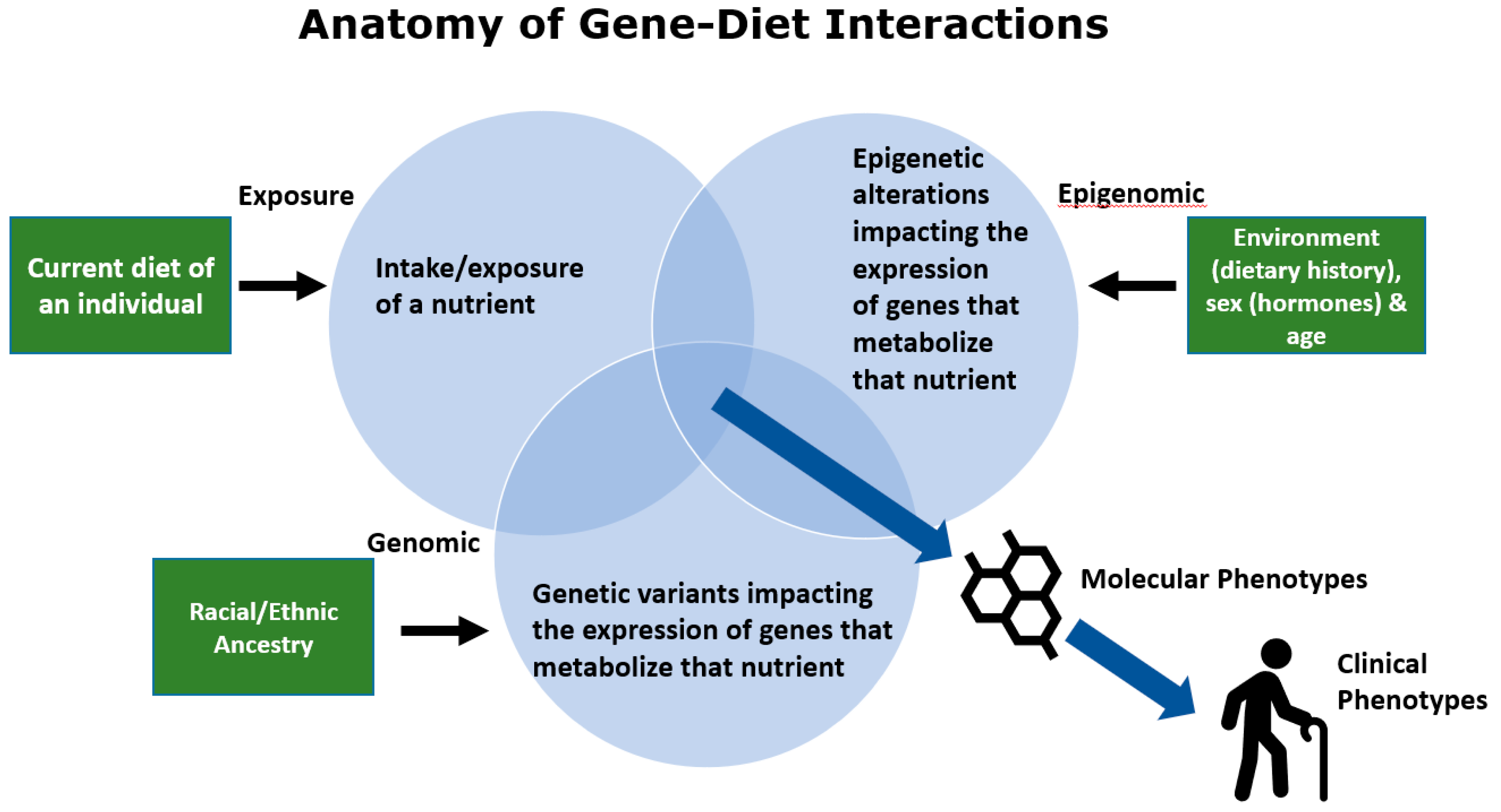
3. The Anatomy of Gene–Diet Interactions
4. Genetic Testing and Nutrigenetics
4.1. Caffeine Sensitivity (CYP1A2 Gene)
4.2. Alcohol Dependence (ADH1B Gene)
4.3. Non-Alcoholic Fatty Liver Disease (PNPLA3 Gene)
4.4. Obesity and Appetite (FTO Gene)
4.5. Cardiovascular and Alzheimer’s Disease (APOE Gene)
4.6. Folate Metabolism (MTHFR Gene)
4.7. Vitamin D Metabolism (GC Gene)
4.8. Long-Chain Fatty Acid Biosynthesis (FADS locus)
5. Direct-to-Consumer Genetic Testing (DTC-GT)
5.1. Scientific Validity, Reliability, and Accuracy
5.2. Ethical Considerations
5.3. Practical Applications
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Kim, J.W. Direct-to-consumer genetic testing. Genom. Inf. 2019, 17, e34. [Google Scholar] [CrossRef]
- Luca, F.; Perry, G.H.; Di Rienzo, A. Evolutionary adaptations to dietary changes. Annu. Rev. Nutr. 2010, 30, 291–314. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Hansen, M.E.; Lo, Y.; Tishkoff, S.A. Going global by adapting local: A review of recent human adaptation. Science 2016, 354, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Heine, R.G.; Alrefaee, F.; Bachina, P.; de Leon, J.C.; Geng, L.; Gong, S.; Madrazo, J.A.; Ngamphaiboon, J.; Ong, C.; Rogacion, J.M. Lactose intolerance and gastrointestinal cow’s milk allergy in infants and children—Common misconceptions revisited. World Allergy Organ. J. 2017, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Lapides, R.A.; Savaiano, D.A. Gender, age, race and lactose intolerance: Is there evidence to support a differential symptom response? A scoping review. Nutrients 2018, 10, 1956. [Google Scholar] [CrossRef] [PubMed]
- Caulfield, T.; Ries, N.M.; Ray, P.N.; Shuman, C.; Wilson, B. Direct-to-consumer genetic testing: Good, bad or benign? Clin. Genet. 2010, 77, 101–105. [Google Scholar] [CrossRef]
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94. [Google Scholar] [CrossRef]
- Larochelle, S. Protein isoforms: More than meets the eye. Nat. Methods 2016, 13, 291. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Belmont, J.W.; Hardenbol, P.; Willis, T.D.; Yu, F.; Yang, H.; Ch’ang, L.Y.; Huang, W.; Liu, B.; Shen, Y.; et al. The International HapMap Project. Nature 2003, 426, 789–796. [Google Scholar] [CrossRef]
- Li, J.Z.; Absher, D.M.; Tang, H.; Southwick, A.M.; Casto, A.M.; Ramachandran, S.; Cann, H.M.; Barsh, G.S.; Feldman, M.; Cavalli-Sforza, L.L.; et al. Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008, 319, 1100–1104. [Google Scholar] [CrossRef]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.R.; Marnellos, G.; Kammerer, S.; Hoyal, C.R.; Shi, M.M.; Cantor, C.R.; Braun, A. Large-scale validation of single nucleotide polymorphisms in gene regions. Genome Res. 2004, 14, 1664–1668. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.G.; Nielsen, R.; Signorovitch, J.; Matise, T.C.; Glanowski, S.; Heil, J.; Winn-Deen, E.S.; Holden, A.L.; Lai, E. Linkage disequilibrium and inference of ancestral recombination in 538 single-nucleotide polymorphism clusters across the human genome. Am. J. Hum. Genet. 2003, 73, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.F.; West, B.; Tuke, M.; Jones, S.E.; Patel, K.; Laver, T.W.; Beaumont, R.N.; Tyrell, J.; Wood, A.R.; Frayling, T.M.; et al. Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. Am. J. Hum. Genet. 2019, 104, 275–286. [Google Scholar] [CrossRef]
- Nikpay, M.; Goel, A.; Won, H.H.; Hall, L.M.; Willenborg, C.; Kanoni, S.; Saleheen, D.; Kyriakou, T.; Nelson, C.P.; Hopewell, J.C.; et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 2015, 47, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Fuchsberger, C.; Flannick, J.; Teslovich, T.M.; Mahajan, A.; Agarwala, V.; Gaulton, K.J.; Ma, C.; Fontanillas, P.; Moutsianas, L.; McCarthy, D.J.; et al. The genetic architecture of type 2 diabetes. Nature 2016, 536, 41–47. [Google Scholar] [CrossRef]
- Marcotte, B.V.; Guénard, F.; Marquis, J.; Charpagne, A.; Vadillo-Ortega, F.; Tejero, M.E.; Binia, A.; Vohl, M.-C. Genetic Risk Score Predictive of the Plasma Triglyceride Response to an Omega-3 Fatty Acid Supplementation in a Mexican Population. Nutrients 2019, 11, 737. [Google Scholar] [CrossRef]
- Solovieff, N.; Cotsapas, C.; Lee, P.H.; Purcell, S.M.; Smoller, J.W. Pleiotropy in complex traits: Challenges and strategies. Nat. Rev. Genet. 2013, 14, 483–495. [Google Scholar] [CrossRef]
- Nagel, R.L. Pleiotropic and epistatic effects in sickle cell anemia. Curr. Opin. Hematol. 2001, 8, 105–110. [Google Scholar] [CrossRef]
- Moore, J.H.; Williams, S.M. Epistasis and its implications for personal genetics. Am. J. Hum. Genet. 2009, 85, 309–320. [Google Scholar] [CrossRef]
- Bordoni, L.; Marchegiani, F.; Piangerelli, M.; Napolioni, V.; Gabbianelli, R. Obesity-related genetic polymorphisms and adiposity indices in a young Italian population. IUBMB Life 2017, 69, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, C.; Felix, J.F.; Estrada, K.; Peters, M.J.; Herrera, L.; Kruithof, C.J.; Duijts, L.; Hofman, A.; van Duijn, C.M.; Uitterlinden, A.G.; et al. Challenges in conducting genome-wide association studies in highly admixed multi-ethnic populations: The Generation R Study. Eur. J. Epidemiol. 2015, 30, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Chilton, F.H.; Dutta, R.; Reynolds, L.M.; Sergeant, S.; Mathias, R.A.; Seeds, M.C. Precision nutrition and omega-3 polyunsaturated fatty acids: A case for personalized supplementation approaches for the prevention and management of human diseases. Nutrients 2017, 9, 1165. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Stone, N.J.; Ballantyne, C.; Bittner, V.; Criqui, M.H.; Ginsberg, H.N.; Goldberg, A.C.; Howard, W.J.; Jacobson, M.S.; Kris-Etherton, P.M.; et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circ. J. 2011, 123, 2292–2333. [Google Scholar] [CrossRef]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef]
- Di Marzo, V.; Melck, D.; Bisogno, T.; de Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998, 21, 521–528. [Google Scholar] [CrossRef]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef]
- Okuyama, H.; Kobayashi, T.; Watanabe, S. Dietary fatty acids—The n-6/n-3 balance and chronic elderly diseases. Excess linoleic acid and relative n-3 deficiency syndrome seen in Japan. Prog. Lipid Res. 1996, 35, 409–457. [Google Scholar] [CrossRef]
- Chilton, F.H.; Murphy, R.C.; Wilson, B.A.; Sergeant, S.; Ainsworth, H.; Seeds, M.C.; Mathias, R.A. Diet-gene interactions and PUFA metabolism: A potential contributor to health disparities and human diseases. Nutrients 2014, 6, 1993–2022. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Pani, V.; Chilton, F.H. Genetic variants in the FADS gene: Implications for dietary recommendations for fatty acid intake. Curr. Nutr. Rep. 2014, 3, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Bersaglieri, T.; Sabeti, P.C.; Patterson, N.; Vanderploeg, T.; Schaffner, S.F.; Drake, J.A.; Rhodes, M.; Reich, D.E.; Hirschhorn, J.N. Genetic signatures of strong recent positive selection at the lactase gene. Am. J. Hum. Genet. 2004, 74, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Sergeant, S.; Ruczinski, I.; Torgerson, D.G.; Hugenschmidt, C.E.; Kubala, M.; Vaidya, D.; Suktitipat, B.; Ziegler, J.T.; Ivester, P.; et al. The impact of FADS genetic variants on omega6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet. 2011, 12, 50. [Google Scholar] [CrossRef]
- Sergeant, S.; Hugenschmidt, C.E.; Rudock, M.E.; Ziegler, J.T.; Ivester, P.; Ainsworth, H.C.; Vaidya, D.; Case, L.D.; Langefeld, C.D.; Freedman, B.I.; et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012, 107, 547–555. [Google Scholar] [CrossRef]
- Harris, D.N.; Ruczinski, I.; Yanek, L.R.; Becker, L.C.; Becker, D.M.; Guio, H.; Cui, T.; Chilton, F.H.; Mathias, R.A.; O’Connor, T.D. Evolution of hominin polyunsaturated fatty acid metabolism: From Africa to the New World. Genome Biol. Evol. 2019, 11, 1417–1430. [Google Scholar] [CrossRef]
- Malcomson, F.C.; Mathers, J.C. Nutrition, epigenetics and health through life. Nutr. Bull. 2017, 42, 254–265. [Google Scholar] [CrossRef]
- Choi, S.W.; Friso, S. Epigenetics: A new bridge between nutrition and health. Adv. Nutr. 2010, 1, 8–16. [Google Scholar] [CrossRef]
- Tobi, E.W.; Slieker, R.C.; Luijk, R.; Dekkers, K.; Stein, A.D.; Xu, K.M. Biobank-based Integrative Omics Studies, Consortium; Slagboom, P.E.; van Zwet, E.W.; Lumey, L.H.; et al. DNA methylation as a mediator of the association between prenatal adversity and risk factors for metabolic disease in adulthood. Sci. Adv. 2018, 4, eaao4364. [Google Scholar] [CrossRef]
- Roseboom, T.J.; Meulen, J.V.D.; Osmond, C.; Barker, D.J.P.; Ravelli, A.C.J.; Bleker, O.P. Plasma lipid profiles in adults after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2000, 72, 1101–1106. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Ahmed, A.; Tollefsbol, T.O. Epigenetic targets of bioactive dietary components for cancer prevention and therapy. Clin. Epigenetics 2010, 1, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Gómez, A.B.; Castillo-Lluva, S.; Sáez-Freire, M.D.M.; Hontecillas-Prieto, L.; Mao, J.H.; Castellanos-Martín, A.; Pérez-Losada, J. Missing heritability of complex diseases: Enlightenment by genetic variants from intermediate phenotypes. BioEssays 2016, 38, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Kvale, M.N.; Hesselson, S.E.; Zhan, Y.; Aquino, C.; Cao, Y.; Cawley, S.; Chung, E.; Connell, S.; Eshragh, J. Next generation genome-wide association tool: Design and coverage of a high-throughput European-optimized SNP array. Genomics 2011, 98, 79–89. [Google Scholar] [CrossRef]
- International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef]
- De Caterina, R.; El-Sohemy, A. Moving toward specific nutrigenetic recommendation algorithms: Caffeine, genetic variation and cardiovascular risk. Lifestyle Genom. 2016, 9, 106–115. [Google Scholar] [CrossRef]
- Frary, C.D.; Johnson, R.K.; Wang, M.Q. Food sources and intakes of caffeine in the diets of persons in the United States. J. Am. Diet Assoc. 2005, 105, 110–113. [Google Scholar] [CrossRef]
- Sachse, C.; Bhambra, U.; Smith, G.; Lightfoot, T.J.; Barrett, J.H.; Scollay, J.; Garner, R.C.; Boobis, A.R.; Wolf, C.R.; Gooderham, N.J. Colorectal Cancer Study Group. Polymorphisms in the cytochrome P450 CYP1A2 gene (CYP1A2) in colorectal cancer patients and controls: Allele frequencies, linkage disequilibrium and influence on caffeine metabolism. Br. J. Clin. Pharmacol. 2003, 55, 68–76. [Google Scholar] [CrossRef]
- Nehlig, A. Interindividual differences in caffeine metabolism and factors driving caffeine consumption. Pharmacol. Rev. 2018, 70, 384–411. [Google Scholar] [CrossRef]
- Womack, C.J.; Saunders, M.J.; Bechtel, M.K.; Bolton, D.J.; Martn, M.; Luden, N.D.; Dunham, W.; Hancock, M. The influence of a CYP1A2 polymorphism on the ergogenic effects of caffeine. J. Int. Soc. Sports Nutr. 2012, 9, 1–6. [Google Scholar] [CrossRef]
- Algrain, H.A.; Thomas, M.E.; Carrillo, A.E.; Ryan, E.J.; Kim, C.H.; LettanII, R.B.; Ryan, E.J. The Effects of a polymorphism in the cytochrome P450 CYP1A2 gene on performance enhancement with caffeine in recreational cyclists. J. Caffeine Res. 2016, 6, 34–39. [Google Scholar] [CrossRef]
- Salinero, J.J.; Lara, B.; Ruiz-Vicente, D.; Areces, F.; Puente-Torres, C.; Gallo-Salazar, C.; Pascual, T.; del Coso, J. CYP1A2 genotype variations do not modify the benefits and drawbacks of caffeine during exercise: A pilot study. Nutrients 2017, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar] [PubMed]
- Hubáček, J.A.; Šedová, L.; Olišarová, V.; Adámková, V.; Adámek, V.; Tóthová, V. Distribution of ADH1B genotypes predisposed to enhanced alcohol consumpton in the Czech Roma/Gypsy population. Cent. Eur. J. Public Health 2018, 26, 284–288. [Google Scholar] [CrossRef]
- Hurley, T.D. Genes encoding enzymes involved in ethanol metabolism. Alcohol Res. Health 2012, 34, 339–344. [Google Scholar]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- William, B.; Salt, M., II. Nonalcoholic fatty liver disease (NAFLD): A comprehensive review. J. Insur. Med. 2004, 36, 27–41. [Google Scholar]
- Chen, L.Z.; Xin, Y.-N.; Geng, N.; Jiang, M.; Zhang, D.D.; Xuan, S.Y. PNPLA3 I148M variant in nonalcoholic fatty liver disease: Demographic and ethnic characteristics and the role of the variant in nonalcoholic fatty liver fibrosis. World J. Gastroenterol. 2015, 21, 794–802. [Google Scholar] [CrossRef]
- Bertot, L.C.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef]
- Mazo, D.F.; Malta, F.M.; Stefano, J.T.; Salles, A.P.M.; Gomes-Gouvea, M.S.; Nastri, A.C.S.; Almeida, J.R.; Pinho, J.R.R.; Carrilho, F.J.; Oliveira, C.P. Validation of PNPLA3 polymorphisms as risk factor for NAFLD and liver fibrosis in an admixed population. Ann. Hepatol. 2019, 18, 466–471. [Google Scholar] [CrossRef]
- Sookoian, S.P.C. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Kumari, M.S.G.; Chitraju, C.; Paar, M.; Cornaciu, I.; Rangrez, A.Y.; Wongsiriroj, N.; Nagy, H.M.; Ivanova, P.T.; Scott, S.A. Adiponutrin functions as a nutritionally regulated lysophosphatidic acid acyltransferase. Cell Metab. 2012, 15, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Trepo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.L.P.; Li, X.; Zhou, X.; He, S. Association between PNPLA3 rs738409 polymorphism and nonalcoholic fatty liver disease (NAFLD) susceptibility and severity: A meta-analysis. Medicine 2019, 98, e14324. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.W.; Belbin, G.M.; Sorokin, E.P.; van Vleck, T.; Wojcik, G.L.; Moscati, A.; Gignoux, C.R.; Cho, J.; Abul-Husn, N.S.; Nadkarni, G.; et al. A common variant in PNPLA3 is associated with age at diagnosis of NAFLD in patients from a multi-ethnic biobank. J. Hepatol. 2020, 72, 1070–1081. [Google Scholar] [CrossRef]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Rustichelli, A.; Dongiovanni, P. Nutrition and genetics in NAFLD: The perfect binomium. Int. J. Mol. Sci. 2020, 21, 2986. [Google Scholar] [CrossRef]
- Peng, S.; Zhu, Y.; Xu, F.; Ren, X.; Li, X.; Lai, M. FTO gene polymorphisms and obesity risk: A meta-analysis. BMC Med. 2011, 9, 71. [Google Scholar] [CrossRef]
- Tanofsky-Kraff, M.; Han, J.C.; Anandalingam, K.; Shomaker, L.B.; Columbo, K.M.; Wolkoff, L.E.; Kozlosky, M.; Elliott, C.; Ranzenhofer, L.M.; Roza, C.A.; et al. The FTO gene rs9939609 obesity-risk allele and loss of control over eating. Am. J. Clin. Nutr. 2009, 90, 1483–1488. [Google Scholar] [CrossRef]
- Agagunduz, D.; Gezmen-Karadag, M. Association of FTO common variant (rs9939609) with body fat in Turkish individuals. Lipids Health Dis. 2019, 18, 212. [Google Scholar] [CrossRef]
- Fawcett, K.A.; Barroso, I. The genetics of obesity: FTO leads the way. Trends Genet. 2010, 26, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.B.; Feis, D.L.; Schilbach, L.; Kracht, L.; Hess, M.E.; Mauer, J.; Bruning, J.C.; Tittgemeyer, M. FTO gene variant modulates the neural correlates of visual food perception. Neuroimage 2016, 128, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Duicu, C.; Marginean, C.O.; Voidazan, S.; Tripon, F.; Banescu, C. FTO rs 9939609 SNP is associated with adiponectin and leptin levels and the risk of obesity in a cohort of Romanian children population. Medicine 2016, 95, e3709. [Google Scholar] [CrossRef] [PubMed]
- Hardy, D.S.; Gavin, J.T.; Mersha, T.B.; Racette, S.B. Ancestry specific associations of FTO gene variant and metabolic syndrome: A longitudinal ARIC study. Medicine 2020, 99, e18820. [Google Scholar] [CrossRef] [PubMed]
- Speakman, J.R.; Rance, K.A.; Johnstone, A.M. Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity 2008, 16, 1961–1965. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Axelsson, T.; Söderberg, S.; Larsson, A.; Ingelsson, E.; Lind, L.; Schiöth, H.B. Fat mass and obesity-associated gene (FTO) is linked to higher plasma levels of the hunger hormone ghrelin and lower serum levels of the satiety hormone leptin in older adults. Diabetes 2014, 63, 3955–3959. [Google Scholar] [CrossRef]
- Wasim, M.; Awan, F.R.; Najam, S.S.; Khan, A.R.; Khan, H.N. Role of leptin deficiency, inefficiency, and leptin receptors in obesity. Biochem. Genet. 2016, 54, 565–572. [Google Scholar] [CrossRef]
- Labayen, I.; Ruiz, J.R.; Ortega, F.B.; Dalongeville, J.; Jiménez-Pavón, D.; Castillo, M.J.; de Henauw, S.; González-Gross, M.; Bueno, G.; Molnár, D.; et al. Association between the FTO rs9939609 polymorphism and leptin in European adolescents: A possible link with energy balance control. The HELENA study. Int. J. Obes. 2010, 35, 66–71. [Google Scholar] [CrossRef]
- Da Silva, T.E.R.; Andrade, N.L.; Cunha, D.O.; Leão-Cordeiro, J.A.B.; Vilanova-Costa, C.A.S.T.; Silva, A.M.T.C. The FTO rs9939609 polymorphism and obesity risk in teens: Evidence-based meta-analysis. Obes. Res. Clin. Pract. 2018, 12, 432–437. [Google Scholar] [CrossRef]
- Martins, I.J.; Hone, E.; Foster, J.K.; Sunram-Lea, S.I.; Gnjec, A.; Fuller, S.J.; Nolan, D.; Gandy, S.E.; Martins, R.N. Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol. Psychiatry 2006, 11, 721–736. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Weng, R.; Gu, X.; Zhong, Z. Apolipoprotein E gene polymorphism and the risk of cardiovascular disease and type 2 diabetes. BMC Cardiovasc. Disord. 2019, 19, 213. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, L.; Gualtieri, P.; Romano, L.; Marrone, G.; Noce, A.; Pujia, A.; Perrone, M.A.; Aiello, V.; Coloca, C.; de Lorenzo, A. Role of Personalized Nutrition in Chronic-Degenerative Diseases. Nutrients 2019, 11, 1707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, H.Q.; Peng, W.J.; Zhang, B.B.; Liu, M. Meta-analysis for the association of apolipoprotein E ε2/ε3/ε4 polymorphism with coronary heart disease. Chin. Med. J. 2015, 128, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Easton, D.F. Apolipoprotein E genetic variation and Alzheimer’s disease. A meta-analysis. Dement. Geriatr. Cogn. Disord. 1999, 10, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Absolute 10-year risk of dementia by age, sex and APOE genotype: A population-based cohort study. Can. Med. Assoc. J. 2018, 190, E1033–E1041. [Google Scholar] [CrossRef]
- Liu, S.; Breitbart, A.; Sun, Y.; Mehta, P.D.; Boutajangout, A.; Scholtzova, H.; Wisniewski, T. Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology. J. Neurochem. 2014, 128, 577–591. [Google Scholar] [CrossRef]
- Liu, S.; Song, Y.; Hu, F.B.; Niu, T.; Ma, J.; Gaziano, M.; Stampfer, M.J. A prospective study of the APOA1 XmnI and APOC3 SstI polymorphisms in the APOA1/C3/A4 gene cluster and risk of incident myocardial infarction in men. Atherosclerosis 2004, 177, 119–126. [Google Scholar] [CrossRef]
- Ornish, D.; Scherwitz, L.W.; Billings, J.H.; Brown, S.E.; Gould, K.L.; Merritt, T.A.; Sparler, S.; Armstrong, W.T.; Ports, T.A.; Kirkeeide, R.L.; et al. Intensive lifestyle changes for reversal of coronary heart disease. JAMA 1998, 280, 2001–2007. [Google Scholar] [CrossRef]
- Arab, L.; Sabbagh, M.N. Are certain lifestyle habits associated with lower Alzheimer’s disease risk? J. Alzheimer’s Dis. 2010, 20, 785–794. [Google Scholar] [CrossRef]
- Abondio, P.; Sazzini, M.; Garagnani, P.; Boattini, A.; Monti, D.; Franceschi, C.; Luiselli, D.; Giuliani, C. The genetic variability of APOE in different human populations and its implications for longevity. Genes 2019, 10, 222. [Google Scholar] [CrossRef]
- Shiao, S.P.K.; Grayson, J.; Lie, A.; Yu, C.H. Personalized nutrition-genes, diet, and related interactive parameters as predictors of cancer in multiethnic colorectal cancer families. Nutrients 2018, 10, 795. [Google Scholar] [CrossRef] [PubMed]
- Sibani, S.; Christensen, B.; O’Ferrall, E.; Saadi, I.; Hiou-Tim, F.; Rosenblatt, D.S.; Rozen, R. Characterization of six novel mutations in the methylenetetrahydrofolate reductase (MTHFR) gene in patients with homocystinuria. Hum. Mutat. 2000, 15, 280–287. [Google Scholar] [CrossRef]
- Kauwell, G.P.; Wilsky, C.E.; Cerda, J.J.; Herrlinger-Garcia, K.; Hutson, A.D.; Theriaque, D.W.; Boddie, A.; Rampersaud, G.C.; Bailey, L.B. Methylenetetrahydrofolate reductase mutation (677C → T) negatively influences plasma homocysteine response to marginal folate intake in elderly women. Metabolism 2000, 49, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Liew, S.C.; Gupta, E.D. Methylenetetrahydrofolate reductase (MTHFR) C677T polymorphism: Epidemiology, metabolism and the associated diseases. Eur. J. Med. Genet. 2015, 58, 1–10. [Google Scholar] [CrossRef]
- Saxena, A.K.; Singh, V.; Agarwal, M.; Tiwari, M.; Kumar, K.; Ramanuj, K.; Aphrodite, C. Pratap Patra and Sanjeev Kumar. Penetrance of MTHFR, MTRR and SHMT gene polymorphism modulate folate metabolism in maternal blood and increases “Risk Factor” in neural tube defects in Eastern India. Hum. Genet. Embryol. 2018, 8, 2. [Google Scholar] [CrossRef]
- Jin, H.; Cheng, H.; Chen, W.; Sheng, X.; Levy, M.A.; Brown, M.J.; Tian, J. An evidence-based approach to globally assess the covariate-dependent effect of the MTHFR single nucleotide polymorphism rs1801133 on blood homocysteine: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2018, 107, 817–825. [Google Scholar] [CrossRef]
- Wang, X.; Wang, K.; Yan, J.; Wu, M. A meta-analysis on associations of FTO, MTHFR and TCF7L2 polymorphisms with polycystic ovary syndrome. Genomics 2020, 112, 1516–1521. [Google Scholar] [CrossRef]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef]
- Pludowski, P.; Holick, M.F.; Grant, W.B.; Konstantynowicz, J.; Mascarenhas, M.R.; Haq, A.; Povoroznyuk, V.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Biol. 2018, 175, 125–135. [Google Scholar] [CrossRef]
- Sepulveda-Villegas, M.; Elizondo-Montemayor, L.; Trevino, V. Identification and analysis of 35 genes associated with vitamin D deficiency: A systematic review to identify genetic variants. J. Steroid Biochem. Mol. Biol. 2020, 196, 105516. [Google Scholar] [CrossRef]
- Bikle, D.D.; Schwartz, J. Vitamin D binding protein, total and free vitamin D levels in different physiological and pathophysiological conditions. Front. Endocrinol. 2019, 10, 317. [Google Scholar] [CrossRef]
- Jolliffe, D.A.; Walton, R.T.; Griffiths, C.J.; Martineau, A.R. Single nucleotide polymorphisms in the vitamin D pathway associating with circulating concentrations of vitamin D metabolites and non-skeletal health outcomes: Review of genetic association studies. J. Steroid Biochem. 2016, 164, 18–29. [Google Scholar] [CrossRef]
- Chun, R.F.; Peercy, B.E.; Orwoll, E.S.; Nielson, C.M.; Adams, J.S.; Hewison, M. Vitamin D and DBP: The free hormone hypothesis revisited. J. Steroid Biochem. 2014, 144, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, V.S.; Jones, K.S.; Schoenmakers, I.; Silver, M.; Prentice, A.; Hennig, B.J. Vitamin D binding protein genotype is associated with plasma 25OHD concentration in West African children. Bone 2015, 74, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Lauridsen, A.L.; Vestergaard, P.; Nexo, E. Mean serum concentration of vitamin D-binding protein (Gc globulin) is related to the Gc phenotype in women. Clin. Chem. 2001, 47, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Jiang, S.; Muyiduli, X.; Wang, S.; Mo, M.; Li, M.; Wang, Z.; Yu, Y. Vitamin D pathway gene polymorphisms influenced vitamin D level among pregnant women. Clin. Nutr. 2018, 37, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Enlund-Cerullo, M.; Koljonen, L.; Holmlund-Suila, E.; Hauta-Alus, H.; Rosendahl, J.; Valkama, S.; Helve, O.; Hytinantti, T.; Viljakainen, H.; Andersson, S.; et al. Genetic variation of the vitamin D Binding protein affects vitamin D status and response to supplementation in infants. J. Clin. Endocrinol. Metab. 2019, 1, 5483–5498. [Google Scholar] [CrossRef] [PubMed]
- SanGiovanni, J.P.; Chew, E.Y. The role of omega-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog. Retin. Eye Res. 2005, 24, 87–138. [Google Scholar] [CrossRef]
- Reynolds, L.M.; Dutta, R.; Seeds, M.C.; Lake, K.N.; Hallmark, B.; Mathias, R.A.; Howard, T.D.; Chilton, F.H. FADS genetic and metabolomic analyses identify the Δ5 desaturase (FADS1) step as a critical control point in the formation of biologically important lipids. Sci. Rep. 2020, 10, 1–15873. [Google Scholar] [CrossRef]
- Park, H.G.; Park, W.J.; Kothapalli, K.S.; Brenna, J.T. The fatty acid desaturase 2 (FADS2) gene product catalyzes Delta4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015, 29, 3911–3919. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Kothapalli, K.S.; Brenna, J.T. Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesis. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, S.; Hallmark, B.; Mathias, R.A.; Mustin, T.L.; Ivester, P.; Bohannon, M.L.; Ruczinski, I.; Johnstone, L.; Seeds, M.C.; Chilton, F.H. Prospective clinical trial examining the impact of genetic variation in FADS1 on the metabolism of linoleic acid- and -linolenic acid-containing botanical oils. Am. J. Clin. Nutr. 2020, 111, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Lemaitre, R.N.; King, I.B.; Song, X.; Huang, H.; Sacks, F.M.; Rimm, E.B.; Wang, M.; Siscovick, D.S. Plasma phospholipid long-chain ω-3 fatty acids and total and cause-specific mortality in older adults: A cohort study. Ann. Intern. Med. 2013, 158, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, S.; Zhao, Y.; Luo, Y.; Tong, H.; Su, L. Correlation analysis of omega-3 fatty acids and mortality of sepsis and sepsis-induced ARDS in adults: Data from previous randomized controlled trials. Nutr. J. 2018, 17, 57. [Google Scholar] [CrossRef]
- Gorusupudi, A.; Liu, A.; Hageman, G.S.; Bernstein, P.S. Associations of human retinal very long-chain polyunsaturated fatty acids with dietary lipid biomarkers. J. Lipid Res. 2016, 57, 499–508. [Google Scholar] [CrossRef]
- Jensen, T.K.; Priskorn, L.; Holmboe, S.A.; Nassan, F.L.; Andersson, A.M.; Dalgård, C.; Petersen, J.H.; Chavarro, J.E.; Jørgensen, N. Associations of fish oil supplement use with testicular function in young men. JAMA Netw. Open 2020, 3, e1919462. [Google Scholar] [CrossRef]
- Guasch-Ferre, M.; Dashti, H.S.; Merino, J. Nutritional genomics and direct-to-consumer genetic testing: An Overview. Adv. Nutr. 2018, 9, 128–135. [Google Scholar] [CrossRef]
- Horton, R.; Crawford, G.; Freeman, L.; Fenwick, A.; Wright, C.F.; Lucassen, A. Direct-to-consumer genetic testing. Brit. Med. J. 2019, 367, l5688. [Google Scholar] [CrossRef]
- Tandy-Connor, S.; Guiltinan, J.; Krempely, K.; LaDuca, H.; Reineke, P.; Gutierrez, S.; Gray, P.; Tippin Davis, B. False-positive results released by direct-to-consumer genetic tests highlight the importance of clinical confirmation testing for appropriate patient care. Genet. Med. 2018, 20, 1515–1521. [Google Scholar] [CrossRef]
- Feero, W.G.; Wicklund, C.A. Consumer genomic testing in 2020. JAMA 2020. [Google Scholar] [CrossRef]
- Salloum, R.G.; George, T.J.; Silver, N.; Markham, M.J.; Hall, J.M.; Guo, Y.; Bian, J.; Shenkman, E.A. Rural-urban and racial-ethnic differences in awareness of direct-to-consumer genetic testing. BMC Public Health 2018, 18, 277. [Google Scholar] [CrossRef] [PubMed]
- Marshe, V.S.; Gorbovskaya, I.; Kanji, S.; Kish, M.; Muller, D.J. Clinical implications of APOE genotyping for late-onset Alzheimer’s disease (LOAD) risk estimation: A review of the literature. J. Neural Transm. 2019, 126, 65–85. [Google Scholar] [CrossRef] [PubMed]
- Allyse, M.A.; Robinson, D.H.; Ferber, M.J.; Sharp, R.R. Direct-to-consumer testing 2.0: Emerging models of direct-to-consumer genetic testing. Mayo Clin. Proc. 2018, 93, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Floris, M.; Cano, A.; Porru, L.; Addis, R.; Cambedda, A.; Idda, M.L.; Steri, M.; Ventura, C.; Maioli, M. Direct-to-Consumer Nutrigenetics Testing: An Overview. Nutrients 2020, 12, 566. [Google Scholar] [CrossRef]
- Celis-Morales, C.; Livingstone, K.M.; Marsaux, C.F.; Macready, A.L.; Fallaize, R.; O’Donovan, C.B.; Woolhead, C.; Forster, H.; Walsh, M.C.; Navas-Carretero, S.; et al. Effect of personalized nutrition on health-related behaviour change: Evidence from the Food4Me European randomized controlled trial. Int. J. Epidemiol. 2017, 46, 578–588. [Google Scholar] [CrossRef]
- Nielsen, D.E.; Carere, D.A.; Wang, C.; Roberts, J.S.; Green, R.C.; Group, P.G.S. Diet and exercise changes following direct-to-consumer personal genomic testing. BMC Med. Genom. 2017, 10, 24. [Google Scholar] [CrossRef]
- Kohlmeier, M.; de Caterina, R.; Ferguson, L.R.; Gorman, U.; Allayee, H.; Prasad, C.; Kang, J.X.; Nicoletti, C.F.; Martinez, J.A. Guide and position of the International Society of Nutrigenetics/Nutrigenomics on personalized nutrition: Part 2—Ethics, Challenges and Endeavors of Precision Nutrition. Lifestyle Genom. 2016, 9, 28–46. [Google Scholar] [CrossRef]
- Camp, K.M.; Trujillo, E. Position of the Academy of Nutrition and Dietetics: Nutritional Genomics. J. Acad. Nutr. Diet. 2014, 114, 299–312. [Google Scholar] [CrossRef]
- Nielsen, R.; Akey, J.M.; Jakobsson, M.; Pritchard, J.K.; Tishkoff, S.; Willerslev, E. Tracing the peopling of the world through genomics. Nature 2017, 541, 302–310. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Albert, C.M.; Gordon, D.; Copeland, T.; et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N. Engl. J. Med. 2019, 380, 23–32. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Effect Size | A measure of the size of a genetic association. Small effect sizes are common |
| Epistasis | When the effect of a variant depends on other genetic variants present (i.e., the genetic background) |
| Genotype | The two DNA bases at a given site, e.g., A/A, A/T or T/T, one from each parent |
| Genotype-Phenotype Map | The relationship between phenotypes and genotypes |
| Heritability | The degree to which a trait is transmitted across generations |
| INDEL | Insertion/deletion polymorphism |
| Linkage Disequilibrium | When nearby variants are passed down together through human lineages |
| Locus | A location in the genome |
| Penetrance | The probability of observing the associated phenotype for a given variant. |
| Phenotype | An observed trait, e.g., weight |
| Pleiotropy | When a single gene or variant controls multiple, sometimes unrelated traits |
| Polygenic | A phenotypic trait that is the result of small contributions from many genes |
| Site | A single DNA base-pair, i.e., A, C, G, or T, where the other half of the base-pair is implied |
| Single nucleotide polymorphism (SNP) | A site at which there are two common DNA base pairs in the population, e.g., A and T occur at 20 and 80% respectively |
| Variant | A DNA polymorphism, often a SNP |
| Causal (functional) SNP | A SNP that is responsible for the observed phenotypic association, e.g., a protein-altering mutation |
| Dietary exposure | The amount of a food or nutrient an individual or population consumes |
| Gene | SNP | Nutrition and Health Issue | Genotype Differences | ||
|---|---|---|---|---|---|
| CYP1A2 | rs762551 | Caffeine Metabolism | C/C slow metabolizer | A/C slow metabolizer | A/A rapid metabolizer |
| ADH1B | rs1229984 | Alcohol Metabolism | G/G | A/G Increased ETOH metabolism | A/A Increased ETOH metabolism |
| rs2066702 | G/G | A/G Increased ETOH metabolism | A/A Increased ETOH metabolism | ||
| PNPLA3 | rs738409 | Non-alcoholic fatty liver disease | C/C | G/C Increased fat accumulation | G/G Increase fat accumulation |
| FTO | rs9939609 | Obesity and Appetite | T/T | A/T Increased adiposity | A/A Increased adiposity |
| APOE | rs7412 | Cardiovascular and Alzheimer’s Disease | T/T Lowest AD risk | C/T | C/C Increased AD risk |
| rs429358 | T/T Lowest AD risk | C/T | C/C Increased AD risk | ||
| MTHFR | rs1801133 | Folate Metabolism | C/C | T/C Diminished enzyme activity | T/T Diminished enzyme activity |
| GC | rs7041 | Vitamin D Transport | TT | TG | GG Lower Serum 25(OH)D |
| rs4588 | CC | C/A | AA Lower Serum 25(OH)D | ||
| FADS1 | rs174537 | Long-Chain Fatty Acid Biosynthesis | G/G Most efficient | T/G Varied efficiency | T/T Inefficient |
| rs429358 | rs7412 | Genotype | Risk | Recommendation |
|---|---|---|---|---|
| C/C | C/C | APOE4/APOE4 | Highest | Low fat, plant-based diet |
| C/T | C/C | APOE3/APOE4 | Increased | Low fat, plant-based diet |
| T/T | C/C | APOE3/APOE3 | Average | Plant-centered diet |
| T/T | C/T | APOE2/APOE3 | Average | Plant-centered diet |
| T/T | T/T | APOE2/APOE2 | Lowest | No related restrictions |
| rs7041 | rs4588 | Haplotype | Risk |
|---|---|---|---|
| T (432D) | C (436T) | GC1f | Average |
| G (432E) | C (436 T) | GC1s | Average |
| T (432D) | A (436K) | GC2 | Highest |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mullins, V.A.; Bresette, W.; Johnstone, L.; Hallmark, B.; Chilton, F.H. Genomics in Personalized Nutrition: Can You “Eat for Your Genes”? Nutrients 2020, 12, 3118. https://doi.org/10.3390/nu12103118
Mullins VA, Bresette W, Johnstone L, Hallmark B, Chilton FH. Genomics in Personalized Nutrition: Can You “Eat for Your Genes”? Nutrients. 2020; 12(10):3118. https://doi.org/10.3390/nu12103118
Chicago/Turabian StyleMullins, Veronica A., William Bresette, Laurel Johnstone, Brian Hallmark, and Floyd H. Chilton. 2020. "Genomics in Personalized Nutrition: Can You “Eat for Your Genes”?" Nutrients 12, no. 10: 3118. https://doi.org/10.3390/nu12103118
APA StyleMullins, V. A., Bresette, W., Johnstone, L., Hallmark, B., & Chilton, F. H. (2020). Genomics in Personalized Nutrition: Can You “Eat for Your Genes”? Nutrients, 12(10), 3118. https://doi.org/10.3390/nu12103118