Urinary TMAO Levels Are Associated with the Taxonomic Composition of the Gut Microbiota and with the Choline TMA-Lyase Gene (cutC) Harbored by Enterobacteriaceae


 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Design and Use of Primers Targeting the cutC Gene
2.2. PCR Detection of cutC Gene in Single Strains
2.3. Detection of Choline-Utilization Activity in Single Strains
2.4. Study Population
2.5. Collection of Fecal and Urine Samples
2.6. Analysis of cutC Gene by Quantitative Real-Time PCR
2.7. Analysis of the Bacterial Taxonomic Composition of Fecal Samples
2.8. TMAO Quantification in Urine Samples
2.9. Statistical Analysis
3. Results
3.1. Distribution of the cutC Gene among Bacterial Taxa
3.2. Bacterial Taxonomic Structure of the Fecal Microbiota
3.3. Putative cutC Genes in Human Fecal Metagenomic DNA
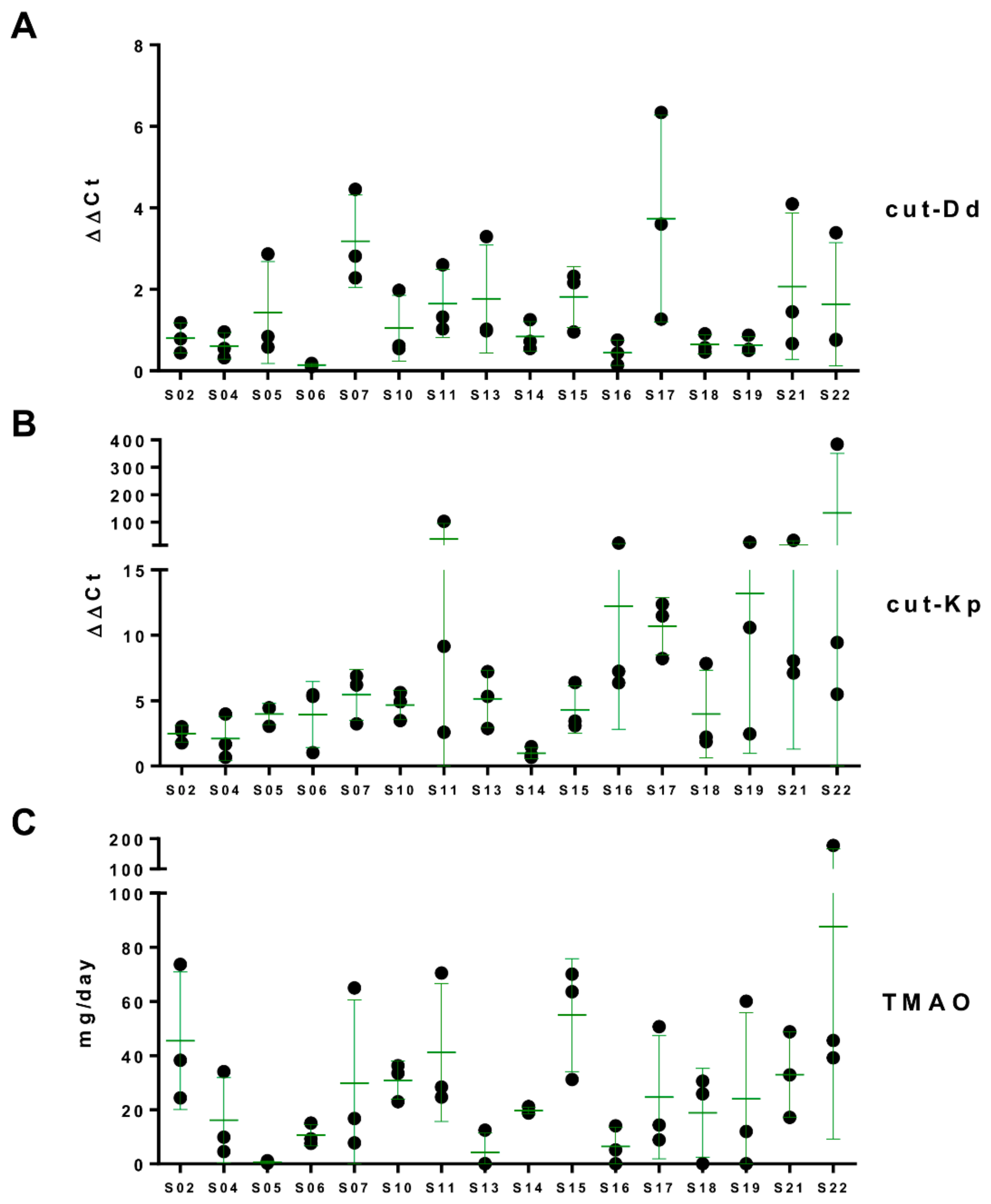
3.4. Daily Urinary Excretion of TMAO
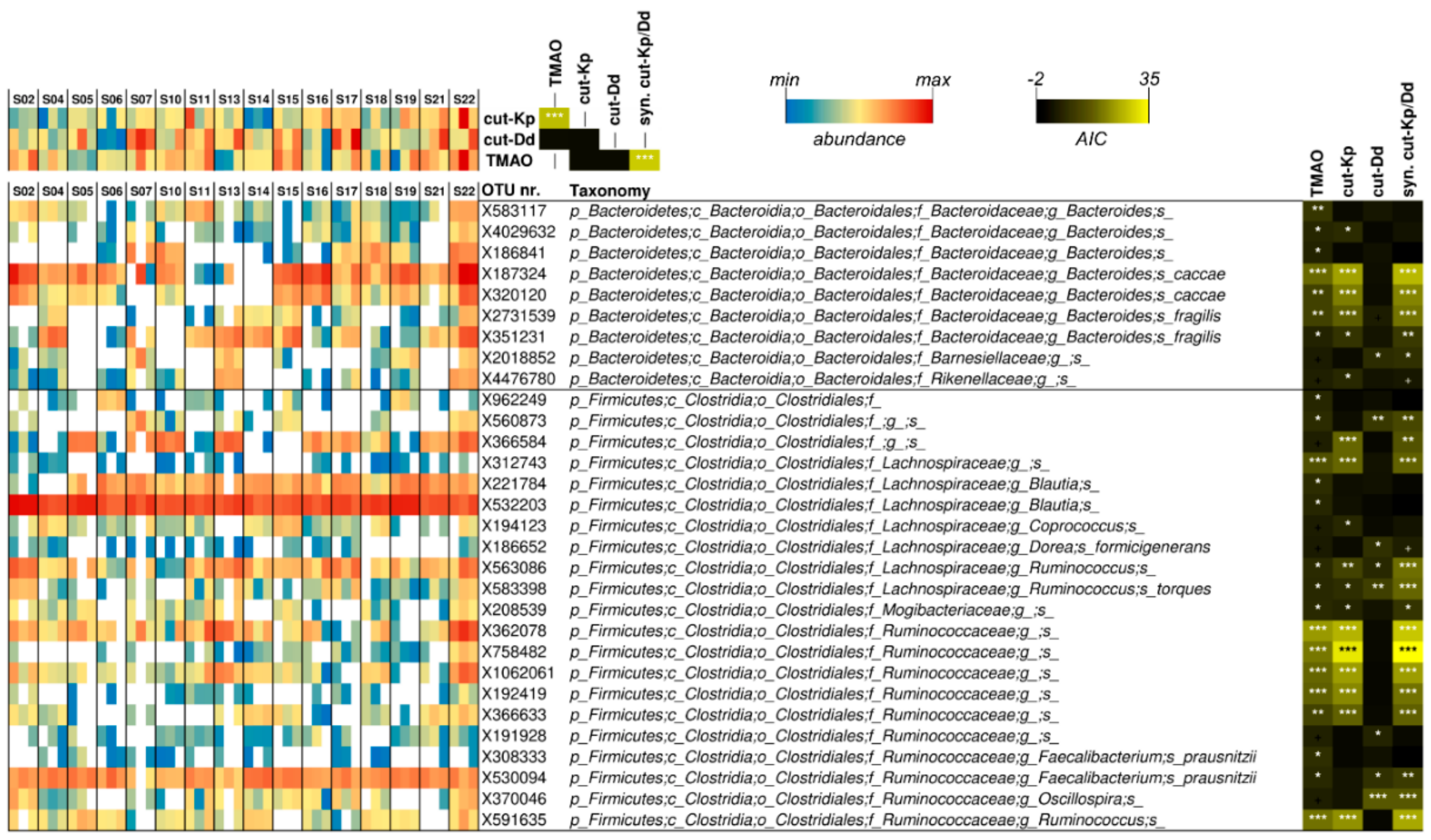
3.5. Associations among Urinary TMAO, Fecal cutC, and Fecal Bacterial Taxa
4. Discussion
- First, we quantified the abundance of a gene of the intestinal microbiome without considering if and how much this gene was expressed. This could therefore limit the possibility of associating the abundance of this gene with its product.
- Furthermore, the production of TMA, in addition to the presence of the bacterial gene that allows its production (cutC), depends on the availability of the choline substrate, which mainly comes from the diet.
- Nonetheless, the contribution to the TMA produced in the intestine and, consequently, to the TMAO generated in the liver, derives from different chemical moieties (mainly choline, betaine, and carnitine) and includes different microbial metabolic pathways, such as those involving the carnitine monooxygenase CntAB and the glycine betaine reductase GrdH, in addition to the choline TMA-lyase CutC [40].
- Finally, TMAO can also be ingested directly from foods such as fish and seafood, which are naturally rich in this molecule [43].
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohajeri, M.H.; Brummer, R.J.M.; Rastall, R.A.; Weersma, R.K.; Harmsen, H.J.M.; Faas, M.; Eggersdorfer, M. The role of the microbiome for human health: From basic science to clinical applications. Eur. J. Nutr. 2018, 57, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Nowinski, A.; Ufnal, M. Trimethylamine N-oxide: A harmful, protective or diagnostic marker in lifestyle diseases? Nutrition 2018, 46, 7–12. [Google Scholar] [CrossRef]
- Huc, T.; Drapala, A.; Gawrys, M.; Konop, M.; Bielinska, K.; Zaorska, E.; Samborowska, E.; Wyczalkowska-Tomasik, A.; Paczek, L.; Dadlez, M.; et al. Chronic, low-dose TMAO treatment reduces diastolic dysfunction and heart fibrosis in hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1805–H1820. [Google Scholar] [CrossRef]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE(-/-) transgenic mice expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Blusztajn, J.K. Choline and human nutrition. Annu. Rev. Nutr. 1994, 14, 269–296. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Mar, M.H.; Howe, J.C.; Holden, J.M. Concentrations of choline-containing compounds and betaine in common foods. J. Nutr. 2003, 133, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Wiedeman, A.M.; Barr, S.I.; Green, T.J.; Xu, Z.; Innis, S.M.; Kitts, D.D. Dietary choline intake: Current state of knowledge across the life cycle. Nutrients 2018, 10, 1513. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; Blusztajn, J.K.; Caudill, M.A.; Klatt, K.C.; Natker, E.; Zeisel, S.H.; Zelman, K.M. Choline: The underconsumed and underappreciated essential nutrient. Nutr. Today 2018, 53, 240–253. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA). Dietary reference values for choline. EFSA J. 2016, 14, 70. [Google Scholar]
- Craciun, S.; Balskus, E.P. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc. Natl. Acad. Sci. USA 2012, 109, 21307–21312. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef]
- Martinez-del Campo, A.; Bodea, S.; Hamer, H.A.; Marks, J.A.; Haiser, H.J.; Turnbaugh, P.J.; Balskus, E.P. Characterization and detection of a widely distributed gene cluster that predicts anaerobic choline utilization by human gut bacteria. mBio 2015, 6, e00042-15. [Google Scholar] [CrossRef]
- Xu, K.Y.; Xia, G.H.; Lu, J.Q.; Chen, M.X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Craciun, S.; Marks, J.A.; Balskus, E.P. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem. Biol. 2014, 9, 1408–1413. [Google Scholar] [CrossRef] [PubMed]
- Kalnins, G.; Kuka, J.; Grinberga, S.; Makrecka-Kuka, M.; Liepinsh, E.; Dambrova, M.; Tars, K. Structure and function of CutC choline lyase from human microbiota bacterium klebsiella pneumoniae. J. Biol. Chem. 2015, 290, 21732–21740. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, C.; Gargari, G.; Koirala, R.; Laureati, M.; Riso, P.; Guglielmetti, S.; Pagliarini, E. New insights into the relationship between taste perception and oral microbiota composition. Sci. Rep. 2019, 9, 3549. [Google Scholar] [CrossRef] [PubMed]
- Gargari, G.; Taverniti, V.; Gardana, C.; Cremon, C.; Canducci, F.; Pagano, I.; Barbaro, M.R.; Bellacosa, L.; Castellazzi, A.M.; Valsecchi, C.; et al. Fecal clostridiales distribution and short-chain fatty acids reflect bowel habits in irritable bowel syndrome. Environ. Microbiol. 2018, 20, 3201–3213. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W. A flow injection electrospray ionization tandem mass spectrometric method for the simultaneous measurement of trimethylamine and trimethylamine N-oxide in urine. J. Mass Spectrom. 2008, 43, 495–499. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. Artic. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Chhibber-Goel, J.; Singhal, V.; Parakh, N.; Bhargava, B.; Sharma, A. The metabolite Trimethylamine-N-Oxide is an emergent biomarker of human health. Curr. Med. Chem. 2017, 24, 3942–3953. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramirez, M.J.; Milagro, F.I.; Martinez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in disease: Potential biomarker or new therapeutic target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef]
- Ufnal, M.; Nowinski, A. Is increased plasma TMAO a compensatory response to hydrostatic and osmotic stress in cardiovascular diseases? Med. Hypotheses 2019, 130, 109271. [Google Scholar] [CrossRef]
- Zeisel, S.H.; Warrier, M. Trimethylamine N-Oxide, the microbiome, and heart and kidney disease. Annu. Rev. Nutr. 2017, 37, 157–181. [Google Scholar] [CrossRef] [PubMed]
- Jameson, E.; Fu, T.; Brown, I.R.; Paszkiewicz, K.; Purdy, K.J.; Frank, S.; Chen, Y. Anaerobic choline metabolism in microcompartments promotes growth and swarming of Proteus mirabilis. Environ. Microbiol. 2016, 18, 2886–2898. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Vieira-Silva, S.; Raes, J. Microbiology meets big data: The case of gut microbiota-derived Trimethylamine. Annu. Rev. Microbiol. 2015, 69, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Zarzycki, J.; Erbilgin, O.; Kerfeld, C.A. Bioinformatic characterization of glycyl radical enzyme-associated bacterial microcompartments. Appl. Environ. Microbiol. 2015, 81, 8315–8329. [Google Scholar] [CrossRef] [PubMed]
- Fracalanzza, S.A.; Scheidegger, E.M.; Santos, P.F.; Leite, P.C.; Teixeira, L.M. Antimicrobial resistance profiles of enterococci isolated from poultry meat and pasteurized milk in Rio de Janeiro, Brazil. Mem. Inst. Oswaldo Cruz 2007, 102, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Franzetti, L.; Frusca, M.; Scarpellini, M. Phenotypic and genotypic characterization of lactic acid bacteria isolated from Artisanal Italian goat cheese. J. Food Prot. 2010, 73, 657–662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Jong, A.; Simjee, S.; Rose, M.; Moyaert, H.; El Garch, F.; Youala, M.; Group, E.S. Antimicrobial resistance monitoring in commensal enterococci from healthy cattle, pigs and chickens across Europe during 2004-14 (EASSA Study). J. Antimicrob. Chemother. 2019, 74, 921–930. [Google Scholar] [CrossRef]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef]
- Zhang, A.Q.; Mitchell, S.; Smith, R. Fish odour syndrome: Verification of carrier detection test. J. Inherit. Metab. Dis. 1995, 18, 669–674. [Google Scholar] [CrossRef]
- Jameson, E.; Quareshy, M.; Chen, Y. Methodological considerations for the identification of choline and carnitine-degrading bacteria in the gut. Methods 2018, 149, 42–48. [Google Scholar] [CrossRef]
- Ufnal, M.; Pham, K. The gut-blood barrier permeability—A new marker in cardiovascular and metabolic diseases? Med. Hypotheses 2017, 98, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Prokopienko, A.J.; West, R.E., 3rd; Nolin, T.D.; Stubbs, J.R. Decreased kidney function is associated with enhanced hepatic flavin monooxygenase activity and increased circulating Trimethylamine N-Oxide concentrations in mice. Drug Metab. Dispos. Biol. Fate Chem. 2018, 46, 1304–1309. [Google Scholar] [CrossRef] [PubMed]
- Seibel, B.A.; Walsh, P.J. Trimethylamine oxide accumulation in marine animals: Relationship to acylglycerol storage. J. Exp. Biol. 2002, 205, 297–306. [Google Scholar] [PubMed]
- Jameson, E.; Doxey, A.C.; Airs, R.; Purdy, K.J.; Murrell, J.C.; Chen, Y. Metagenomic data-mining reveals contrasting microbial populations responsible for trimethylamine formation in human gut and marine ecosystems. Microb. Genom. 2016, 2, e000080. [Google Scholar] [CrossRef]
- Wilson, M. The indigenous microbiota of the gastrointestinal tract. In The Human Microbiota in Health and Disease: An Ecological and Community-Based Approach, 1st ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2018; 472p. [Google Scholar]
- Gosalbes, M.J.; Durban, A.; Pignatelli, M.; Abellan, J.J.; Jimenez-Hernandez, N.; Perez-Cobas, A.E.; Latorre, A.; Moya, A. Metatranscriptomic approach to analyze the functional human gut microbiota. PLoS ONE 2011, 6, e17447. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalla Via, A.; Gargari, G.; Taverniti, V.; Rondini, G.; Velardi, I.; Gambaro, V.; Visconti, G.L.; De Vitis, V.; Gardana, C.; Ragg, E.; et al. Urinary TMAO Levels Are Associated with the Taxonomic Composition of the Gut Microbiota and with the Choline TMA-Lyase Gene (cutC) Harbored by Enterobacteriaceae. Nutrients 2020, 12, 62. https://doi.org/10.3390/nu12010062
Dalla Via A, Gargari G, Taverniti V, Rondini G, Velardi I, Gambaro V, Visconti GL, De Vitis V, Gardana C, Ragg E, et al. Urinary TMAO Levels Are Associated with the Taxonomic Composition of the Gut Microbiota and with the Choline TMA-Lyase Gene (cutC) Harbored by Enterobacteriaceae. Nutrients. 2020; 12(1):62. https://doi.org/10.3390/nu12010062
Chicago/Turabian StyleDalla Via, Alessandro, Giorgio Gargari, Valentina Taverniti, Greta Rondini, Ilaria Velardi, Veniero Gambaro, Giacomo Luca Visconti, Valerio De Vitis, Claudio Gardana, Enzio Ragg, and et al. 2020. "Urinary TMAO Levels Are Associated with the Taxonomic Composition of the Gut Microbiota and with the Choline TMA-Lyase Gene (cutC) Harbored by Enterobacteriaceae" Nutrients 12, no. 1: 62. https://doi.org/10.3390/nu12010062
APA StyleDalla Via, A., Gargari, G., Taverniti, V., Rondini, G., Velardi, I., Gambaro, V., Visconti, G. L., De Vitis, V., Gardana, C., Ragg, E., Pinto, A., Riso, P., & Guglielmetti, S. (2020). Urinary TMAO Levels Are Associated with the Taxonomic Composition of the Gut Microbiota and with the Choline TMA-Lyase Gene (cutC) Harbored by Enterobacteriaceae. Nutrients, 12(1), 62. https://doi.org/10.3390/nu12010062