The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention

Abstract
1. Introduction
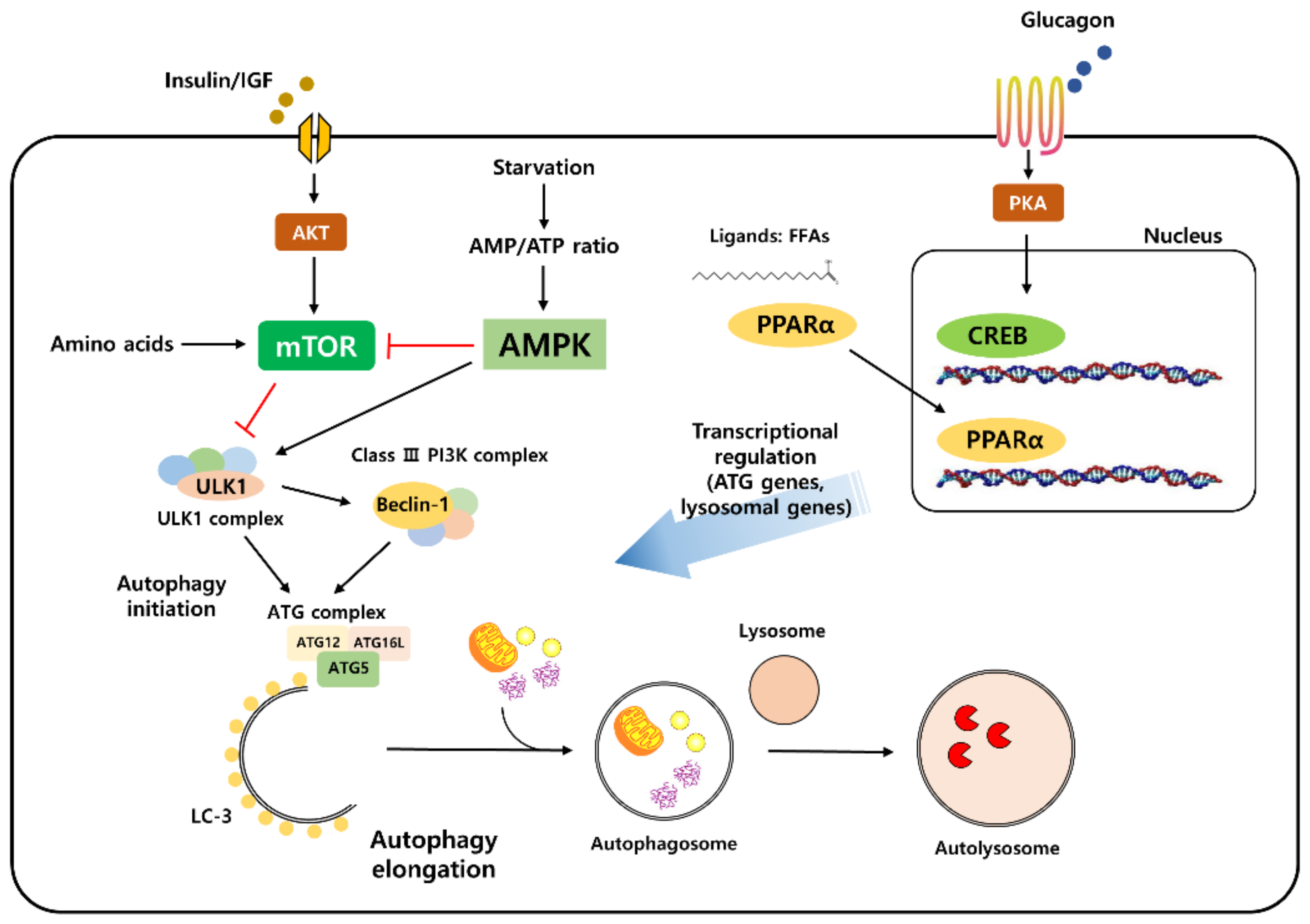
1.1. The Autophagy Process
1.2. Molecular Machinery of the Autophagy Process
1.3. Autophagy Is Regulated by Nutrient-Sensing Signaling
1.4. Autophagy and Aging
2. Calorie Restriction (CR) Modulates Autophagy Processes
2.1. Introduction to Calorie Restriction
2.2. Evidence for the Beneficial Effects of CR-Mediated Autophagy
3. Protective Effects of CR-Induced Autophagy on Different Organs
3.1. Liver
3.2. Muscle
3.3. Adipose Tissue
3.4. Kidney
4. Benefits of Intermeal Fasting in Autophagy: Is CR the Only Solution?
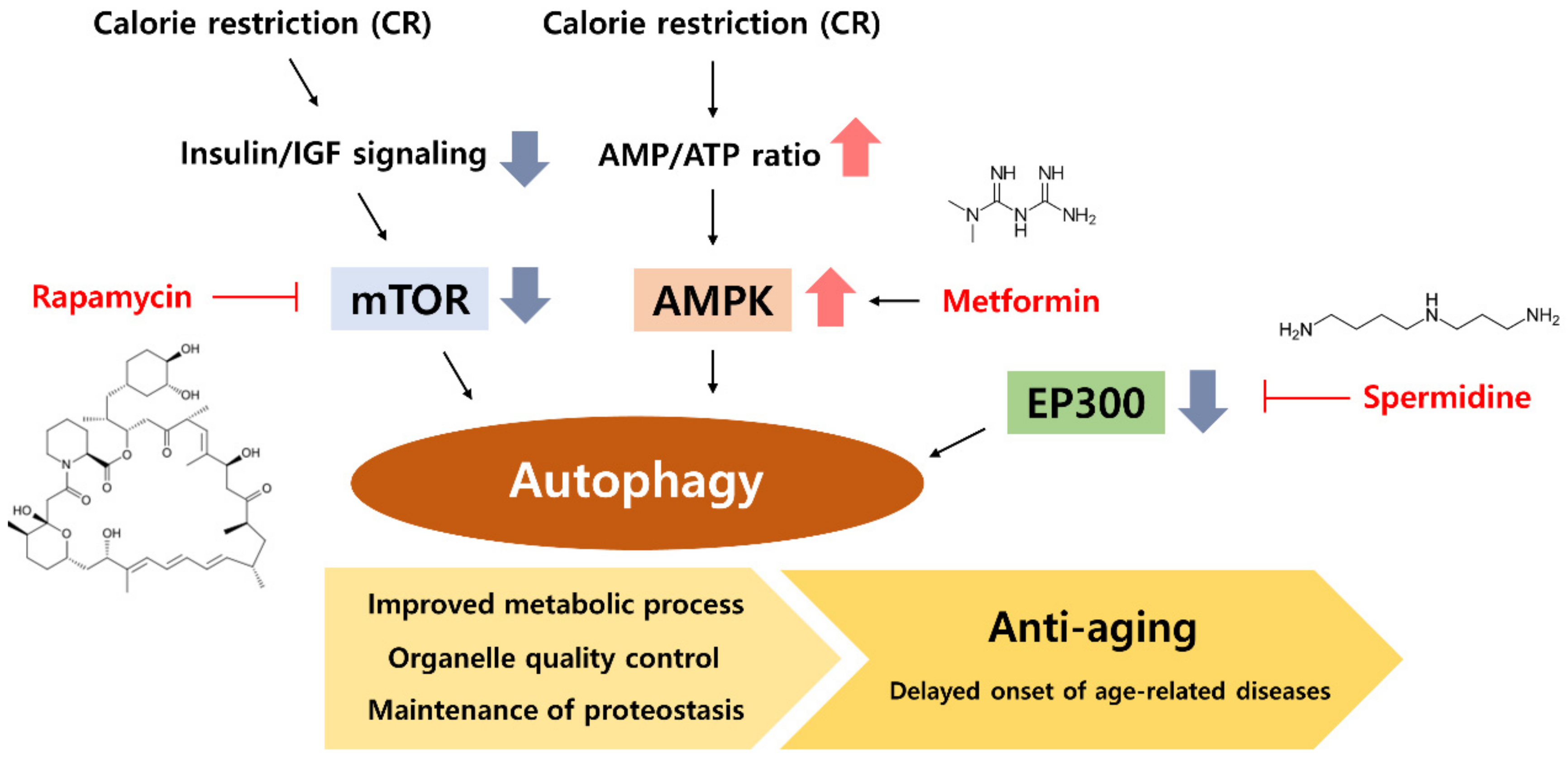
5. CR Mimetic as an Autophagy Inducer
5.1. Rapamycin, an mTOR Inhibitor
5.2. Metformin, an AMPK Activator
5.3. Spermidine
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Zhang, J.; Zhao, J.; Ma, N.; Kim, S.W.; Qiao, S.; Ma, X. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv. Nutr. 2018, 9, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin. Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Bond, P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr. 2016, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Yao, Y.; Jones, E.; Inoki, K. Lysosomal Regulation of mTORC1 by Amino Acids in Mammalian Cells. Biomolecules 2017, 7, 51. [Google Scholar] [CrossRef]
- Puertollano, R. mTOR and lysosome regulation. F1000Prime Rep. 2014, 6, 52. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef] [PubMed]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Singh, R. Autophagy and aging. Adv. Exp. Med. Biol. 2015, 847, 73–87. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef]
- Sarkis, G.J.; Ashcom, J.D.; Hawdon, J.M.; Jacobson, L.A. Decline in protease activities with age in the nematode Caenorhabditis elegans. Mech. Ageing Dev. 1988, 45, 191–201. [Google Scholar] [CrossRef]
- Bai, H.; Kang, P.; Hernandez, A.M.; Tatar, M. Activin signaling targeted by insulin/dFOXO regulates aging and muscle proteostasis in Drosophila. PLoS Genet. 2013, 9, e1003941. [Google Scholar] [CrossRef]
- Kaushik, S.; Arias, E.; Kwon, H.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; Pessin, J.E.; Singh, R. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012, 13, 258–265. [Google Scholar] [CrossRef]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Dice, J.F. Age-related decline in chaperone-mediated autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T.; Kumsta, C.; Hellman, A.B.; Adams, L.M.; Hansen, M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Jiao, K.; Liu, X.; Sun, Q.; Wang, K.; Xu, H.; Zhang, S.; Wu, Y.; Wu, L.; Liu, D.; et al. Omi/HtrA2 Participates in Age-Related Autophagic Deficiency in Rat Liver. Aging Dis. 2018, 9, 1031–1042. [Google Scholar] [CrossRef]
- Matecic, M.; Smith, D.L.; Pan, X.; Maqani, N.; Bekiranov, S.; Boeke, J.D.; Smith, J.S. A microarray-based genetic screen for yeast chronological aging factors. PLoS Genet. 2010, 6, e1000921. [Google Scholar] [CrossRef]
- Toth, M.L.; Sigmond, T.; Borsos, E.; Barna, J.; Erdelyi, P.; Takacs-Vellai, K.; Orosz, L.; Kovacs, A.L.; Csikos, G.; Sass, M.; et al. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Stroikin, Y.; Dalen, H.; Loof, S.; Terman, A. Inhibition of autophagy with 3-methyladenine results in impaired turnover of lysosomes and accumulation of lipofuscin-like material. Eur. J. Cell Biol. 2004, 83, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013, 4, 2300. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef]
- Seok, S.; Fu, T.; Choi, S.E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARα and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Sung, B.; Park, S.; Yu, B.P.; Chung, H.Y. Modulation of PPAR in aging, inflammation, and calorie restriction. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Morris, K.A.; Gold, P.E. Age-related impairments in memory and in CREB and pCREB expression in hippocampus and amygdala following inhibitory avoidance training. Mech. Ageing Dev. 2012, 133, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Kim, D.H.; Park, M.H.; Choi, Y.J.; Kim, N.D.; Lee, J.; Yu, B.P.; Chung, H.Y. Recent advances in calorie restriction research on aging. Exp. Gerontol. 2013, 48, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective with Translational Potential. EBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Libert, S.; Guarente, L. Metabolic and neuropsychiatric effects of calorie restriction and sirtuins. Annu. Rev. Physiol. 2013, 75, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Redman, L.M.; Smith, S.R.; Burton, J.H.; Martin, C.K.; Il’yasova, D.; Ravussin, E. Metabolic Slowing and Reduced Oxidative Damage with Sustained Caloric Restriction Support the Rate of Living and Oxidative Damage Theories of Aging. Cell Metab. 2018, 27, 805–815.e4. [Google Scholar] [CrossRef] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef]
- Kume, S.; Uzu, T.; Horiike, K.; Chin-Kanasaki, M.; Isshiki, K.; Araki, S.; Sugimoto, T.; Haneda, M.; Kashiwagi, A.; Koya, D. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J. Clin. Investig. 2010, 120, 1043–1055. [Google Scholar] [CrossRef]
- Ruckenstuhl, C.; Netzberger, C.; Entfellner, I.; Carmona-Gutierrez, D.; Kickenweiz, T.; Stekovic, S.; Gleixner, C.; Schmid, C.; Klug, L.; Sorgo, A.G.; et al. Lifespan extension by methionine restriction requires autophagy-dependent vacuolar acidification. PLoS Genet. 2014, 10, e1004347. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef]
- Madeo, F.; Zimmermann, A.; Maiuri, M.C.; Kroemer, G. Essential role for autophagy in life span extension. J. Clin. Investig. 2015, 125, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, S.E.; Julian, D.; Akin, D.E.; Fried, J.; Toscano, K.; Leeuwenburgh, C.; Dunn, W.A., Jr. Autophagy in the heart and liver during normal aging and calorie restriction. Rejuvenation Res. 2007, 10, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Donati, A.; Cavallini, G.; Bergamini, E. Effects of aging, antiaging calorie restriction and in vivo stimulation of autophagy on the urinary excretion of 8OHdG in male Sprague-Dawley rats. Age 2013, 35, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Luevano-Martinez, L.A.; Forni, M.F.; Peloggia, J.; Watanabe, I.S.; Kowaltowski, A.J. Calorie restriction promotes cardiolipin biosynthesis and distribution between mitochondrial membranes. Mech. Ageing Dev. 2017, 162, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Derous, D.; Mitchell, S.E.; Wang, L.; Green, C.L.; Wang, Y.; Chen, L.; Han, J.J.; Promislow, D.E.L.; Lusseau, D.; Douglas, A.; et al. The effects of graded levels of calorie restriction: XI. Evaluation of the main hypotheses underpinning the life extension effects of CR using the hepatic transcriptome. Aging 2017, 9, 1770–1824. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, S.E.; Seo, A.Y.; Marzetti, E.; Lees, H.A.; Leeuwenburgh, C. Skeletal muscle autophagy and apoptosis during aging: Effects of calorie restriction and life-long exercise. Exp. Gerontol. 2010, 45, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Licastro, D.; Cava, E.; Veronese, N.; Spelta, F.; Rizza, W.; Bertozzi, B.; Villareal, D.T.; Hotamisligil, G.S.; Holloszy, J.O.; et al. Long-Term Calorie Restriction Enhances Cellular Quality-Control Processes in Human Skeletal Muscle. Cell Rep. 2016, 14, 422–428. [Google Scholar] [CrossRef]
- Gutierrez-Casado, E.; Khraiwesh, H.; Lopez-Dominguez, J.A.; Montero-Guisado, J.; Lopez-Lluch, G.; Navas, P.; de Cabo, R.; Ramsey, J.J.; Gonzalez-Reyes, J.A.; Villalba, J.M. The Impact of Aging, Calorie Restriction and Dietary Fat on Autophagy Markers and Mitochondrial Ultrastructure and Dynamics in Mouse Skeletal Muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 760–769. [Google Scholar] [CrossRef]
- Nunez, C.E.; Rodrigues, V.S.; Gomes, F.S.; Moura, R.F.; Victorio, S.C.; Bombassaro, B.; Chaim, E.A.; Pareja, J.C.; Geloneze, B.; Velloso, L.A.; et al. Defective regulation of adipose tissue autophagy in obesity. Int. J. Obes. 2013, 37, 1473–1480. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Mau, T.; O’Brien, M.; Garg, S.; Yung, R. Impaired autophagy activity is linked to elevated ER-stress and inflammation in aging adipose tissue. Aging 2016, 8, 2525–2537. [Google Scholar] [CrossRef]
- Ning, Y.C.; Cai, G.Y.; Zhuo, L.; Gao, J.J.; Dong, D.; Cui, S.; Feng, Z.; Shi, S.Z.; Bai, X.Y.; Sun, X.F.; et al. Short-term calorie restriction protects against renal senescence of aged rats by increasing autophagic activity and reducing oxidative damage. Mech. Ageing Dev. 2013, 134, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.W.; Kim, K.M.; Choi, Y.J.; An, H.J.; Lee, B.; Kim, D.H.; Lee, E.K.; Im, E.; Lee, J.; Im, D.S.; et al. The critical role played by endotoxin-induced liver autophagy in the maintenance of lipid metabolism during sepsis. Autophagy 2017, 13, 1113–1129. [Google Scholar] [CrossRef] [PubMed]
- Zirin, J.; Nieuwenhuis, J.; Perrimon, N. Role of autophagy in glycogen breakdown and its relevance to chloroquine myopathy. PLoS Biol. 2013, 11, e1001708. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Tarabra, E.; Toledo, M.; Garcia-Macia, M.; Sahu, S.; Coletto, L.; Batista-Gonzalez, A.; Barzilai, N.; Pessin, J.E.; Schwartz, G.J.; et al. System-wide Benefits of Intermeal Fasting by Autophagy. Cell Metab. 2017, 26, 856–871. [Google Scholar] [CrossRef]
- Chen, L.; Wang, K.; Long, A.; Jia, L.; Zhang, Y.; Deng, H.; Li, Y.; Han, J.; Wang, Y. Fasting-induced hormonal regulation of lysosomal function. Cell Res. 2017, 27, 748–763. [Google Scholar] [CrossRef]
- Neel, B.A.; Lin, Y.; Pessin, J.E. Skeletal muscle autophagy: A new metabolic regulator. Trends Endocrinol. Metab. 2013, 24, 635–643. [Google Scholar] [CrossRef]
- Meijer, A.J.; Lorin, S.; Blommaart, E.F.; Codogno, P. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015, 47, 2037–2063. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Demontis, F. Skeletal muscle autophagy and its role in sarcopenia and organismal aging. Curr. Opin. Pharmacol. 2017, 34, 1–6. [Google Scholar] [CrossRef]
- Carter, H.N.; Kim, Y.; Erlich, A.T.; Zarrin-Khat, D.; Hood, D.A. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J. Physiol. 2018, 596, 3567–3584. [Google Scholar] [CrossRef] [PubMed]
- Di Rienzo, M.; Antonioli, M.; Fusco, C.; Liu, Y.; Mari, M.; Orhon, I.; Refolo, G.; Germani, F.; Corazzari, M.; Romagnoli, A.; et al. Autophagy induction in atrophic muscle cells requires ULK1 activation by TRIM32 through unanchored K63-linked polyubiquitin chains. Sci. Adv. 2019, 5, eaau8857. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.E.; Bareja, A.; Bartlett, D.B.; White, J.P. Autophagy as a Therapeutic Target to Enhance Aged Muscle Regeneration. Cells 2019, 8, 183. [Google Scholar] [CrossRef]
- Cerletti, M.; Jang, Y.C.; Finley, L.W.; Haigis, M.C.; Wagers, A.J. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell 2012, 10, 515–519. [Google Scholar] [CrossRef]
- Miller, K.N.; Burhans, M.S.; Clark, J.P.; Howell, P.R.; Polewski, M.A.; DeMuth, T.M.; Eliceiri, K.W.; Lindstrom, M.J.; Ntambi, J.M.; Anderson, R.M. Aging and caloric restriction impact adipose tissue, adiponectin, and circulating lipids. Aging Cell 2017, 16, 497–507. [Google Scholar] [CrossRef]
- Ferhat, M.; Funai, K.; Boudina, S. Autophagy in Adipose Tissue Physiology and Pathophysiology. Antioxid. Redox Signal. 2019, 31, 487–501. [Google Scholar] [CrossRef]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef]
- Cai, J.; Pires, K.M.; Ferhat, M.; Chaurasia, B.; Buffolo, M.A.; Smalling, R.; Sargsyan, A.; Atkinson, D.L.; Summers, S.A.; Graham, T.E.; et al. Autophagy Ablation in Adipocytes Induces Insulin Resistance and Reveals Roles for Lipid Peroxide and Nrf2 Signaling in Adipose-Liver Crosstalk. Cell Rep. 2018, 25, 1708–1717.e5. [Google Scholar] [CrossRef]
- Haim, Y.; Bluher, M.; Slutsky, N.; Goldstein, N.; Kloting, N.; Harman-Boehm, I.; Kirshtein, B.; Ginsberg, D.; Gericke, M.; Guiu Jurado, E.; et al. Elevated autophagy gene expression in adipose tissue of obese humans: A potential non-cell-cycle-dependent function of E2F1. Autophagy 2015, 11, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.J.; van Essen, P.; Koenen, T.; Joosten, L.A.; Netea, M.G.; Tack, C.J.; Stienstra, R. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012, 153, 5866–5874. [Google Scholar] [CrossRef] [PubMed]
- Soussi, H.; Reggio, S.; Alili, R.; Prado, C.; Mutel, S.; Pini, M.; Rouault, C.; Clement, K.; Dugail, I. DAPK2 Downregulation Associates With Attenuated Adipocyte Autophagic Clearance in Human Obesity. Diabetes 2015, 64, 3452–3463. [Google Scholar] [CrossRef] [PubMed]
- Bobulescu, I.A. Renal lipid metabolism and lipotoxicity. Curr. Opin. Nephrol. Hypertens. 2010, 19, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Stumvoll, M.; Meyer, C.; Mitrakou, A.; Gerich, J.E. Important role of the kidney in human carbohydrate metabolism. Med. Hypotheses 1999, 52, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Gerich, J.E. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabet. Med. 2010, 27, 136–142. [Google Scholar] [CrossRef]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Quiroga, A.; et al. Emerging role of autophagy in kidney function, diseases and aging. Autophagy 2012, 8, 1009–1031. [Google Scholar] [CrossRef]
- Deng, X.; Xie, Y.; Zhang, A. Advance of autophagy in chronic kidney diseases. Ren. Fail. 2017, 39, 306–313. [Google Scholar] [CrossRef]
- Xu, X.M.; Cai, G.Y.; Bu, R.; Wang, W.J.; Bai, X.Y.; Sun, X.F.; Chen, X.M. Beneficial Effects of Caloric Restriction on Chronic Kidney Disease in Rodent Models: A Meta-Analysis and Systematic Review. PLoS ONE 2015, 10, e0144442. [Google Scholar] [CrossRef]
- Stekovic, S.; Hofer, S.J.; Tripolt, N.; Aon, M.A.; Royer, P.; Pein, L.; Stadler, J.T.; Pendl, T.; Prietl, B.; Url, J.; et al. Alternate Day Fasting Improves Physiological and Molecular Markers of Aging in Healthy, Non-obese Humans. Cell Metab. 2019, 30, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Shintani, H.; Shintani, T.; Ashida, H.; Sato, M. Calorie Restriction Mimetics: Upstream-Type Compounds for Modulating Glucose Metabolism. Nutrients 2018, 10, 1821. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Escobar, K.A.; Cole, N.H.; Mermier, C.M.; VanDusseldorp, T.A. Autophagy and aging: Maintaining the proteome through exercise and caloric restriction. Aging Cell 2019, 18, e12876. [Google Scholar] [CrossRef]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef]
- Ehninger, D.; Neff, F.; Xie, K. Longevity, aging and rapamycin. Cell. Mol. Life Sci. 2014, 71, 4325–4346. [Google Scholar] [CrossRef]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. eLife 2016, 5, e16351. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Pallet, N.; Legendre, C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2013, 12, 177–186. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 294. [Google Scholar] [CrossRef] [PubMed]
- Valencia, W.M.; Palacio, A.; Tamariz, L.; Florez, H. Metformin and ageing: Improving ageing outcomes beyond glycaemic control. Diabetologia 2017, 60, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M.; Mote, P.L.; Fahy, G.M.; Spindler, S.R. Identification of potential caloric restriction mimetics by microarray profiling. Physiol. Genom. 2005, 23, 343–350. [Google Scholar] [CrossRef]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cocheme, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef]
- Shi, W.Y.; Xiao, D.; Wang, L.; Dong, L.H.; Yan, Z.X.; Shen, Z.X.; Chen, S.J.; Chen, Y.; Zhao, W.L. Therapeutic metformin/AMPK activation blocked lymphoma cell growth via inhibition of mTOR pathway and induction of autophagy. Cell Death Dis. 2012, 3, e275. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Wang, G.Y.; Bi, Y.G.; Liu, X.D.; Zhao, Y.; Han, J.F.; Wei, M.; Zhang, Q.Y. Autophagy was involved in the protective effect of metformin on hyperglycemia-induced cardiomyocyte apoptosis and Connexin43 downregulation in H9c2 cells. Int. J. Med. Sci. 2017, 14, 698–704. [Google Scholar] [CrossRef]
- Madeo, F.; Bauer, M.A.; Carmona-Gutierrez, D.; Kroemer, G. Spermidine: A physiological autophagy inducer acting as an anti-aging vitamin in humans? Autophagy 2019, 15, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Yue, F.; Li, W.; Zou, J.; Jiang, X.; Xu, G.; Huang, H.; Liu, L. Spermidine Prolongs Lifespan and Prevents Liver Fibrosis and Hepatocellular Carcinoma by Activating MAP1S-Mediated Autophagy. Cancer Res. 2017, 77, 2938–2951. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Minois, N.; Carmona-Gutierrez, D.; Bauer, M.A.; Rockenfeller, P.; Eisenberg, T.; Brandhorst, S.; Sigrist, S.J.; Kroemer, G.; Madeo, F. Spermidine promotes stress resistance in Drosophila melanogaster through autophagy-dependent and -independent pathways. Cell Death Dis. 2012, 3, e401. [Google Scholar] [CrossRef]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Criollo, A.; Maiuri, M.C.; Tavernarakis, N.; Madeo, F.; Kroemer, G. Autophagy mediates pharmacological lifespan extension by spermidine and resveratrol. Aging 2009, 1, 961–970. [Google Scholar] [CrossRef]
- Pietrocola, F.; Lachkar, S.; Enot, D.P.; Niso-Santano, M.; Bravo-San Pedro, J.M.; Sica, V.; Izzo, V.; Maiuri, M.C.; Madeo, F.; Marino, G.; et al. Spermidine induces autophagy by inhibiting the acetyltransferase EP300. Cell Death Differ. 2015, 22, 509–516. [Google Scholar] [CrossRef]
- Minois, N.; Carmona-Gutierrez, D.; Madeo, F. Polyamines in aging and disease. Aging 2011, 3, 716–732. [Google Scholar] [CrossRef]
- Kiechl, S.; Pechlaner, R.; Willeit, P.; Notdurfter, M.; Paulweber, B.; Willeit, K.; Werner, P.; Ruckenstuhl, C.; Iglseder, B.; Weger, S.; et al. Higher spermidine intake is linked to lower mortality: A prospective population-based study. Am. J. Clin. Nutr. 2018, 108, 371–380. [Google Scholar] [CrossRef]
- Pietrocola, F.; Castoldi, F.; Markaki, M.; Lachkar, S.; Chen, G.; Enot, D.P.; Durand, S.; Bossut, N.; Tong, M.; Malik, S.A.; et al. Aspirin Recapitulates Features of Caloric Restriction. Cell Rep. 2018, 22, 2395–2407. [Google Scholar] [CrossRef]
- Pietrocola, F.; Castoldi, F.; Maiuri, M.C.; Kroemer, G. Aspirin-another caloric-restriction mimetic. Autophagy 2018, 14, 1162–1163. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Liver Autophagy | |||
|---|---|---|---|
| Species | CR Methods | Main Results | Ref |
| Fisher Rats | 40% calorie restriction (Life-long) | CR had no substantial effect on the expression of autophagic proteins | [62] |
| SD Rats | Alternative day fasting (10 months) | Alternative day fasting increased autophagy at high levels | [63] |
| Rat | 40% calorie restriction (4 months) | CR increased autophagy flux (LC3-II/LC3-I ratio) especially in the mitochondrial membrane | [64] |
| Mice | 0%–40% calorie restriction (4 months) | A significant increase in autophagy was detected | [65] |
| Muscle Autophagy | |||
| Fisher Rats | 8% calorie restriction (Life-long) | Mild CR attenuated the impairment of autophagy in rodent muscle during aging | [66] |
| Human | Up to 30% calorie restriction (3–15 years) | Autophagy-related genes were significantly increased in response to CR | [67] |
| Mice | 40% calorie restriction (6–18 months) | Autophagy and mitochondrial integrity was significantly increased | [68] |
| Adipose Tissue Autophagy | |||
| Mice | 40% calorie restriction (15 days) | Autophagy was significantly induced in lean mice (but not in obese mice) | [69] |
| Mice | 40% calorie restriction (Life-long) | Autophagy activity was enhanced in CR mice compare to aged mice | [70] |
| Kidney Autophagy | |||
| Mice | 40% calorie restriction (12 month) | Autophagy flux and LC3 conversion were higher in CR mice | [58] |
| SD Rats | 40% calorie restriction (2 month) | Short-term CR increased LC3-II/LC3-I ratio and beclin-1 expression | [71] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chung, K.W.; Chung, H.Y. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923. https://doi.org/10.3390/nu11122923
Chung KW, Chung HY. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients. 2019; 11(12):2923. https://doi.org/10.3390/nu11122923
Chicago/Turabian StyleChung, Ki Wung, and Hae Young Chung. 2019. "The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention" Nutrients 11, no. 12: 2923. https://doi.org/10.3390/nu11122923
APA StyleChung, K. W., & Chung, H. Y. (2019). The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients, 11(12), 2923. https://doi.org/10.3390/nu11122923