Mens sana in corpore sano: Does the Glycemic Index Have a Role to Play?

Abstract
1. Introduction
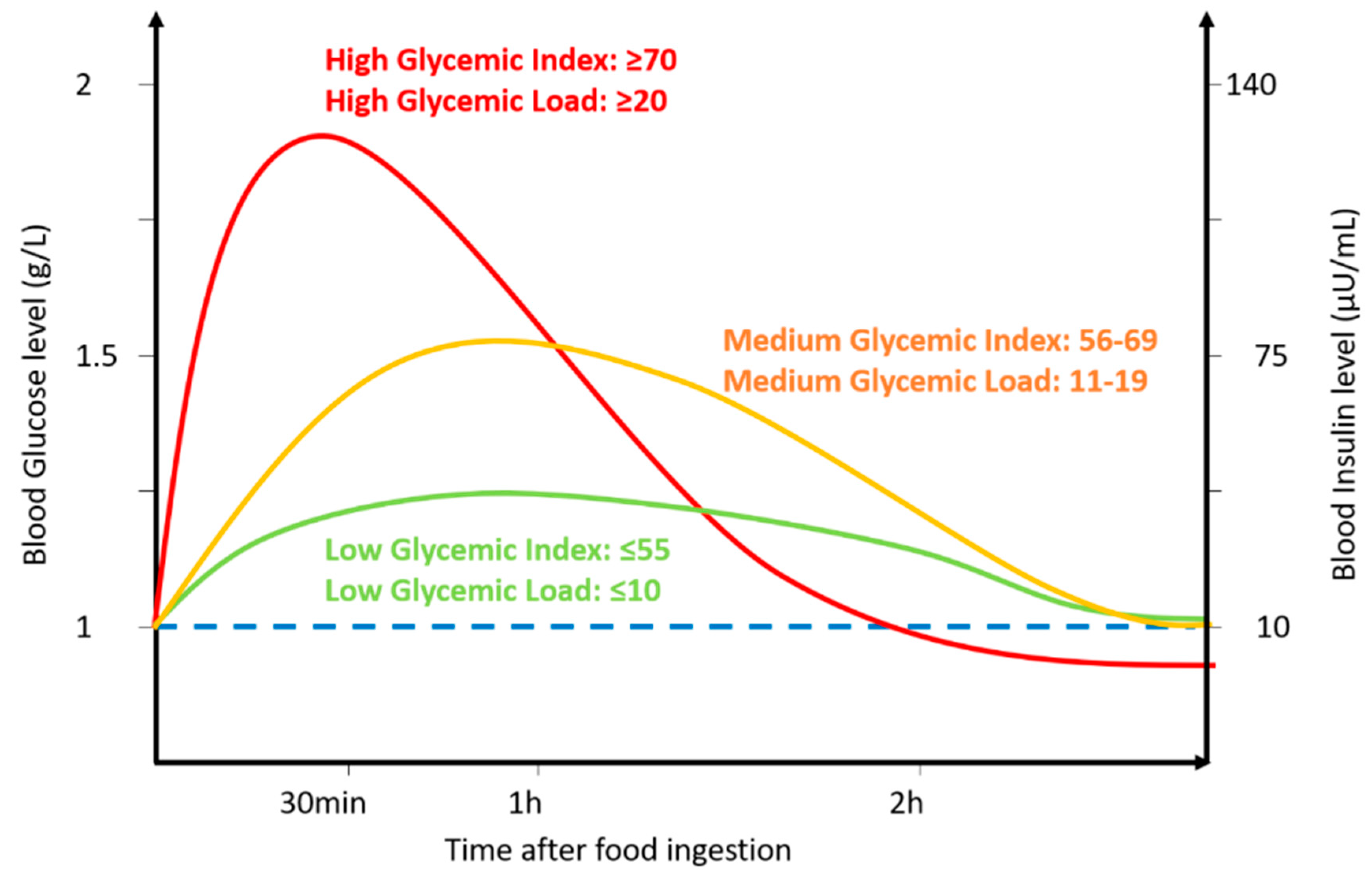
2. Effect of a Glycemic Index Diet on Brain Function
Effect of the Glycemic Index on Cognitive Function in Healthy People
3. Low-GI Diet and Neurological Dysfunctions
3.1. Epilepsy
3.2. Stroke
3.3. Alzheimer’s Disease (AD)
3.4. Others: Dementia, Depression, Mental Health, etc.
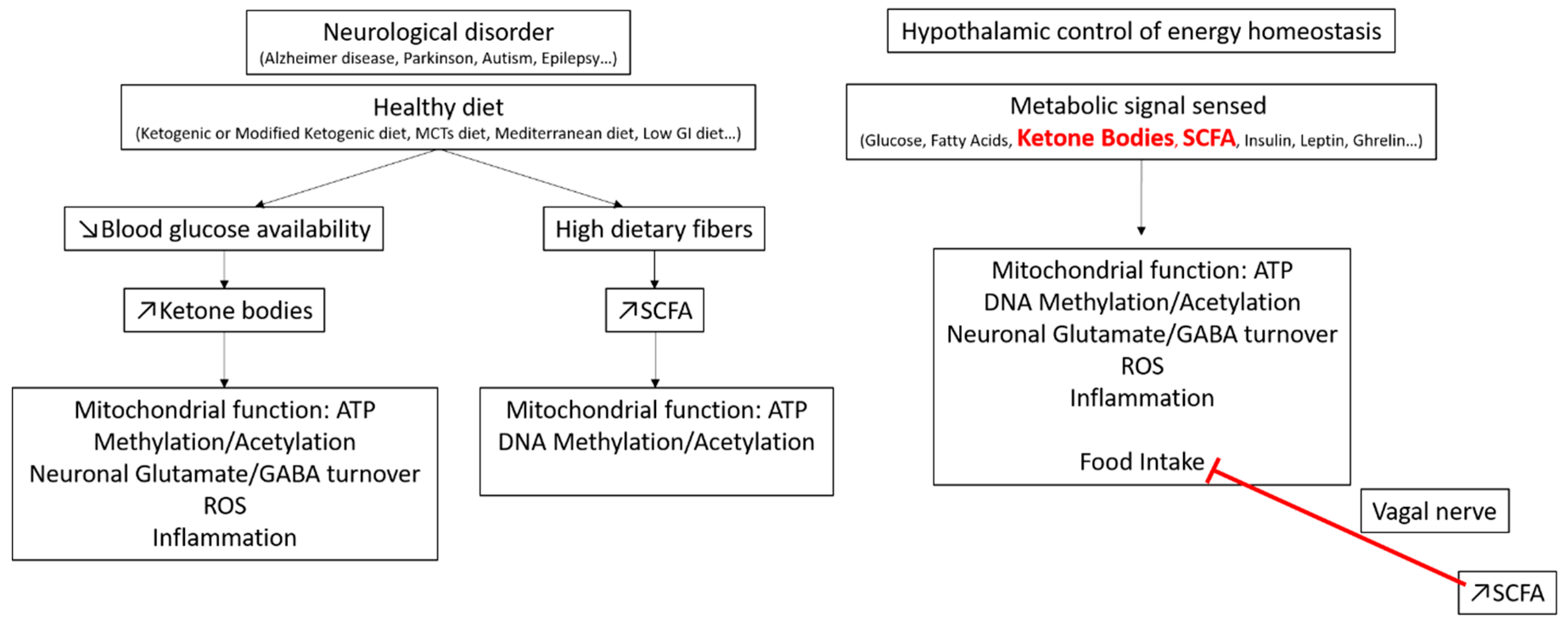
4. Glycemic Index and Brain Regulation of Energy Homeostasis
GI/GL and Brain Glucose Detection
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Jenkins, D.J.A.; Wolever, T.M.S.; Jenkins, A.L.; Thorne, M.J.; Lee, R.; Kalmusky, J.; Reichew, R.; Wong, G.S. The Glycaemic Index of Foods Tested in Diabetic Patients: A New Basis for Carbohydrate Exchange Favouring the Use of Legumes. Diabetologia 1983, 24, 257–264. [Google Scholar] [CrossRef]
- Venn, B.J.; Green, T.J. Glycemic index and glycemic load: Measurement issues and their effect on diet–disease relationships. Eur. J. Clin. Nutr. 2007, 61, S122–S131. [Google Scholar] [CrossRef]
- Galgani, J.; Aguirre, C.; Díaz, E. Acute effect of meal glycemic index and glycemic load on blood glucose and insulin responses in humans. Nutr. J. 2006, 5. [Google Scholar] [CrossRef]
- Butler, T.; Kerley, C.P.; Altieri, N.; Alvarez, J.; Green, J.; Hinchliffe, J.; Stanford, D.; Paterson, K. Optimum nutritional strategies for cardiovascular disease prevention and rehabilitation (BACPR). Heart 2020, 106, 724–731. [Google Scholar] [CrossRef]
- Williams, T.J.; Cervenka, M.C. The role for ketogenic diets in epilepsy and status epilepticus in adults. Clin. Neurophysiol. Pract. 2017, 2, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Vergati, M.; Krasniqi, E.; Monte, G.D.; Riondino, S.; Vallone, D.; Guadagni, F.; Ferroni, P.; Roselli, M. Ketogenic Diet and Other Dietary Intervention Strategies in the Treatment of Cancer. Curr. Med. Chem. 2017, 24. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Pinilla, F. Brain foods: The effects of nutrients on brain function. Nat. Rev. Neurosci. 2008, 9, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Wolever, T.M.; Jenkins, D.J. The Use of the Glycemic Index in Predicting the Blood Glucose Response to Mixed Meals. Am. J. Clin. Nutr. 1986, 43, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Ballance, S.; Knutsen, S.H.; Fosvold, Ø.W.; Fernandez, A.S.; Monro, J. Predicting mixed-meal measured glycaemic index in healthy subjects. Eur. J. Nutr. 2019, 58, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Peters, A. The selfish brain: Competition for energy resources. Am. J. Hum. Biol. 2011, 23, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Blouet, C.; Schwartz, G.J. Hypothalamic nutrient sensing in the control of energy homeostasis. Behav. Brain Res. 2010, 209, 1–12. [Google Scholar] [CrossRef] [PubMed]
- López-Gambero, A.J.; Martínez, F.; Salazar, K.; Cifuentes, M.; Nualart, F. Brain Glucose-Sensing Mechanism and Energy Homeostasis. Mol. Neurobiol. 2019, 56, 769–796. [Google Scholar] [CrossRef] [PubMed]
- Makki, K.; Deehan, E.C.; Walter, J.; Bäckhed, F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe 2018, 23, 705–715. [Google Scholar] [CrossRef]
- Shieh, J.C.C.; Huang, P.T.; Lin, Y.F. Alzheimer’s Disease and Diabetes: Insulin Signaling as the Bridge Linking Two Pathologies. Mol. Neurobiol. 2020, 57, 1966–1977. [Google Scholar] [CrossRef]
- Kulas, J.A.; Weigel, T.K.; Ferris, H.A. Insulin resistance and impaired lipid metabolism as a potential link between diabetes and Alzheimer’s disease. Drug Dev. Res. 2020, 81, 194–205. [Google Scholar] [CrossRef]
- Toth, C. Diabetes and neurodegeneration in the brain. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 126, pp. 489–511. [Google Scholar]
- Akhtar, A.; Sah, S.P. Insulin signaling pathway and related molecules: Role in neurodegeneration and Alzheimer’s disease. Neurochem. Int. 2020, 135, 104707. [Google Scholar] [CrossRef]
- Vega-López, S.; Venn, B.J.; Slavin, J.L. Relevance of the glycemic index and glycemic load for body weight, diabetes, and cardiovascular disease. Nutrients 2018, 10, 1361. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Dye, L.; Lluch, A.; Blundell, J.E. Macronutrients and Mental Performance. Nutrition 2000, 1, 1021–1034. [Google Scholar] [CrossRef]
- Power, S.E.; O’Connor, E.M.; Ross, R.P.; Stanton, C.; O’Toole, P.W.; Fitzgerald, G.F.; Jeffery, I.B. Dietary glycaemic load associated with cognitive performance in elderly subjects. Eur. J. Nutr. 2015, 54, 557–568. [Google Scholar] [CrossRef]
- Simeon, V.; Chiodini, P.; Mattiello, A.; Sieri, S.; Panico, C.; Brighenti, F.; Krogh, V.; Panico, S. Dietary glycemic load and risk of cognitive impairment in women: Findings from the EPIC-Naples cohort. Eur. J. Epidemiol. 2015, 30, 425–433. [Google Scholar] [CrossRef]
- Seetharaman, S.; Andel, R.; McEvoy, C.; Aslan, A.K.D.; Finkel, D.; Pedersen, N.L. Blood glucose, diet-based glycemic load and cognitive aging among dementia-free older adults. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 471–479. [Google Scholar] [CrossRef]
- Francis, H.; Stevenson, R. The longer-term impacts of Western diet on human cognition and the brain. Appetite 2013, 63, 119–128. [Google Scholar] [CrossRef]
- Kanoski, S.E.; Davidson, T.L. Western diet consumption and cognitive impairment: Links to hippocampal dysfunction and obesity. Physiol. Behav. 2011, 103, 59–68. [Google Scholar] [CrossRef]
- Torres, S.J.; Lautenschlager, N.T.; Wattanapenpaiboon, N.; Greenop, K.R.; Beer, C.; Flicker, L.; Alfonso, H.; Nowson, C.A. Dietary patterns are associated with cognition among older people with mild cognitive impairment. Nutrients 2012, 4, 1542–1551. [Google Scholar] [CrossRef]
- Garber, A.; Csizmadi, I.; Friedenreich, C.M.; Sajobi, T.T.; Longman, R.S.; Tyndall, A.V.; Drogos, L.L.; Davenport, M.H.; Poulin, M.J. Association between glycemic load and cognitive function in community-dwelling older adults: Results from the Brain in Motion study. Clin. Nutr. 2018, 37, 1690–1699. [Google Scholar] [CrossRef]
- Schothorst, E.M.; Bunschoten, A.; Schrauwen, P.; Mensink, R.P.; Keijer, J. Effects of a high-fat, low- versus high-glycemic index diet: Retardation of insulin resistance involves adipose tissue modulation. FASEB J. 2009, 23, 1092–1101. [Google Scholar] [CrossRef]
- Hamer, J.A.; Testani, D.; Mansur, R.B.; Lee, Y.; Subramaniapillai, M.; McIntyre, R.S. Brain insulin resistance: A treatment target for cognitive impairment and anhedonia in depression. Exp. Neurol. 2019, 315, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Philippou, E.; Constantinou, M. The influence of glycemic index on cognitive functioning: A systematic review of the evidence. Adv. Nutr. 2014, 5, 119–130. [Google Scholar] [CrossRef]
- Philippou, E.; Pot, G.K.; Heraclides, A.; Richards, M.; Bendayan, R. Dietary glycaemic index and cognitive function: Prospective associations in adults of the 1946 British birth cohort. Public Health Nutr. 2019, 22, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Micha, R.; Rogers, P.J.; Nelson, M. The glycaemic potency of breakfast and cognitive function in school children. Eur. J. Clin. Nutr. 2010, 64, 948–957. [Google Scholar] [CrossRef]
- Wesnes, K.A.; Pincock, C.; Scholey, A. Breakfast is associated with enhanced cognitive function in schoolchildren. An internet based study. Appetite 2012, 59, 646–649. [Google Scholar] [CrossRef]
- Micha, R.; Rogers, P.J.; Nelson, M. Glycaemic index and glycaemic load of breakfast predict cognitive function and mood in school children: A randomised controlled trial. Br. J. Nutr. 2011, 106, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.B.; Bandelow, S.; Nute, M.L.; Morris, J.G.; Nevill, M.E. Breakfast glycaemic index and cognitive function in adolescent school children. Br. J. Nutr. 2012, 107, 1823–1832. [Google Scholar] [CrossRef]
- Cooper, S.B.; Bandelow, S.; Nute, M.L.; Morris, J.G.; Nevill, M.E. Breakfast glycaemic index and exercise: Combined effects on adolescents’ cognition. Physiol. Behav. 2015, 139, 104–111. [Google Scholar] [CrossRef]
- Edefonti, V.; Rosato, V.; Parpinel, M.; Nebbia, G.; Fiorica, L.; Fossali, E.; Ferraroni, M.; Decarli, A.; Agostoni, C. The effect of breakfast composition and energy contribution on cognitive and academic performance: A systematic review. Am. J. Clin. Nutr. 2014, 100, 626–656. [Google Scholar] [CrossRef]
- Scarpina, F.; Tagini, S. The stroop color and word test. Front. Psychol. 2017, 8, 557. [Google Scholar] [CrossRef]
- Cooper, S.B.; Bandelow, S.; Nevill, M.E. Breakfast consumption and cognitive function in adolescent schoolchildren. Physiol. Behav. 2011, 103, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, A.; Radeborg, K.; Björck, I. Effects of differences in postprandial glycaemia on cognitive functions in healthy middle-aged subjects. Eur. J. Clin. Nutr. 2009, 63, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Leigh Gibson, E.; Green, M.W. Nutritional influences on cognitive function: Mechanisms of susceptibility. Nutr. Res. Rev. 2002, 15, 169. [Google Scholar] [CrossRef] [PubMed]
- Rohleder, N.; Kirschbaum, C. Effects of nutrition on neuro-endocrine stress responses. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 504–510. [Google Scholar] [CrossRef]
- Scholey, A.B.; Laing, S.; Kennedy, D.O. Blood glucose changes and memory: Effects of manipulating emotionality and mental effort. Biol. Psychol. 2006, 71, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Lamport, D.J.; Lawton, C.L.; Mansfield, M.W.; Dye, L. Impairments in glucose tolerance can have a negative impact on cognitive function: A systematic research review. Neurosci. Biobehav. Rev. 2009, 33, 394–413. [Google Scholar] [CrossRef]
- Homanics, G.E.; Delorey, T.M.; Firestone, L.L.; Quinlan, J.J.; Handforth, A.; Harrison, N.L.; Krasowski, M.D.; Rick, C.E.M.; Korpi, E.R.; Brilliant, M.H.; et al. Mice Devoid of-Aminobutyrate Type A Receptor 3 Subunit Have Epilepsy, Cleft Palate, and Hypersensitive Behavior. Proc. Natl. Acad. Sci. USA 1997, 94, 4143–4148. [Google Scholar] [CrossRef]
- Sander, D.; Kearney, M.T. Reducing the risk of stroke in type 2 diabetes: Pathophysiological and therapeutic perspectives. J. Neurol. 2009, 256, 1603–1619. [Google Scholar] [CrossRef]
- Lee, K.J.; Lee, J.S.; Jung, K.H. Interactive effect of acute and chronic glycemic indexes for severity in acute ischemic stroke patients. BMC Neurol. 2018, 18. [Google Scholar] [CrossRef]
- González-Moreno, E.I.; Cámara-Lemarroy, C.R.; González-González, J.G.; Góngora-Rivera, F. Glycemic Variability and Acute Ischemic Stroke: The Missing Link? Transl. Stroke Res. 2014, 5, 638–646. [Google Scholar] [CrossRef]
- Quast, H.J.; Wei, J.; Huang, C.; Brunder, D.G.; Sell, L.; Gonzalez, J.M.; Hillman, R.; Kent, T.A. Perfusion Deficit Parallels Exacerbation of Cerebral IschemiaIReperfusion Injury in Hyperglycemic Rats. J. Cereb. Blood Flow Metab. 1997, 17, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Bevers, M.B.; Vaishnav, N.H.; Pham, L.; Battey, T.W.K.; Kimberly, W.T. Hyperglycemia is associated with more severe cytotoxic injury after stroke. J. Cereb. Blood Flow Metab. 2017, 37, 2577–2583. [Google Scholar] [CrossRef] [PubMed]
- Power, R.; Prado-Cabrero, A.; Mulcahy, R.; Howard, A.; Nolan, J.M. The Role of Nutrition for the Aging Population: Implications for Cognition and Alzheimer’s Disease. Annu. Rev. Food Sci. Technol. 2019. [Google Scholar] [CrossRef]
- Otaegui-Arrazola, A.; Amiano, P.; Elbusto, A.; Urdaneta, E.; Martínez-Lage, P. Diet, cognition, and Alzheimer’s disease: Food for thought. Eur. J. Nutr. 2014, 53, 1–23. [Google Scholar] [CrossRef]
- Samadi, M.; Moradi, S.; Moradinazar, M.; Mostafai, R.; Pasdar, Y. Dietary pattern in relation to the risk of Alzheimer’s disease: A systematic review. Neurol. Sci. 2019, 40, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Sastre, M.; Klockgether, T.; Heneka, M.T. Contribution of inflammatory processes to Alzheimer’s disease: Molecular mechanisms. Int. J. Dev. Neurosci. 2006, 24, 167–176. [Google Scholar] [CrossRef]
- Taylor, M.K.; Swerdlow, R.H.; Sullivan, D.K. Dietary neuroketotherapeutics for Alzheimer’s disease: An evidence update and the potential role for diet quality. Nutrients 2019, 11, 1910. [Google Scholar] [CrossRef]
- Prins, M.L. Cerebral metabolic adaptation and ketone metabolism after brain injury. J. Cereb. Blood Flow Metab. 2008, 28, 1–16. [Google Scholar] [CrossRef]
- Hajebrahimi, B.; Kiamanesh, A.; Asgharnejad Farid, A.A.; Asadikaram, G. Type 2 diabetes and mental disorders; A plausible link with inflammation. Cell. Mol. Biol. 2016, 62, 71–77. [Google Scholar] [CrossRef]
- Pervanidou, P.; Bastaki, D.; Chouliaras, G.; Papanikolaou, K.; Laios, E.; Kanaka-Gantenbein, C.; Chrousos, G.P. Circadian cortisol profiles, anxiety and depressive symptomatology, and body mass index in a clinical population of obese children. Stress 2013, 16, 34–43. [Google Scholar] [CrossRef]
- Kandeel, W.A.; Meguid, N.A.; Bjørklund, G.; Eid, E.M.; Farid, M.; Mohamed, S.K.; Wakeel, K.E.; Chirumbolo, S.; Elsaeid, A.; Hammad, D.Y. Impact of Clostridium Bacteria in Children with Autism Spectrum Disorder and Their Anthropometric Measurements. J. Mol. Neurosci. 2020, 70, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Naushad, S.M.; Md, J.; Jain, N.; Prasad, C.K.; Naik, U.; Rama, R.; Akella, D. Autistic Children Exhibit Distinct Plasma Amino Acid Profile. Indian J. Biochem. Biophys. 2013, 50, 474–478. [Google Scholar] [PubMed]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.J. Re: Biomarkers of Environmental Toxicity and Susceptibility in Autism. J. Neurol. Sci. 2009, 280, 127–128. [Google Scholar] [CrossRef]
- Peirce, J.M.; Alviña, K. The role of inflammation and the gut microbiome in depression and anxiety. J. Neurosci. Res. 2019, 97, 1223–1241. [Google Scholar] [CrossRef]
- Macpherson, H.; Roberstson, B.; Sünram-Lea, S.; Stough, C.; Kennedy, D.; Scholey, A. Glucose administration and cognitive function: Differential effects of age and effort during a dual task paradigm in younger and older adults. Psychopharmacology 2015, 232, 1135–1142. [Google Scholar] [CrossRef]
- Donohoe, R.T.; Benton, D. Cognitive Functioning Is Susceptible to the Level of blood glucose. Psychopharmacology 1999, 145, 378–385. [Google Scholar] [CrossRef]
- Nilsson, A.; Radeborg, K.; Björck, I. Effects on cognitive performance of modulating the postprandial blood glucose profile at breakfast. Eur. J. Clin. Nutr. 2012, 66, 1039–1043. [Google Scholar] [CrossRef]
- Benton, D.; Ruffin, M.P.; Lassel, T.; Nabb, S.; Messaoudi, M.; Vinoy, S.; Desor, D.; Lang, V. The delivery rate of dietary carbohydrates affects cognitive performance in both rats and humans. Psychopharmacology 2003, 166, 86–90. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Rizkalla, S.W.; Taghrid, L.; Laromiguiere, M.; Huet, D.; Boillot, J.; Rigoir, A.; Elgrably, F.; Slama, G. Improved Plasma Glucose Control, Whole-Body Glucose Utilization, and Lipid Profile on a Low-Glycemic Index Diet in Type 2 Diabetic Men A Randomized Controlled Trial. Diabetes Care. 2004, 27, 1866–1872. [Google Scholar] [CrossRef] [PubMed]
- Sadeghifar, F.; Penry, V.B. Mechanisms and Uses of Dietary Therapy as a Treatment for Epilepsy: A Review. Glob. Adv. Health Med. 2019, 8, 216495611987478. [Google Scholar] [CrossRef] [PubMed]
- Muzykewicz, D.A.; Lyczkowski, D.A.; Memon, N.; Conant, K.D.; Pfeifer, H.H.; Thiele, E.A. Efficacy, safety, and tolerability of the low glycemic index treatment in pediatric epilepsy. Epilepsia 2009, 50, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Guzmiirp, M.; Geefen, M.J. Regulation of Fatty Acid Oxidation in Mammalian Liver. Biochim. Biophys. Acta 1993, 1167, 227–241. [Google Scholar] [CrossRef]
- Hartman, A.L.; Gasior, M.; Vining, E.P.G.; Rogawski, M.A. The Neuropharmacology of the Ketogenic Diet. Pediatr. Neurol. 2007, 36, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Olsen, R.W.; Avoli, M. GABA and epileptogenesis. Epilepsia 1997, 38, 399–407. [Google Scholar] [CrossRef]
- Petroff, O.A.C.; Rothman, D.L.; Behar, K.L.; Mattson, R.H. Low brain GABA level is associated with poor seizure control. Ann. Neurol. 1996, 40, 908–911. [Google Scholar] [CrossRef]
- Chuang, S.H.; Reddy, D.S. Isobolographic Analysis of Antiseizure Activity of the GABA Type A Receptor-Modulating Synthetic Neurosteroids Brexanolone and Ganaxolone with Tiagabine and Midazolam. J. Pharmacol. Exp. Ther. 2020, 372, 285–298. [Google Scholar] [CrossRef]
- Sills, G.J.; Rogawski, M.A. Mechanisms of action of currently used antiseizure drugs. Neuropharmacology 2020, 168, 197966. [Google Scholar] [CrossRef]
- Treiman, D.M. GABAergic mechanisms in epilepsy. Epilepsia 2001, 42, 8–12. [Google Scholar] [CrossRef]
- Dahlin, M.; Elfving, Å.; Ungerstedt, U.; Åmark, P. The ketogenic diet influences the levels of excitatory and inhibitory amino acids in the CSF in children with refractory epilepsy. Epilepsy Res. 2005, 64, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Berg, J.; Yellen, G. Ketogenic diet metabolites reduce firing in central neurons by opening KATP channels. J. Neurosci. 2007, 27, 3618–3625. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; O’Leary, E.I.; Tanner, G.R. The ketogenic diet metabolite beta-hydroxybutyrate (β-HB) reduces incidence of seizure-like activity (SLA) in a K atp- and GABA b-dependent manner in a whole-animal Drosophila melanogaster model. Epilepsy Res. 2017, 133, 6–9. [Google Scholar] [CrossRef]
- Omote, H.; Miyaji, T.; Juge, N.; Moriyama, Y. Vesicular neurotransmitter transporter: Bioenergetics and regulation of glutamate transport. Biochemistry 2011, 50, 5558–5565. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic Control of Vesicular Glutamate Transport and Release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Izzo, V.; Bravo-San Pedro, J.M.; Sica, V.; Kroemer, G.; Galluzzi, L. Mitochondrial Permeability Transition: New Findings and Persisting Uncertainties. Trends Cell Biol. 2016, 26, 655–667. [Google Scholar] [CrossRef]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef]
- Zhou, Z.; Austin, G.; Young, L.; Johnson, L.; Sun, R. Mitochondrial Metabolism in Major Neurological Diseases. Cells 2018, 7, 229. [Google Scholar] [CrossRef]
- Cooper, M.A.; McCoin, C.; Pei, D.; Thyfault, J.P.; Koestler, D.; Wright, D.E. Reduced mitochondrial reactive oxygen species production in peripheral nerves of mice fed a ketogenic diet. Exp. Physiol. 2018, 103, 1206–1212. [Google Scholar] [CrossRef]
- Pearson-Smith, J.N.; Patel, M. Metabolic dysfunction and oxidative stress in epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. [Google Scholar] [CrossRef]
- Knowles, S.; Budney, S.; Deodhar, M.; Matthews, S.A.; Simeone, K.A.; Simeone, T.A. Ketogenic diet regulates the antioxidant catalase via the transcription factor PPARγ2. Epilepsy Res. 2018, 147, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Simeone, K.A.; Rho, J.M. Ketone Bodies as Anti-Seizure Agents. Neurochem. Res. 2017, 42, 2011–2018. [Google Scholar] [CrossRef] [PubMed]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Lang, B.; Aronica, E. Immunity and inflammation in epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The b-hydroxybutyrate receptor HCA 2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Spence, J.D.; Tangney, C. Lower risk of stroke with a vegetarian diet. Neurology 2020, 94, 463–464. [Google Scholar] [CrossRef]
- Waldmann, A.; Ströhle, A.; Koschizke, J.W.; Leitzmann, C.; Hahn, A. Overall glycemic index and glycemic load of vegan diets in relation to plasma lipoproteins and triacylglycerols. Ann. Nutr. Metab. 2007, 51, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Corredor, P.A.; Gutiérrez-Vargas, J.A.; Ciro-Ramírez, L.; Balcazar, N.; Cardona-Gómez, G.P. High fructose diet-induced obesity worsens post-ischemic brain injury in the hippocampus of female rats. Nutr. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Robbins, N.M.; Swanson, R.A. Opposing effects of glucose on stroke and reperfusion injury: Acidosis, oxidative stress, and energy metabolism. Stroke 2014, 45, 1881–1886. [Google Scholar] [CrossRef]
- Song, T.J.; Chang, Y.; Chun, M.Y.; Lee, C.Y.; Kim, A.R.; Kim, Y.; Kim, Y.J. High dietary glycemic load is associated with poor functional outcome in patients with acute cerebral infarction. J. Clin. Neurol. 2018, 14, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Luitse, M.J.A.; Velthuis, B.K.; Kappelle, L.J.; van der Graaf, Y.; Biessels, G.J. Chronic hyperglycemia is related to poor functional outcome after acute ischemic stroke. Int. J. Stroke 2017, 12, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Kamouchi, M.; Matsuki, T.; Hata, J.; Kuwashiro, T.; Ago, T.; Sambongi, Y.; Fukushima, Y.; Sugimori, H.; Kitazono, T. Prestroke glycemic control is associated with the functional outcome in acute ischemic stroke: The fukuoka stroke registry. Stroke 2011, 42, 2788–2794. [Google Scholar] [CrossRef]
- Anderson, R.E.; Tan, W.K.; Martin, H.S.; Meyer, F.B. Effects of Glucose and PaO 2 Modulation on Cortical Intracellular Acidosis, NADH Redox State, and Infarction in the Ischemic Penumbra. Stroke 1999, 30, 160–170. [Google Scholar] [CrossRef]
- Ceriello, A.; Esposito, K.; Piconi, L.; Ihnat, M.A.; Thorpe, J.E.; Testa, R.; Boemi, M.; Giugliano, D. Oscillating glucose is more deleterious to endothelial function and oxidative stress than mean glucose in normal and type 2 diabetic patients. Diabetes 2008, 57, 1349–1354. [Google Scholar] [CrossRef]
- Santos-García, D.; Blanco, M.; Serena, J.; Arias, S.; Millán, M.; Rodríguez-Yáñez, M.; Leira, R.; Dávalos, A.; Castillo, J. Brachial arterial flow mediated dilation in acute ischemic stroke. Eur. J. Neurol. 2009, 16, 684–690. [Google Scholar] [CrossRef]
- Raynaud, E.; Pérez-Martin, A.; Brun, J.F.; Aïssaaïssa-Benhaddad, A.; Fédou, C.; Mercier, J. Relationships Between Fibrinogen and Insulin Resistance. Atherosclerosis 2000, 150, 365–370. [Google Scholar] [CrossRef]
- Meigs, J.B.; Mittleman, M.A.; Nathan, D.M.; Tofler, G.H.; Singer, D.E.; Murphy-Sheehy, P.M.; Lipinska, I.; D’agostino, R.B.; Wilson, P.W.F. Hyperinsulinemia, Hyperglycemia, and Impaired Hemostasis the Framingham Offspring Study. JAMA 2000, 283, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Song, T.J.; Chang, Y.; Kim, A.R.; Kim, Y.; Kim, Y.J. High dietary glycemic load was associated with the presence and burden of cerebral small vessel diseases in acute ischemic stroke patients. Nutr. Res. 2018, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Zhang, X.; Shu, X.O.; Cai, H.; Li, H.; Ding, D.; Hong, Z.; Xiang, Y.B.; Gao, Y.T.; Zheng, W.; et al. Dietary glycemic index, glycemic load, and refined carbohydrates are associated with risk of stroke: A prospective cohort study in urban Chinese women. Am. J. Clin. Nutr. 2016, 104, 1345–1351. [Google Scholar] [CrossRef]
- Spence, J.D. Nutrition and risk of stroke. Nutrients 2019, 11, 647. [Google Scholar] [CrossRef]
- Lim, J.S.; Kim, C.; Oh, M.S.; Lee, J.H.; Jung, S.; Jang, M.U.; Lee, S.H.; Kim, Y.J.; Kim, Y.; Suh, S.W.; et al. Effects of glycemic variability and hyperglycemia in acute ischemic stroke on post-stroke cognitive impairments. J. Diabetes Complicat. 2018, 32, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Campos-Peña, V.; Toral-Rios, D.; Becerril-Pérez, F.; Sánchez-Torres, C.; Delgado-Namorado, Y.; Torres-Ossorio, E.; Franco-Bocanegra, D.; Carvajal, K. Metabolic Syndrome as a Risk Factor for Alzheimer’s Disease: Is Aβ a Crucial Factor in Both Pathologies? Antioxid. Redox Signal. 2017, 26, 542–560. [Google Scholar] [CrossRef]
- Taylor, M.K.; Sullivan, D.K.; Swerdlow, R.H.; Vidoni, E.D.; Morris, J.K.; Mahnken, J.D.; Burns, J.M. A high-glycemic diet is associated with cerebral amyloid burden in cognitively normal older adults. Am. J. Clin. Nutr. 2017, 106, 1463–1470. [Google Scholar] [CrossRef]
- Hascup, E.R.; Broderick, S.O.; Russell, M.K.; Fang, Y.; Bartke, A.; Boger, H.A.; Hascup, K.N. Diet-Induced Insulin Resistance Elevates Hippocampal Glutamate as well as VGLUT1 and GFAP Expression in AβPP/PS1 Mice HHS Public Access. J. Neurochem. 2019, 148, 219–237. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Yamaguchi, K.; Matsui, K.; Sano, T.; Kubota, T.; Hashimoto, T.; Mano, A.; Yamada, K.; Matsuo, Y.; Kubota, N.; et al. Differential effects of diet- and genetically-induced brain insulin resistance on amyloid pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2019, 14. [Google Scholar] [CrossRef]
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef]
- Castellano, C.A.; Nugent, S.; Paquet, N.; Tremblay, S.; Bocti, C.; Lacombe, G.; Imbeault, H.; Turcotte, É.; Fulop, T.; Cunnane, S.C. Lower brain 18F-fluorodeoxyglucose uptake but normal 11C-acetoacetate metabolism in mild Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rippy, N.A.; Dorenbos, K.; Concepcion, R.C.; Agarwal, A.K.; Rho, J.M. The Ketogenic Diet Increases Mitochondrial Uncoupling Protein Levels and Activity. Ann. Neurol. 2004, 55, 576–580. [Google Scholar] [CrossRef] [PubMed]
- Klaus, S.; Ost, M. Mitochondrial uncoupling and longevity—A role for mitokines? Exp. Gerontol. 2020, 130, 110796. [Google Scholar] [CrossRef]
- Peixoto, L.; Abel, T. The role of histone acetylation in memory formation and cognitive impairments. Neuropsychopharmacology 2013, 38, 62–76. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, S.; Yu, L.; Jin, J.; Ye, X.; Liu, Y.; Xu, Y. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell 2017, 16, 1073–1082. [Google Scholar] [CrossRef]
- Yamada, K.; Nabeshima, T. Brain-Derived Neurotrophic Factor/TrkB Signaling in Memory Processes. J. Pharmacol. Sci. 2003, 91, 267–270. [Google Scholar] [CrossRef]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef]
- Koppel, I.; Timmusk, T. Differential regulation of Bdnf expression in cortical neurons by class-selective histone deacetylase inhibitors. Neuropharmacology 2013, 75, 106–115. [Google Scholar] [CrossRef]
- Omar, S.H. Mediterranean and MIND diets containing olive biophenols reduces the prevalence of Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 2797. [Google Scholar] [CrossRef] [PubMed]
- Trichopoulou, A.; Costacou, T.; Bamia, C.; Trichopoulos, D. Adherence to a Mediterranean Diet and Survival in a Greek Population. N. Engl. J. Med. 2003, 348, 2599–2608. [Google Scholar] [CrossRef]
- Becerra-Tomás, N.; Blanco Mejía, S.; Viguiliouk, E.; Khan, T.; Kendall, C.W.C.; Kahleova, H.; Rahelić, D.; Sievenpiper, J.L.; Salas-Salvadó, J. Mediterranean diet, cardiovascular disease and mortality in diabetes: A systematic review and meta-analysis of prospective cohort studies and randomized clinical trials. Crit. Rev. Food Sci. Nutr. 2020, 60, 1207–1227. [Google Scholar] [CrossRef] [PubMed]
- Petersson, S.D.; Philippou, E. Mediterranean diet, cognitive function, and dementia: A systematic review of the evidence. Adv. Nutr. 2016, 7, 889–904. [Google Scholar] [CrossRef] [PubMed]
- Bozzetto, L.; Alderisio, A.; Giorgini, M.; Barone, F.; Giacco, A.; Riccardi, G.; Rivellese, A.A.; Annuzzi, G. Extra-virgin olive oil reduces glycemic response to a high-glycemic index meal in patients with type 1 diabetes: A randomized controlled trial. Diabetes Care 2016, 39, 518–524. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef]
- Martínez Leo, E.E.; Segura Campos, M.R. Effect of ultra-processed diet on gut microbiota and thus its role in neurodegenerative diseases. Nutrition 2020, 71, 110609. [Google Scholar] [CrossRef]
- Van de Sande, M.M.H.; van Buul, V.J.; Brouns, F.J.P.H. Autism and nutrition: The role of the gut-brain axis. Nutr. Res. Rev. 2014, 27, 199–214. [Google Scholar] [CrossRef]
- Craft, S.; Watson, S.C. Insulin and Neurodegenerative Disease: Shared and Specific Mechanisms. Lancet Neurol. 2004, 3, 169–178. [Google Scholar] [CrossRef]
- Murakami, K.; Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; et al. Dietary glycemic index is inversely associated with the risk of Parkinson’s disease: A case-control study in Japan. Nutrition 2010, 26, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Dohrmann, D.D.; Putnik, P.; Bursać Kovačević, D.; Simal-Gandara, J.; Lorenzo, J.M.; Barba, F.J. Japanese, Mediterranean and Argentinean diets and their potential roles in neurodegenerative diseases. Food Res. Int. 2019, 120, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.C.; Wei, W.Y.; Tsai, K.J.; Wang, L.C. High fat diet suppresses peroxisome proliferator-activated receptors and reduces dopaminergic neurons in the Substantia nigra. Int. J. Mol. Sci. 2020, 21, 207. [Google Scholar] [CrossRef]
- Jackson, A.; Forsyth, C.B.; Shaikh, M.; Voigt, R.M.; Engen, P.A.; Ramirez, V.; Keshavarzian, A. Diet in Parkinson’s Disease: Critical Role for the Microbiome. Front. Neurol. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Unger, M.M.; Spiegel, J.; Dillmann, K.U.; Grundmann, D.; Philippeit, H.; Bürmann, J.; Faßbender, K.; Schwiertz, A.; Schäfer, K.H. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat. Disord. 2016, 32, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Shin, C.; Lim, Y.; Lim, H.; Ahn, T.B. Plasma Short-Chain Fatty Acids in Patients with Parkinson’s Disease. Mov. Disord. 2020, 35, 1021–1027. [Google Scholar] [CrossRef]
- Salari-Moghaddam, A.; Saneei, P.; Larijani, B.; Esmaillzadeh, A. Glycemic index, glycemic load, and depression: A systematic review and meta-analysis. Eur. J. Clin. Nutr. 2019, 73, 356–365. [Google Scholar] [CrossRef]
- Zemdegs, J.; Martin, H.; Pintana, H.; Bullich, S.; Manta, S.; Marqués, M.A.; Moro, C.; Laye, S.; Ducrocq, F.; Chattipakorn, N.; et al. Metformin promotes anxiolytic and antidepressant-like responses in insulin-resistant mice by decreasing circulating branched-chain amino acids. J. Neurosci. 2019, 39, 5935–5948. [Google Scholar] [CrossRef]
- Zemdegs, J.; Quesseveur, G.; Jarriault, D.; Pénicaud, L.; Fioramonti, X.; Guiard, B.P. Themed Section: Updating Neuropathology and Neuropharmacology of Monoaminergic Systems High-fat diet-induced metabolic disorders impairs 5-HT function and anxiety-like behavior in mice LINKED ARTICLES. Br. J. Pharmacol. 2016, 173, 2095. [Google Scholar] [CrossRef]
- Quesseveur, G.; Portal, B.; Basile, J.A.; Ezan, P.; Mathou, A.; Halley, H.; Leloup, C.; Fioramonti, X.; Déglon, N.; Giaume, C.; et al. Attenuated levels of hippocampal connexin 43 and its phosphorylation correlate with antidepressant-and anxiolytic-like activities in mice. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef]
- Palaiologos, G.; Philippidis, H.; Chomatas, H.; Iakovou, D.; Linardou, A. Effects of Branched Chain Amino Acids, Pyruvate, or Ketone Bodies on the Free Amino Acid Pool and Release from Brain Cortex Slices of Normal and Streptozotocin-Diabetic Rats. Neurochem. Res. 1987, 12, 1–7. [Google Scholar] [CrossRef]
- Sonnet, D.S.; O’Leary, M.N.; Gutierrez, M.A.; Nguyen, S.M.; Mateen, S.; Hsu, Y.; Mitchell, K.P.; Lopez, A.J.; Vockley, J.; Kennedy, B.K.; et al. Metformin inhibits Branched Chain Amino Acid (BCAA) derived ketoacidosis and promotes metabolic homeostasis in MSUD. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Hahad, O.; Prochaska, J.H.; Daiber, A.; Muenzel, T. Environmental Noise-Induced Effects on Stress Hormones, Oxidative Stress, and Vascular Dysfunction: Key Factors in the Relationship between Cerebrocardiovascular and Psychological Disorders. Oxidative Med. Cell. Longev. 2019, 2019, 4623109. [Google Scholar] [CrossRef] [PubMed]
- Mathews, D.C.; Henter, I.D.; Zarate, C.A. Targeting the glutamatergic system to treat major depressive disorder: Rationale and progress to date. Drugs 2012, 72, 1313–1333. [Google Scholar] [CrossRef]
- Krakowiak, P.; Walker, C.K.; Bremer, A.A.; Baker, A.S.; Ozonoff, S.; Hansen, R.L.; Hertz-Picciotto, I. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012, 129. [Google Scholar] [CrossRef]
- Lyall, K.; Pauls, D.L.; Santangelo, S.; Spiegelman, D.; Ascherio, A. Maternal early life factors associated with hormone levels and the risk of having a child with an autism spectrum disorder in the nurses health study II. J. Autism Dev. Disord. 2011, 41, 618–627. [Google Scholar] [CrossRef]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain Res. 2009, 204, 313–321. [Google Scholar] [CrossRef]
- Michel, M.; Schmidt, M.J.; Mirnics, K. Immune system gene dysregulation in autism and schizophrenia. Dev. Neurobiol. 2012, 72, 1277–1287. [Google Scholar] [CrossRef]
- Neuhouser, M.L.; Schwarz, Y.; Wang, C.; Breymeyer, K.; Coronado, G.; Wang, C.Y.; Noar, K.; Song, X.; Lampe, J.W. A low-glycemic load diet reduces serum C-reactive protein and modestly increases adiponectin in overweight and obese adults. J. Nutr. 2012, 142, 369–374. [Google Scholar] [CrossRef]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1–13. [Google Scholar] [CrossRef]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process—A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Farrokhi, C.; Dargusch, R.; Goujon-Svrzic, M.; Maher, P. Dietary glycemic index modulates the behavioral and biochemical abnormalities associated with autism spectrum disorder. Mol. Psychiatry 2016, 21, 426–436. [Google Scholar] [CrossRef] [PubMed]
- Ruskin, D.N.; Svedova, J.; Cote, J.L.; Sandau, U.; Rho, J.M.; Kawamura, M.; Boison, D.; Masino, S.A. Ketogenic Diet Improves Core Symptoms of Autism in BTBR Mice. PLoS ONE 2013, 8, e65021. [Google Scholar] [CrossRef]
- Sumathi, T.; Manivasagam, T.; Thenmozhi, A.J. The Role of Gluten in Autism. Adv. Neurobiol. 2020, 24, 469–479. [Google Scholar] [CrossRef]
- Karhu, E.; Zukerman, R.; Eshraghi, R.S.; Mittal, J.; Deth, R.C.; Castejon, A.M.; Trivedi, M.; Mittal, R.; Eshraghi, A.A. Nutritional interventions for autism spectrum disorder. Nutr. Rev. 2020, 78, 515–531. [Google Scholar] [CrossRef]
- Berding, K.; Donovan, S.M. Dietary Patterns Impact Temporal Dynamics of Fecal Microbiota Composition in Children with Autism Spectrum Disorder. Front. Nutr. 2020, 6. [Google Scholar] [CrossRef]
- Waye, M.M.Y.; Cheng, H.Y. Genetics and epigenetics of autism: A Review. Psychiatry Clin. Neurosci. 2018, 72, 228–244. [Google Scholar] [CrossRef]
- Bhandari, R.; Paliwal, J.K.; Kuhad, A. Dietary Phytochemicals as Neurotherapeutics for Autism Spectrum Disorder: Plausible Mechanism and Evidence. Adv. Neurobiol. 2020, 24, 615–646. [Google Scholar] [CrossRef]
- Liu, H.; Zimmerman, A.W.; Singh, K.; Connors, S.L.; Diggins, E.; Stephenson, K.K.; Dinkova-Kostova, A.T.; Fahey, J.W. Biomarker Exploration in Human Peripheral Blood Mononuclear Cells for Monitoring Sulforaphane Treatment Responses in Autism Spectrum Disorder. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef]
- Mitsiogianni, M.; Trafalis, D.T.; Franco, R.; Zoumpourlis, V.; Pappa, A.; Panayiotidis, M.I. Sulforaphane and iberin are potent epigenetic modulators of histone acetylation and methylation in malignant melanoma. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klomparens, E.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Connors, S.L.; Macklin, E.A.; Smith, K.D.; Fahey, J.W.; Talalay, P.; Zimmerman, A.W. Sulforaphane treatment of autism spectrum disorder (ASD). Proc. Natl. Acad. Sci. USA 2014, 111, 15550–15555. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.P.J.; Haus, J.M.; Kelly, K.R.; Cook, M.D.; Filion, J.; Rocco, M.; Kashyap, S.R.; Watanabe, R.M.; Barkoukis, H.; Kirwan, J.P. A low-glycemic index diet combined with exercise reduces insulin resistance, postprandial hyperinsulinemia, and glucose-dependent insulinotropic polypeptide responses in obese, prediabetic humans. Am. J. Clin. Nutr. 2010, 92, 1359–1368. [Google Scholar] [CrossRef]
- Radulian, G.; Rusu, E.; Dragomir, A.; Posea, M. Metabolic effects of low glycaemic index diets. Nutr. J. 2009, 8, 5. [Google Scholar] [CrossRef]
- Role, T.H.E.; Nutrient, O.F. Supply and Demand in Cerebral Energy Metabolism. Blood 2007, 27, 1766–1791. [Google Scholar]
- Klip, A.; Tsakiridis, T.; Marette, A.; Ortiz, P.A. Regulation of expression of glucose transporters by glucose: A review of studies in vivo and in cell cultures. FASEB J. 1994, 8, 43–53. [Google Scholar] [CrossRef]
- Silver’, I.A.; Ereciaska, M. Extracellular Glucose Concentration in Mammalian Brain: Continuous Monitoring of Changes during Increased Neuronal Activity and upon Limitation in Oxygen Supply in Normo-, Hypo-, and Hyperglycemic Animals. J. Neurosci. 1994, 14, 5068–5076. [Google Scholar] [CrossRef]
- Meierhans, R.; Béchir, M.; Ludwig, S.; Sommerfeld, J.; Brandi, G.; Haberthür, C.; Stocker, R.; Stover, J.F. Brain metabolism is significantly impaired at blood glucose below 6 mM and brain glucose below 1 mM in patients with severe traumatic brain injury. Crit. Care 2010, 14. [Google Scholar] [CrossRef]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef]
- Kim, K.S.; Seeley, R.J.; Sandoval, D.A. Signalling from the periphery to the brain that regulates energy homeostasis. Nat. Rev. Neurosci. 2018, 19, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.I.; Mills, K.E.; Zheng, J.; Regmi, A.; Hu, S.Q.; Gou, L.; Chen, L.L. Low-glycemic index diets as an intervention for diabetes: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2019, 110, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Abete, I.; Parra, D.; Martinez, J.A. Energy-restricted diets based on a distinct food selection affecting the glycemic index induce different weight loss and oxidative response. Clin. Nutr. 2008, 27, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.J.; Smart, C.E.; Steil, G.M.; Brand-Miller, J.C.; King, B.; Wolpert, H.A. Impact of fat, protein, and glycemic index on postprandial glucose control in type 1diabetes: Implications for intensive diabetes management in the continuous glucose monitoring era. Diabetes Care 2015, 38, 1008–1015. [Google Scholar] [CrossRef] [PubMed]
- Vrolix, R.; van Meijl, L.E.C.; Mensink, R.P. The metabolic syndrome in relation with the glycemic index and the glycemic load. Physiol. Behav. 2008, 94, 293–299. [Google Scholar] [CrossRef]
- Wood, R.J.; Fernandez, M.L. Carbohydrate-restricted versus low-glycemic-index diets for the treatment of insulin resistance and metabolic syndrome. Nutr. Rev. 2009, 67, 179–183. [Google Scholar] [CrossRef]
- Shimazu, T.; Minokoshi, Y. Systemic glucoregulation by glucose-sensing neurons in the ventromedial hypothalamic nucleus (VMH). J. Endocr. Soc. 2017, 1, 449–459. [Google Scholar] [CrossRef]
- Stanley, S.; Moheet, A.; Seaquist, E.R. Central Mechanisms of Glucose Sensing and Counterregulation in Defense of Hypoglycemia. Endocr. Rev. 2018, 40, 768–788. [Google Scholar] [CrossRef]
- Ludwig, D.S. The glycemic index: Physiological mechanisms relating to obesity, diabetes, and cardiovascular disease. J. Am. Med. Assoc. 2002, 287, 2414–2423. [Google Scholar] [CrossRef]
- Leloup, C.; Magnan, C.; Benani, A.; Bonnet, E.; Alquier, T.; Offer, G.; Carriere, A.; Périquet, A.; Fernandez, Y.; Ktorza, A.; et al. Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes 2006, 55, 2084–2090. [Google Scholar] [CrossRef]
- Carneiro, L.; Allard, C.; Guissard, C.; Fioramonti, X.; Tourrel-Cuzin, C.; Bailbé, D.; Barreau, C.; Offer, G.; Nédelec, E.; Salin, B.; et al. Importance of mitochondrial dynamin-related protein 1 in hypothalamic glucose sensitivity in rats. Antioxid. Redox Signal. 2012, 17, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Colombani, A.L.; Carneiro, L.; Benani, A.; Galinier, A.; Jaillard, T.; Duparc, T.; Offer, G.; Lorsignol, A.; Magnan, C.; Casteilla, L.; et al. Enhanced hypothalamic glucose sensing in obesity: Alteration of redox signaling. Diabetes 2009, 58, 2189–2197. [Google Scholar] [CrossRef] [PubMed]
- Leloup, C.; Casteilla, L.; Carrière, A.; Galinier, A.; Benani, A.; Carneiro, L.; Pénicaud, L. Balancing Mitochondrial redox signaling: A key point in metabolic regulation. Antioxid. Redox Signal. 2011, 14, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Desmoulins, L.; Chrétien, C.; Paccoud, R.; Collins, S.; Cruciani-Guglielmacci, C.; Galinier, A.; Liénard, F.; Quinault, A.; Grall, S.; Allard, C.; et al. Mitochondrial Dynamin-Related Protein 1 (DRP1) translocation in response to cerebral glucose is impaired in a rat model of early alteration in hypothalamic glucose sensing. Mol. Metab. 2019, 20, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Fioramonti, X.; Deak, A.; Deshpande, S.; Carneiro, L.; Zhou, C.; Sayed, N.; Orban, B.; Berlin, J.R.; Pénicaud, L.; Leloup, C.; et al. Hypothalamic S-Nitrosylation Contributes to the Counter-Regulatory Response Impairment following Recurrent Hypoglycemia. PLoS ONE 2013, 8, e68709. [Google Scholar] [CrossRef] [PubMed]
- De Guia, R.M.; Hassing, A.S.; Skov, L.J.; Ratner, C.; Plucińska, K.; Madsen, S.; Diep, T.A.; dela Cruz, G.V.; Trammell, S.A.J.; Sustarsic, E.G.; et al. Fasting- and ghrelin-induced food intake is regulated by NAMPT in the hypothalamus. Acta Physiol. 2020, 228. [Google Scholar] [CrossRef]
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef]
- Timper, K.; Paeger, L.; Sánchez-Lasheras, C.; Varela, L.; Jais, A.; Nolte, H.; Vogt, M.C.; Hausen, A.C.; Heilinger, C.; Evers, N.; et al. Mild Impairment of Mitochondrial OXPHOS Promotes Fatty Acid Utilization in POMC Neurons and Improves Glucose Homeostasis in Obesity. Cell Rep. 2018, 25, 383–397. [Google Scholar] [CrossRef]
- Gyengesi, E.; Paxinos, G.; Andrews, Z.B. Oxidative Stress in the Hypothalamus: The Importance of Calcium Signaling and Mitochondrial ROS in Body Weight Regulation. Curr. Neuropharmacol. 2012, 10, 344–353. [Google Scholar] [CrossRef]
- Jaillard, T.; Roger, M.; Galinier, A.; Guillou, P.; Benani, A.; Leloup, C.; Casteilla, L.; Pénicaud, L.; Lorsignol, A. Hypothalamic reactive oxygen species are required for insulin-induced food intake inhibition: An NADPH oxidase-dependent mechanism. Diabetes 2009, 58, 1544–1549. [Google Scholar] [CrossRef]
- Carneiro, L.; Pellerin, L. Monocarboxylate transporters: New players in body weight regulation. Obes. Rev. 2015, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.; Geller, S.; Fioramonti, X.; Hébert, A.; Repond, C.; Leloup, C.; Pellerin, L. Evidence for hypothalamic ketone body sensing: Impact on food intake and peripheral metabolic responses in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E103–E115. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, L.; Geller, S.; Hébert, A.; Repond, C.; Fioramonti, X.; Leloup, C.; Pellerin, L. Hypothalamic sensing of ketone bodies after prolonged cerebral exposure leads to metabolic control dysregulation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Le Foll, C. Hypothalamic Fatty Acids and Ketone Bodies Sensing and Role of FAT/CD36 in the Regulation of Food Intake. Front. Physiol. 2019, 10, 1036. [Google Scholar] [CrossRef]
- Le Foll, C.; Levin, B.E.; Levin, B.E. Fatty acid-induced astrocyte ketone production and the control of food intake. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, 1186–1192. [Google Scholar] [CrossRef]
- le Foll, C.; Dunn-Meynell, A.A.; Miziorko, H.M.; Levin, B.E. Role of VMH ketone bodies in adjusting caloric intake to increased dietary fat content in DIO and DR rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, 872–878. [Google Scholar] [CrossRef]
- le Foll, C.; Dunn-Meynell, A.A.; Miziorko, H.M.; Levin, B.E. Regulation of hypothalamic neuronal sensing and food intake by ketone bodies and fatty acids. Diabetes 2014, 63, 1259–1269. [Google Scholar] [CrossRef]
- Blázquez, M.G. and C. Is There an Astrocyte-Neuron Ketone Body Shuttle. Trends Endocrinol. Metab. 2001, 12, 169–172. [Google Scholar]
- Balasse, E.O.; Féry, F. Ketone body production and disposal: Effects of fasting, diabetes, and exercise. Diabetes Metab. Rev. 1989, 5, 247–270. [Google Scholar] [CrossRef]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef]
- Drouin, J. Transcriptional and epigenetic regulation of POMC gene expression. J. Mol. Endocrinol. 2016, 56, T99–T112. [Google Scholar] [CrossRef] [PubMed]
- Obri, A.; Claret, M. The role of epigenetics in hypothalamic energy balance control: Implications for obesity. Cell Stress 2019, 3, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, T.J. Environmental and hormonal regulation of epigenetic enzymes in the hypothalamus. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.E.; Dunn-Meynell, A.A.; Routh, V.H. Brain Glucosensing and the K(ATP) Channel. Nat. Neurosci. 2001, 4, 459–460. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.; Turovsky, E.; Christie, I.N.; Hosford, P.S.; Hadjihambi, A.; Korsak, A.; Ang, R.; Mastitskaya, S.; Sheikhbahaei, S.; Theparambil, S.M.; et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. GLIA 2018, 66, 1185–1199. [Google Scholar] [CrossRef]
- Leloup, C.; Allard, C.; Carneiro, L.; Fioramonti, X.; Collins, S.; Pénicaud, L. Glucose and hypothalamic astrocytes: More than a fueling role? Neuroscience 2016, 323, 110–120. [Google Scholar] [CrossRef]
- Gao, Y.; Layritz, C.; Legutko, B.; Eichmann, T.O.; Laperrousaz, E.; Moullé, V.S.; Cruciani-Guglielmacci, C.; Magnan, C.; Luquet, S.; Woods, S.C.; et al. Disruption of lipid uptake in astroglia exacerbates diet-induced obesity. Diabetes 2017, 66, 2555–2563. [Google Scholar] [CrossRef]
- Frago, L.M.; Chowen, J.A. Involvement of astrocytes in mediating the central effects of ghrelin. Int. J. Mol. Sci. 2017, 18, 536. [Google Scholar] [CrossRef]
- Chowen, J.A.; Frago, L.M.; Fernández-Alfonso, M.S. Physiological and pathophysiological roles of hypothalamic astrocytes in metabolism. J. Neuroendocrinol. 2019, 31, e12671. [Google Scholar] [CrossRef]
- Yasumoto, Y.; Miyazaki, H.; Ogata, M.; Kagawa, Y.; Yamamoto, Y.; Islam, A.; Yamada, T.; Katagiri, H.; Owada, Y. Glial Fatty Acid-Binding Protein 7 (FABP7) Regulates Neuronal Leptin Sensitivity in the Hypothalamic Arcuate Nucleus. Mol. Neurobiol. 2018, 55, 9016–9028. [Google Scholar] [CrossRef]
- Wang, D.; Zhao, L.; Zheng, H.; Dong, M.; Pan, L.; Zhang, X.; Zhang, H.; Gao, H. Time-Dependent Lactate Production and Amino Acid Utilization in Cultured Astrocytes Under High Glucose Exposure. Mol. Neurobiol. 2018, 55, 1112–1122. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.H.; Sa, M.; Hong, Y.R.; Lee, C.J.; Koo, J.H. Fatty acid increases cAMP-dependent lactate and MAO-B-dependent GABA production in mouse Astrocytes by activating a Gαs protein-coupled receptor. Exp. Neurobiol. 2018, 27, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Allard, C.; Carneiro, L.; Grall, S.; Cline, B.H.; Fioramonti, X.; Chrétien, C.; Baba-Aissa, F.; Giaume, C.; Pénicaud, L.; Leloup, C. Hypothalamic astroglial connexins are required for brain glucose sensing-induced insulin secretion. J. Cereb. Blood Flow Metab. 2014, 34, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Allard, C.; Carneiro, L.; Collins, S.C.; Chrétien, C.; Grall, S.; Pénicaud, L.; Leloup, C. Alteration of hypothalamic glucose and lactate sensing in 48h hyperglycemic rats. Neurosci. Lett. 2013, 534, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Gowd, V.; Xie, L.; Zheng, X.; Chen, W. Dietary fibers as emerging nutritional factors against diabetes: Focus on the involvement of gut microbiota. Crit. Rev. Biotechnol. 2019, 39, 524–540. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of dietary fiber consumption on insulin resistance and the prevention of type 2 diabetes. J. Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef]
- Kerimi, A.; Nyambe-Silavwe, H.; Gauer, J.S.; Tomás-Barberán, F.A.; Williamson, G. Pomegranate juice, but not an extract, confers a lower glycemic response on a high–glycemic index food: Randomized, crossover, controlled trials in healthy subjects. Am. J. Clin. Nutr. 2017, 106, 1384–1393. [Google Scholar] [CrossRef]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Breton, J.; Tennoune, N.; Lucas, N.; Francois, M.; Legrand, R.; Jacquemot, J.; Goichon, A.; Guérin, C.; Peltier, J.; Pestel-Caron, M.; et al. Gut commensal E. coli proteins activate host satiety pathways following nutrient-induced bacterial growth. Cell Metab. 2016, 23, 324–334. [Google Scholar] [CrossRef]
- Schéle, E.; Grahnemo, L.; Anesten, F.; Halleń, A.; Bäckhed, F.; Jansson, J.O. The gut microbiota reduces leptin sensitivity and the expression of the obesity-suppressing neuropeptides proglucagon (Gcg) and brain-derived neurotrophic factor (Bdnf) in the central nervous system. Endocrinology 2013, 154, 3643–3651. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Food | Serving Size (g) | GI | Carbohydrates Per Serving (g) | GL | Food | Serving Size (g) | GI | Carbohydrates Per Serving (g) | GL | Food | Serving Size (g) | GI | Carbohydrates Per Serving (g) | GL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tuna | 100 | 0 | 0 | 0 | Fructose | 10 | 23 | 10 | 2 | Wheat | 200 | 45 | 137 | 62 |
| Salmon | 100 | 0 | 0 | 0 | Blackberry | 60 | 25 | 4 | 2 | Carrot Juice | 250 | 45 | 24 | 11 |
| Sardine | 100 | 0 | 0 | 0 | Grapefruit | 120 | 25 | 11 | 3 | Pineapple Juice | 250 | 46 | 33 | 15 |
| Mackerel | 100 | 0 | 0 | 0 | Milk, full fat | 250 | 27 | 12 | 3 | Banana | 120 | 47 | 24 | 11 |
| Crab | 85 | 0 | 0 | 0 | American Cheese | 28 | 27 | 2 | <1 | Lasagna | 125 | 47 | 19 | 9 |
| Eggs (chicken) | 50 | 0 | 1 | 0 | Cottage | 28 | 27 | 6 | 2 | Penne | 125 | 47 | 94 | 44 |
| Beef | 100 | 0 | 0 | 0 | Chickpeas, boiled | 150 | 28 | 30 | 8 | Butter | 5 | 50 | 0 | 0 |
| Chicken | 140 | 0 | 6 | 0 | Lentil | 200 | 28 | 40 | 11 | Mayonnaise | 15 | 50 | 0 | 0 |
| Goat | 30 | 0 | 0 | 0 | Beans, kidney | 150 | 28 | 25 | 7 | Mango | 120 | 51 | 15 | 8 |
| Pork | 85 | 0 | 0 | 0 | Garlic | 3 | 30 | 1 | <1 | Tortilla | 50 | 52 | 24 | 12 |
| Lamb | 85 | 0 | 1 | 0 | Vanilla extract | 4 | 30 | 3 | 0 | Blueberry | 150 | 53 | 18 | 7 |
| Ham | 85 | 0 | 0 | 0 | Buttermilk | 245 | 31 | 12 | 4 | Kiwi fruit | 150 | 53 | 16 | 9 |
| Turkey | 85 | 0 | 0 | 0 | Lime | 67 | 32 | 7 | <1 | Date | 60 | 54 | 33 | 21 |
| Duck | 140 | 0 | 0 | 0 | Broccoli | 80 | 32 | 4 | 1 | Orange juice | 250 | 55 | 26 | 14 |
| Rabbit | 85 | 0 | 0 | 0 | Artichoke | 150 | 32 | 14 | 4 | Corn, Sweet | 150 | 55 | 32 | 18 |
| Macadamia | 28 | 10 | 4 | <1 | Cauliflower | 100 | 32 | 5 | 2 | Cranberry Juice | 250 | 55 | 33 | 18 |
| Pecan | 28 | 10 | 4 | <1 | Green Bean | 55 | 32 | 4 | 1 | Honey | 25 | 55 | 20 | 11 |
| Almond | 28 | 10 | 6 | <1 | Asparagus | 130 | 32 | 5 | 2 | Brown Rice | 150 | 55 | 33 | 18 |
| Mushrooms | 75 | 10 | 4 | 1 | Radish | 100 | 32 | 7 | 2 | Ketchup | 17 | 55 | 5 | 3 |
| Cabbage | 80 | 10 | 5 | 1 | Mustard | 5 | 32 | 1 | <1 | Apricots | 120 | 57 | 9 | 5 |
| Peanut Butter | 55 | 14 | 5 | 6 | Milk, skim | 250 | 32 | 13 | 4 | Potato | 75 | 60 | 12 | 7 |
| Peanut | 28 | 14 | 6 | 1 | Raspberries | 150 | 32 | 8 | 3 | Coca-Cola | 250 | 60 | 26 | 16 |
| Avocado | 80 | 15 | 3 | 1 | Ice cream | 250 | 32 | 3 | 1 | Fig (dried) | 100 | 61 | 26 | 16 |
| Zucchini | 120 | 15 | 4 | 1 | Pear | 120 | 33 | 13 | 3 | Beetroot | 80 | 64 | 8 | 5 |
| Cucumber | 80 | 15 | 4 | 0 | Apricot | 120 | 34 | 9 | 3 | Cantaloupe | 120 | 65 | 6 | 4 |
| Eggplant | 100 | 15 | 6 | 2 | Low Fat Milk | 250 | 35 | 13 | 5 | Sucrose | 10 | 65 | 10 | 7 |
| Tomato | 100 | 15 | 4 | 1 | Carrot | 60 | 39 | 6 | 2 | White rice | 150 | 65 | 35 | 23 |
| Celery | 80 | 15 | 2 | 1 | Plums | 150 | 39 | 15 | 6 | Couscous, boiled | 150 | 65 | 35 | 23 |
| Lettuce | 100 | 15 | 3 | 1 | Apple | 120 | 40 | 16 | 6 | Pineapple | 120 | 66 | 10 | 6 |
| Spinach | 100 | 15 | 4 | 1 | Orange | 120 | 40 | 11 | 4 | Sweet potato | 130 | 70 | 17 | 12 |
| Onion | 10 | 15 | 1 | <1 | Strawberry | 120 | 40 | 3 | 1 | Crepe | 30 | 71 | 7 | 5 |
| Hazelnuts | 28 | 15 | 5 | <1 | Pepper | 2 | 40 | 1 | <1 | White bread | 30 | 71 | 13 | 10 |
| Red wine | 150 | 15 | 4 | <1 | Apple Juice | 250 | 40 | 30 | 12 | Whole wheat bread | 30 | 71 | 13 | 13 |
| White wine | 150 | 15 | 3 | <1 | Squash | 80 | 41 | 30 | 8 | Watermelon | 120 | 72 | 6 | 4 |
| Ginger | 11 | 15 | 2 | <1 | Peach | 120 | 42 | 11 | 5 | Bagel | 70 | 72 | 30 | 22 |
| Yogurt, low fat | 200 | 15 | 9 | 1 | Beans, black-eyed | 150 | 42 | 30 | 13 | Goat milk | 244 | 72 | 11 | 8 |
| Soybean | 190 | 16 | 56 | 9 | Coconut | 100 | 42 | 17 | 7 | Rutabagas | 385 | 72 | 33 | 24 |
| Pistachios | 28 | 18 | 8 | 1 | Spaghetti | 125 | 42 | 94 | 40 | Popcorn | 30 | 72 | 16 | 12 |
| Walnut | 28 | 20 | 4 | 1 | Chocolate | 28 | 43 | 16 | 7 | Pumpkin | 100 | 75 | 4 | 3 |
| Cherries | 100 | 20 | 16 | 5 | Tagliatelle | 125 | 44 | 90 | 40 | Cornflakes | 50 | 85 | 42 | 36 |
| Lemon | 60 | 20 | 5.5 | 1 | Cranberry | 110 | 45 | 8 | 1 | Baguette | 30 | 95 | 11 | 15 |
| Pea | 100 | 22 | 14 | 3 | Endive | 100 | 45 | 3 | 1 | Glucose | 10 | 100 | 10 | 10 |
| Low GL Diet | Regular Diet | Keto Diet | Modified Keto Diet | MCT Diet | Japanese Diet | Mediterranean Diet | Low GI Diet | Western Diet | High GI Diet | High GL Diet | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Carbohydrates | 45% | 45–55% | 5–10% | 15% | 5–10% | 45–55% | 50–60% | 15–20% | 50% | 45% | 55% |
| Fat | 35% | 20–35% | 70–75% | 55% | 30% MCTs | 20–35% | 25–35% | 60% | 35% | 35% | 30% |
| 30% LCFA | |||||||||||
| 10–15% others | |||||||||||
| Proteins | 20% | 10–35% | 20–25% | 30% | 20–25% | 10–35% | 5–25% | 20–25% | 15% | 30% | 15% |
| Kcal | 2200 | 2200 | 2200 | 2200 | 2200 | ~80% of regular | 2200 | 2200 | ~120% of regular | 2200 | 2200 |
| Food | low GL foods | Fresh food, low processed food | Low carbs food, High Fat, fish, meat, eggs, vegetables, fruits, nuts, berries… | Keto diet with increased amount of carbs | Keto diet enriched in MCT rich food such as coconut oil | Fish, Fruits, seasonal food, green tea, soy, rice (brown)… | Olive oil, fruits, vegetables and legumes, low amount of meat and fish, moderate wine | Low GI foods enriched, high non digestible fibers… | Junk foods, processed food with added sugar, saturated fats, high GI food… | High GI food, low non digestible fibers | high GL foods |
| Group | Diet | Method | Results | Limitation | Ref |
|---|---|---|---|---|---|
| Cognitive Healthy Elderly | No specific diet | Correlation between GI and cognitive score both assessed via questionnaire | Improved cognition in blood glucose regulation defect people | Different diets, background, food habits, medical history Questionnaire assessment of cognition only | [26,27,28,32] |
| Schoolchildren | Low GI breakfast vs. High GI breakfast vs. no breakfast Low GI/low GL vs. low GI/high GL vs. high GI/low GL vs. high GI/high GL | Cognitive test for learning and memory, accuracy and speed score, stress, hunger and thirst assessment | Low GI improves cognition and accuracy and decrease stress | Schoolchildren tested only during the morning for the GI breakfast. No effect measured after lunch or on a long time period. | [37,38,39,40,41,42,43,46] |
| Adults | No specific diet | Correlation between the GI of the diet and cognitive score | No effect | Only study, group compared to elderly No adults group with high GI diet | [47] |
| Epilepsy | KD, modified KD, low GI | Pediatric patients, number of seizure | 50% decrease in the number of seizure | Observational studies, No interventional studies, no controlled diet, no longitudinal studies, no mechanistic studies, only hypothesis | [8,48] |
| Stroke | Vegetarian diets, Mediterranean diet High GI/GL diet | Stroke occurrence | Decreased risk of stroke with vegetarian diets Poor outcome following stroke with High GI/GL | [49,50,51,52,53,54,55] | |
| Alzheimer’s disease | High GI Diet, Low GI, healthy diet, KD, MCT diet, Mediterranean diet | Post mortem brain analysis, memory test | High GI associated with accumulation of Aβ Healthy diet decrease Aβ, improves memory and verbal communication | [56,57,58,59] | |
| Parkinson’s disease | Japanese diet | PD rate | Low PD rate | [60,61] | |
| Autism Spectrum Disorder | High GI or low GI diet | Animal studies, social behavior analysis | High GI increase ASD phenotype while low GI improve social behavior | [62,63,64,65] | |
| Depression and Anxiety | High GI/GL | Rate of disease in a population | Increased depression and anxiety rate | [66] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carneiro, L.; Leloup, C. Mens sana in corpore sano: Does the Glycemic Index Have a Role to Play? Nutrients 2020, 12, 2989. https://doi.org/10.3390/nu12102989
Carneiro L, Leloup C. Mens sana in corpore sano: Does the Glycemic Index Have a Role to Play? Nutrients. 2020; 12(10):2989. https://doi.org/10.3390/nu12102989
Chicago/Turabian StyleCarneiro, Lionel, and Corinne Leloup. 2020. "Mens sana in corpore sano: Does the Glycemic Index Have a Role to Play?" Nutrients 12, no. 10: 2989. https://doi.org/10.3390/nu12102989
APA StyleCarneiro, L., & Leloup, C. (2020). Mens sana in corpore sano: Does the Glycemic Index Have a Role to Play? Nutrients, 12(10), 2989. https://doi.org/10.3390/nu12102989