Human Gut Microbiome Response Induced by Fermented Dairy Product Intake in Healthy Volunteers

 ,
,  ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Preparation and Microbiome Data Analysis
2.3. Analysis of Responders
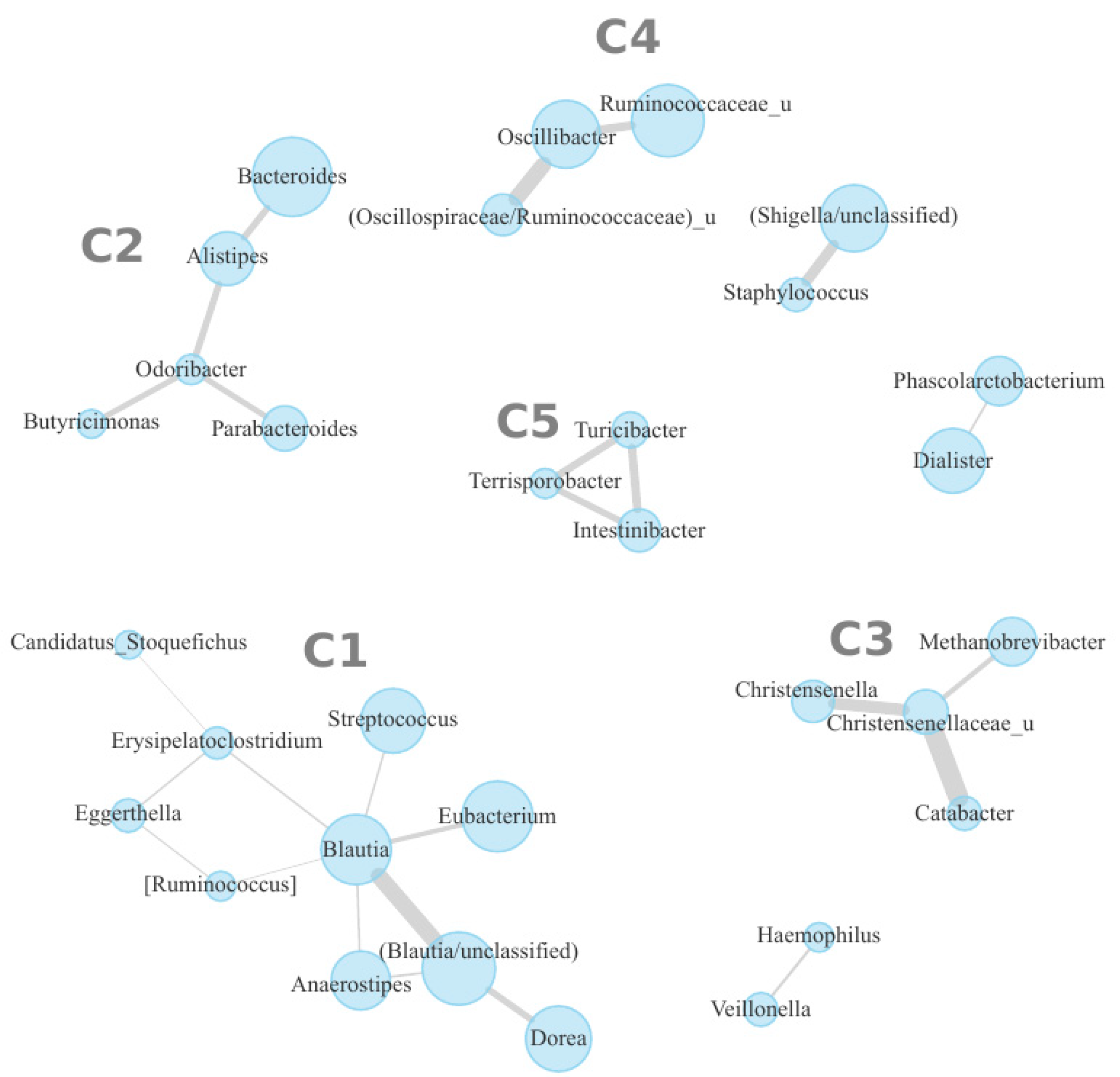
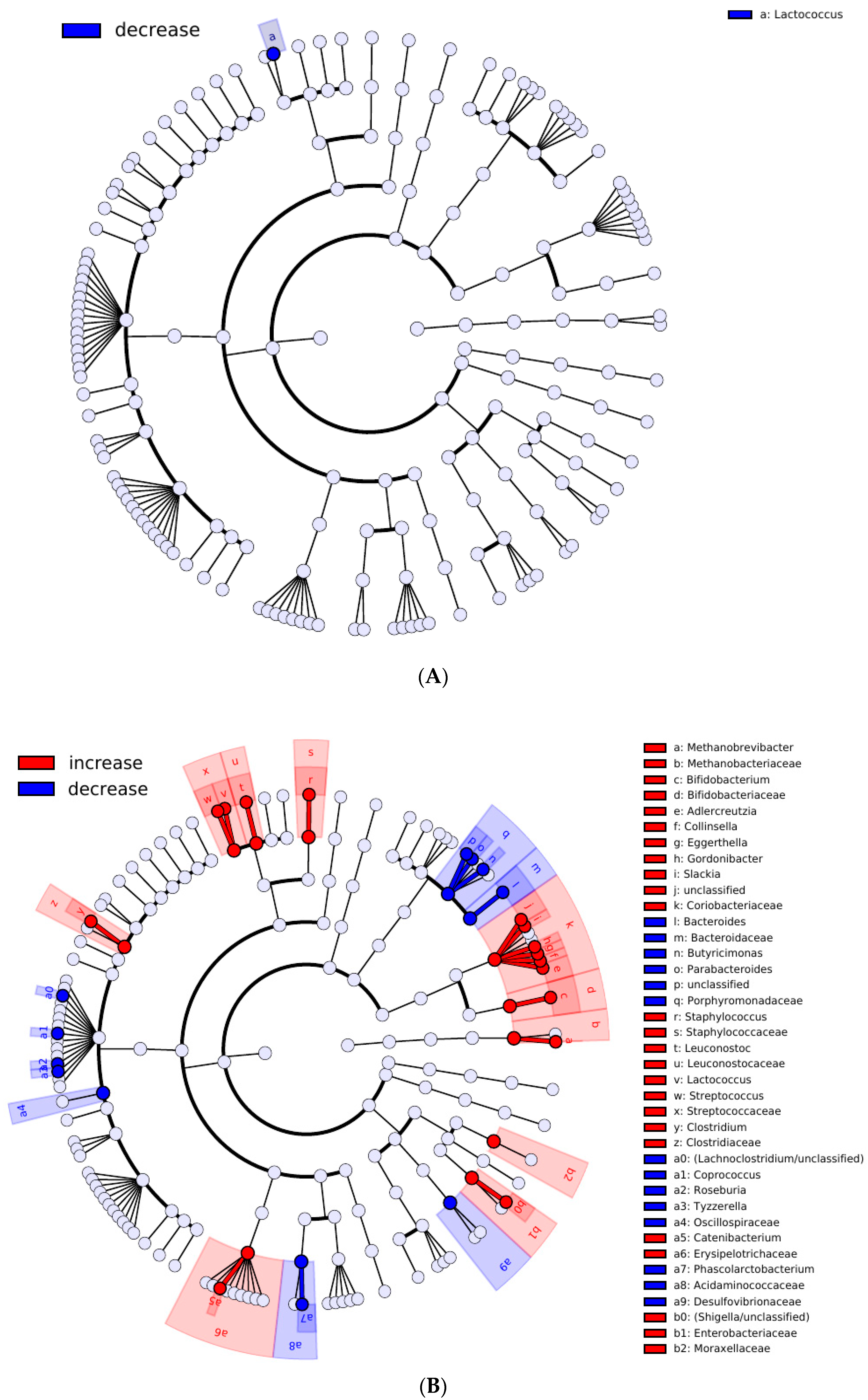
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tuohy, K.; Del Rio, D. Diet-Microbe Interactions in the Gut: Effects on Human Health and Disease; Elsevier Science: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood Microbiota Dysbiosis Is Associated with the Onset of Cardiovascular Events in a Large General Population: The D.E.S.I.R. Study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, N.; Burton, J.P.; Suppiah, P.; Reid, G.; Stebbings, S. The role of the microbiome in rheumatic diseases. Curr. Rheumatol. Rep. 2013, 15, 314. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Wu, Z.; Xu, W.; Yang, J.; Chen, Y.; Li, L. Intestinal microbiota was assessed in cirrhotic patients with hepatitis B virus infection. Intestinal microbiota of HBV cirrhotic patients. Microb. Ecol. 2011, 61, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Stanton, C.; Ross, R.P.; Fitzgerald, G.F.; Van Sinderen, D. Fermented functional foods based on probiotics and their biogenic metabolites. Curr. Opin. Biotechnol. 2005, 16, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Orrhage, K.; Brismar, B.; Nord, C.E. Effect of supplements with Bifidobacterium longum and Lactobacillus acidophilus on the intestinal microbiota during administration of clindamycin. Microb. Ecol. Health Disease 1994, 7, 17–25. [Google Scholar] [CrossRef]
- Plummer, S.; Weaver, M.A.; Harris, J.C.; Dee, P.; Hunter, J. Clostridium difficile pilot study: Effects of probiotic supplementation on the incidence of C. difficile diarrhoea. Int. Microbiol. 2004, 7, 59–62. [Google Scholar] [PubMed]
- Lin, H.C.; Hsu, C.H.; Chen, H.L.; Chung, M.Y.; Hsu, J.F.; Lien, R.I.; Tsao, L.Y.; Chen, C.H.; Su, B.H. Oral probiotics prevent necrotizing enterocolitis in very low birth weight preterm infants: A multicenter, randomized, controlled trial. Pediatrics 2008, 122, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Gionchetti, P.; Rizzello, F.; Venturi, A.; Brigidi, P.; Matteuzzi, D.; Bazzocchi, G.; Poggioli, G.; Miglioli, M.; Campieri, M. Oral bacteriotherapy as maintenance treatment in patients with chronic pouchitis: A double-blind, placebo-controlled trial. Gastroenterology 2000, 119, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Isolauri, E.; Arvola, T.; Sütas, Y.; Moilanen, E.; Salminen, S. Probiotics in the management of atopic eczema. Clin. Exp. Allergy 2000, 30, 1605–1610. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kwon, J.H.; Ahn, S.H.; Lee, S.I.; Han, Y.S.; Choi, Y.O.; Lee, S.Y.; Ahn, K.M.; Ji, G.E. Effect of probiotic mix (Bifidobacterium bifidum, Bifidobacterium lactis, Lactobacillus acidophilus) in the primary prevention of eczema: A double-blind, randomized, placebo-controlled trial. Pediat. Allergy Immunol. 2010, 21, e386–e393. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Hacini-Rachinel, F.; Gosoniu, M.L.; Bourdeau, T.; Holvoet, S.; Doucet-Ladeveze, R.; Beaumont, M.; Mercenier, A.; Nutten, S. Immune-modulatory effect of probiotic Bifidobacterium lactis NCC2818 in individuals suffering from seasonal allergic rhinitis to grass pollen: An exploratory, randomized, placebo-controlled clinical trial. Eur. J. Clin. Nutr. 2013, 67, 161. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Fan, S.W.; Zhu, W.Y. Development of gut microbiota in a mouse model of ovalbumin-induced allergic diarrhea under sub-barrier system. Asian-Australas. J. Anim. Sci. 2013, 26, 545. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Xue, W.; Luo, G.; Deng, Z.; Qin, P.; Guo, R.; Sun, H.; Xia, Y.; Liang, S.; Dai, Y.; Wan, D.; et al. 1520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat. Biotechnol. 2009, 37, 179. [Google Scholar] [CrossRef] [PubMed]
- Veiga, P.; Pons, N.; Agrawal, A.; Oozeer, R.; Guyonnet, D.; Brazeilles, R.; Faurie, J.-M.; van Hylckama Vlieg, J.E.T.; Houghton, L.A.; Whorwell, P.J.; et al. Changes of the human gut microbiome induced by a fermented milk product. Sci. Rep. 2014, 4, 6328. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Akedo, I.; Umesaki, Y.; Tanaka, R.; Imaoka, A.; Otani, T. Randomized controlled trial of the effect of bifidobacteria-fermented milk on ulcerative colitis. J. Am. Coll. Nutr. 2003, 22, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Derrien, M.; Levenez, F.; Brazeilles, R.; Ballal, S.A.; Kim, J.; Degivry, M.-C.; Quéré, G.; Garault, P.; van Hylckama Vlieg, J.E.T.; et al. Ecological robustness of the gut microbiota in response to ingestion of transient food-borne microbes. ISME J. 2016, 10, 2235. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.J.; Gross, R.; Bittinger, K.; Sherrill-Mix, S.; Lewis, J.D.; Collman, R.G.; Bushman, F.D.; Hongzhe, L. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015, 31, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Pevzner, M.I. Osnovy Lechebnogo Pitaniya [The Basics of Clinical Nutrition]; Gosudarstvennoe Izdatel’stvo Literatury: Moscow, Russia, 1949. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Ritari, J.; Salojärvi, J.; Lahti, L.; de Vos, W.M. Improved taxonomic assignment of human intestinal 16S rRNA sequences by a dedicated reference database. BMC Genom. 2015, 16, 1056. [Google Scholar] [CrossRef] [PubMed]
- Brandt, B.W.; Bonder, M.J.; Huse, S.M.; Zaura, E. TaxMan: A server to trim rRNA reference databases and inspect taxonomic coverage. Nucleic Acids Res. 2012, 40, W82–W87. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Efimova, D.; Tyakht, A.; Popenko, A.; Vasilyev, A.; Altukhov, I.; Dovidchenko, N.; Odintsova, V.; Klimenko, N.; Loshkarev, R.; Pashkova, M.; et al. Knomics-Biota—A system for exploratory analysis of human gut microbiota data. BioData Min. 2018, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Basic Report for Fermented Dairy Product Intervention Study, Project ID 302. Available online: https://biota.knomics.ru/public-report?key=uTxTb4qIfy9BFjQ65oR_oA-V1N9MxOSv (accessed on 1 March 2019).
- Paired Analysis Report for Fermented Dairy Product Intervention Study, Project ID 302. Available online: https://biota.knomics.ru/public-report?key=LeEcGCARriO2b4TNAXfdMLfWoXFjKXAV (accessed on 1 March 2019).
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial interactions: From networks to models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.N.; Stine, O.C.; Bravo, H.C.; Pop, M. Robust methods for differential abundance analysis in marker gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed]
- Tyakht, A.V.; Alexeev, D.G.; Popenko, A.S.; Kostryukova, E.S.; Govorun, V.M. Rural and urban microbiota: To be or not to be? Gut Microbes 2014, 5, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Slavin, J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients 2013, 5, 1417–1435. [Google Scholar] [CrossRef] [PubMed]
- Tyakht, A.V.; Kostryukova, E.S.; Popenko, A.S.; Belenikin, M.S.; Pavlenko, A.V.; Larin, A.K.; Karpova, I.Y.; Selezneva, O.V.; Semashko, T.A.; Ospanova, E.A. Human gut microbiota community structures in urban and rural populations in Russia. Nat. Commun. 2013, 4, 2469. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, S.; Suda, W.; Oshima, K.; Kim, S.W.; Hirose, Y.; Morita, H.; Hattori, M. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 2016, 23, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, A.; Bingham, S.; Setchell, K.D. Biological effects of a diet of soy protein rich in isoflavones on the menstrual cycle of premenopausal women. Am. J. Clin. Nutr. 1994, 60, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Bowen, R.; Cai, Q.; Barnes, S.; Wang, Y. Antioxidant and antipromotional effects of the soybean isoflavone genistein. Proc. Soc. Exp. Biol. Med. 1995, 208, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Lamartiniere, C.A. Protection against breast cancer with genistein: A component of soy. Am. J. Clin. Nutr. 2000, 71, 1705S–1707S. [Google Scholar] [CrossRef] [PubMed]
- Matthies, A.; Blaut, M.; Braune, A. Isolation of a human intestinal bacterium capable of daidzein and genistein conversion. Appl. Environ. Microbiol. 2009, 75, 1740–1744. [Google Scholar] [CrossRef] [PubMed]
- Joy, S.; Siow, R.C.; Rowlands, D.J.; Becker, M.; Wyatt, A.W.; Aaronson, P.I.; Coen, C.W.; Kallo, I.; Jacob, R.; Mann, G.E. The Isoflavone Equol Mediates Rapid Vascular Relaxation Ca2+-independent activation of endothelial nitric-oxide synthase/hsp90 involving erk1/2 and akt phosphorylation in human endothelial cell. J. Biol. Chem. 2006, 281, 27335–27345. [Google Scholar] [CrossRef] [PubMed]
- Itsumi, M.; Shiota, M.; Takeuchi, A.; Kashiwagi, E.; Inokuchi, J.; Tatsugami, K.; Kajioka, S.; Uchiumi, T.; Naito, S.; Eto, M.; et al. Equol inhibits prostate cancer growth through degradation of androgen receptor by S-phase kinase-associated protein 2. Cancer Sci. 2016, 107, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Insights into the Role of Erysipelotrichaceae in the Human Host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.L.; Greiwe, J.S.; Desai, P.B.; Schwen, R.J. Single-dose and steady-state pharmacokinetic studies of S-equol, a potent nonhormonal, estrogen receptor β-agonist being developed for the treatment of menopausal symptoms. Menopause 2011, 18, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Klimenko, N.S.; Tyakht, A.V.; Popenko, A.S.; Vasiliev, A.S.; Altukhov, I.A.; Ischenko, D.S.; Shashkova, T.I.; Efimova, D.A.; Nikogosov, D.A.; Osipenko, D.A.; et al. Microbiome Responses to an Uncontrolled Short-Term Diet Intervention in the Frame of the Citizen Science Project. Nutrients 2018, 10, 576. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| (A) Increased in responders. | ||||
| Taxon | Rank | p | Adjusted p | Linear Regression Coefficient |
| Cooperative #2 | Cooperative | 0.0038 | 0.0097 | 0.0692 |
| Bacteroidaceae | Family | 0.003 | 0.0393 | 0.068 |
| Oxalobacteraceae | Family | 0.0033 | 0.0393 | 0.0031 |
| Bacteroidales | Order | 0 | 0.0007 | 0.1074 |
| Bacteroidia | Class | 0 | 0.0005 | 0.1074 |
| (B) decreased in responders | ||||
| Taxon | Rank | p | Adjusted p | Linear Regression Coefficient |
| Cooperative #1 | Cooperative | 0.0003 | 0.0013 | −0.0954 |
| Streptococcaceae | Family | 0.0001 | 0.0417 | −0.0433 |
| Coriobacteriaceae | Family | 0.0046 | 0.0375 | −0.0192 |
| Peptostreptococcaceae | Family | 0.0024 | 0.0392 | −0.0204 |
| (Lachnospiraceae/unclassified) | Family | 0.0052 | 0.0417 | −0.0215 |
| Lactococcus | Genus | 0 | 0 | −0.0222 |
| Blautia | Genus | 0 | 0.0013 | −0.0543 |
| Lactococcus_lactis | Species | 0 | 0.0004 | −0.0185 |
| OTU1383 (Blautia)/OTU1528 (Blautia)/OTU819 (Blautia) | Species | 0 | 0.0014 | −0.0425 |
| OTU262 (Ruminococcus)/OTU286 (Ruminococcus) | Species | 0 | 0.0098 | −0.0530 |
| OTU513 (Blautia)/OTU661 (Blautia) | Species | 0.0003 | 0.0324 | −0.0244 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volokh, O.; Klimenko, N.; Berezhnaya, Y.; Tyakht, A.; Nesterova, P.; Popenko, A.; Alexeev, D. Human Gut Microbiome Response Induced by Fermented Dairy Product Intake in Healthy Volunteers. Nutrients 2019, 11, 547. https://doi.org/10.3390/nu11030547
Volokh O, Klimenko N, Berezhnaya Y, Tyakht A, Nesterova P, Popenko A, Alexeev D. Human Gut Microbiome Response Induced by Fermented Dairy Product Intake in Healthy Volunteers. Nutrients. 2019; 11(3):547. https://doi.org/10.3390/nu11030547
Chicago/Turabian StyleVolokh, Olesya, Natalia Klimenko, Yulia Berezhnaya, Alexander Tyakht, Polina Nesterova, Anna Popenko, and Dmitry Alexeev. 2019. "Human Gut Microbiome Response Induced by Fermented Dairy Product Intake in Healthy Volunteers" Nutrients 11, no. 3: 547. https://doi.org/10.3390/nu11030547
APA StyleVolokh, O., Klimenko, N., Berezhnaya, Y., Tyakht, A., Nesterova, P., Popenko, A., & Alexeev, D. (2019). Human Gut Microbiome Response Induced by Fermented Dairy Product Intake in Healthy Volunteers. Nutrients, 11(3), 547. https://doi.org/10.3390/nu11030547