Lotus Seedpod Extracts Reduced Lipid Accumulation and Lipotoxicity in Hepatocytes



Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Preparation of LSE
2.2. Cell Culture
2.3. Trypan Blue Dye Exclusion Assay
2.4. Oil Red O Staining
2.5. Nile Red Stain
2.6. Reactive Oxygen Species (ROS) Assay
2.7. Acridine Orange (AO) Staining
2.8. 4,6-Diamidino-2-Phenylindole (DAPI) Staining
2.9. Annexin V-Fluorescein Isothiocyanate (FITC) and 7-Amino-Actinomycin (7-AAD) Double Staining
2.10. Mitochondrial Membrane Depolarization Assay
2.11. Western Blot Analysis
2.12. Statistical Analysis
3. Results
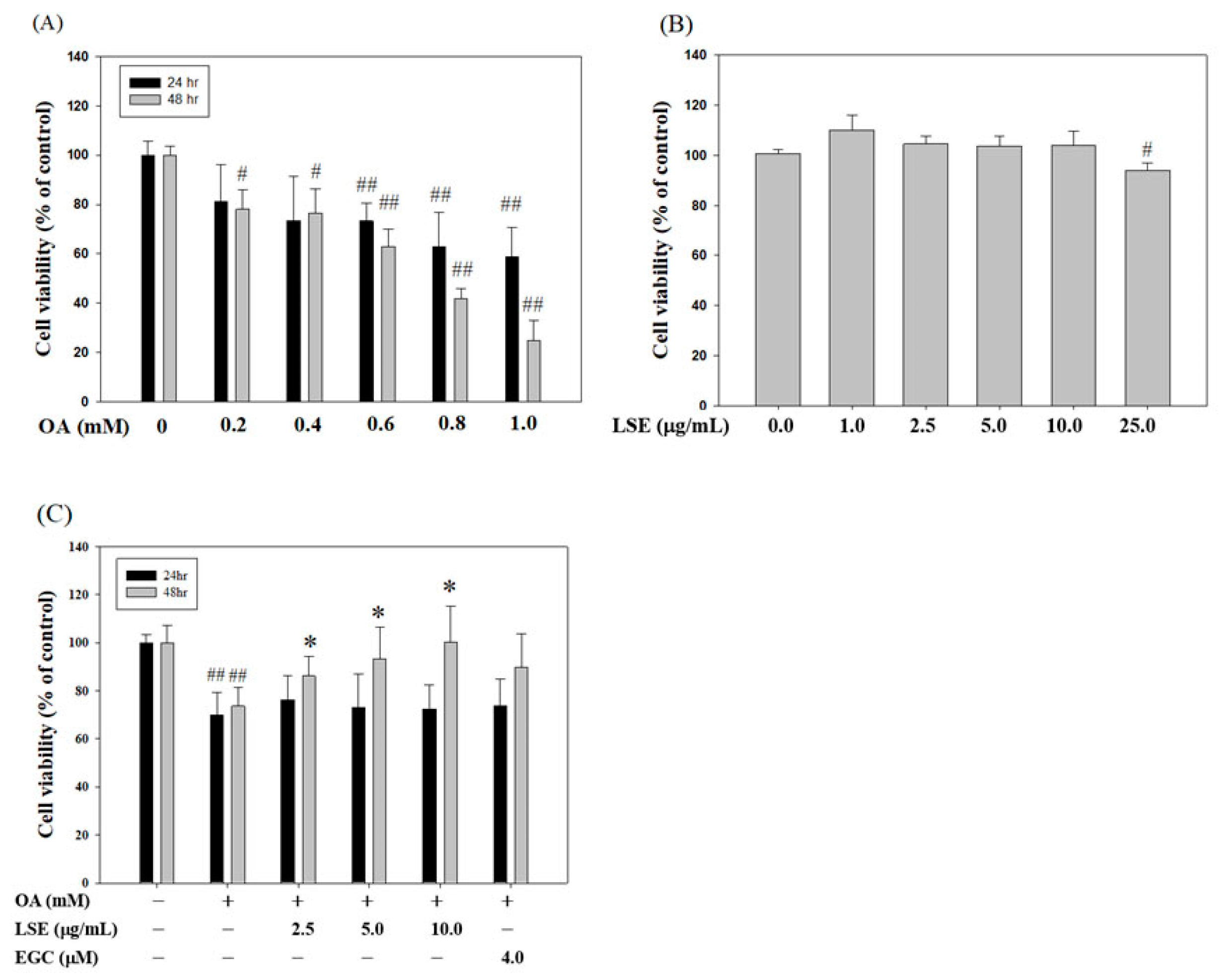
3.1. LSE Attenuated the Cytotoxic Effect of OA in HepG2 cell
3.2. Effects of LSE on the OA-Induced Intracellular Lipid Accumulation
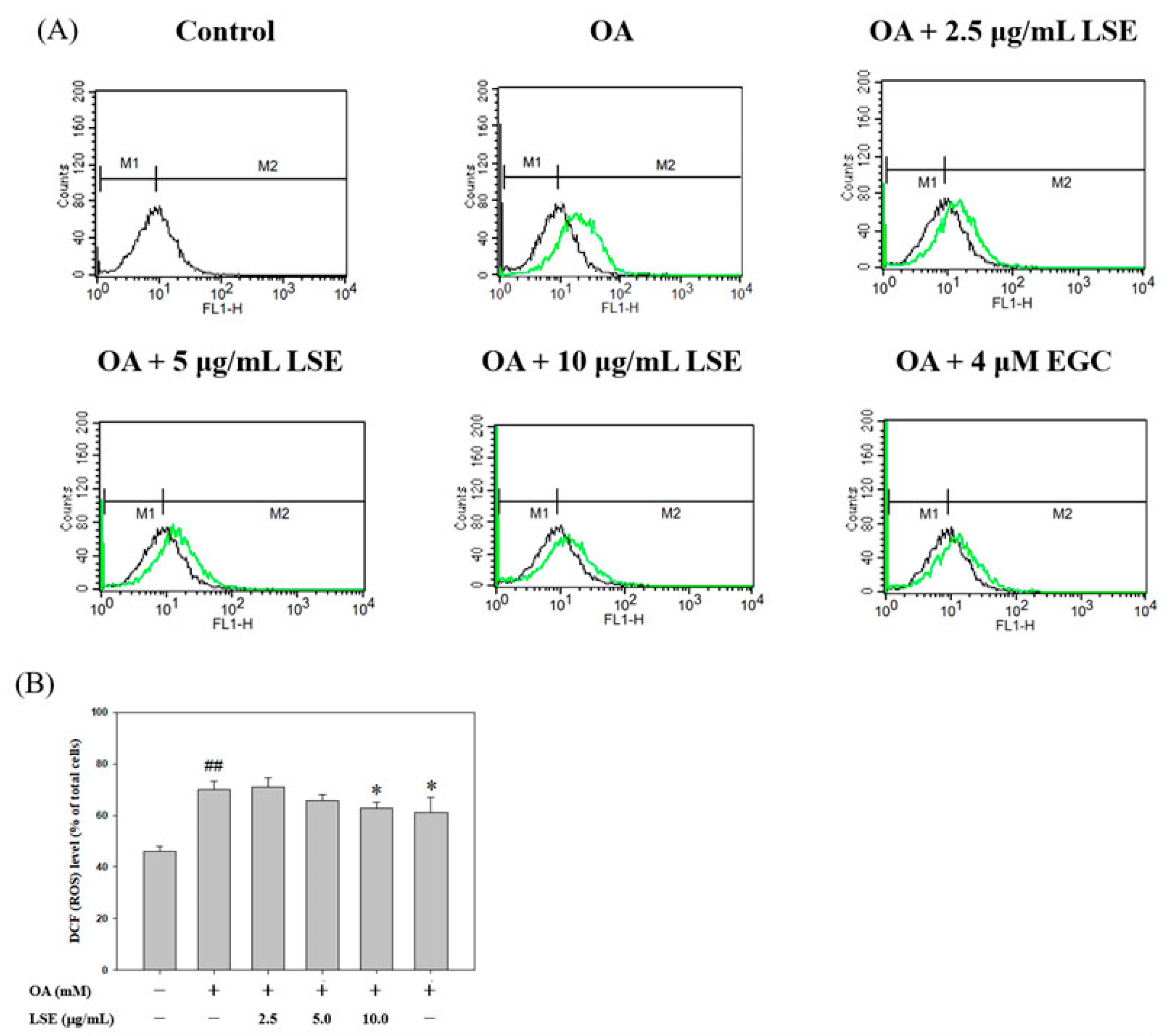
3.3. Effects of LSE on OA-Induced an Increase in ROS Content in HepG2 cells
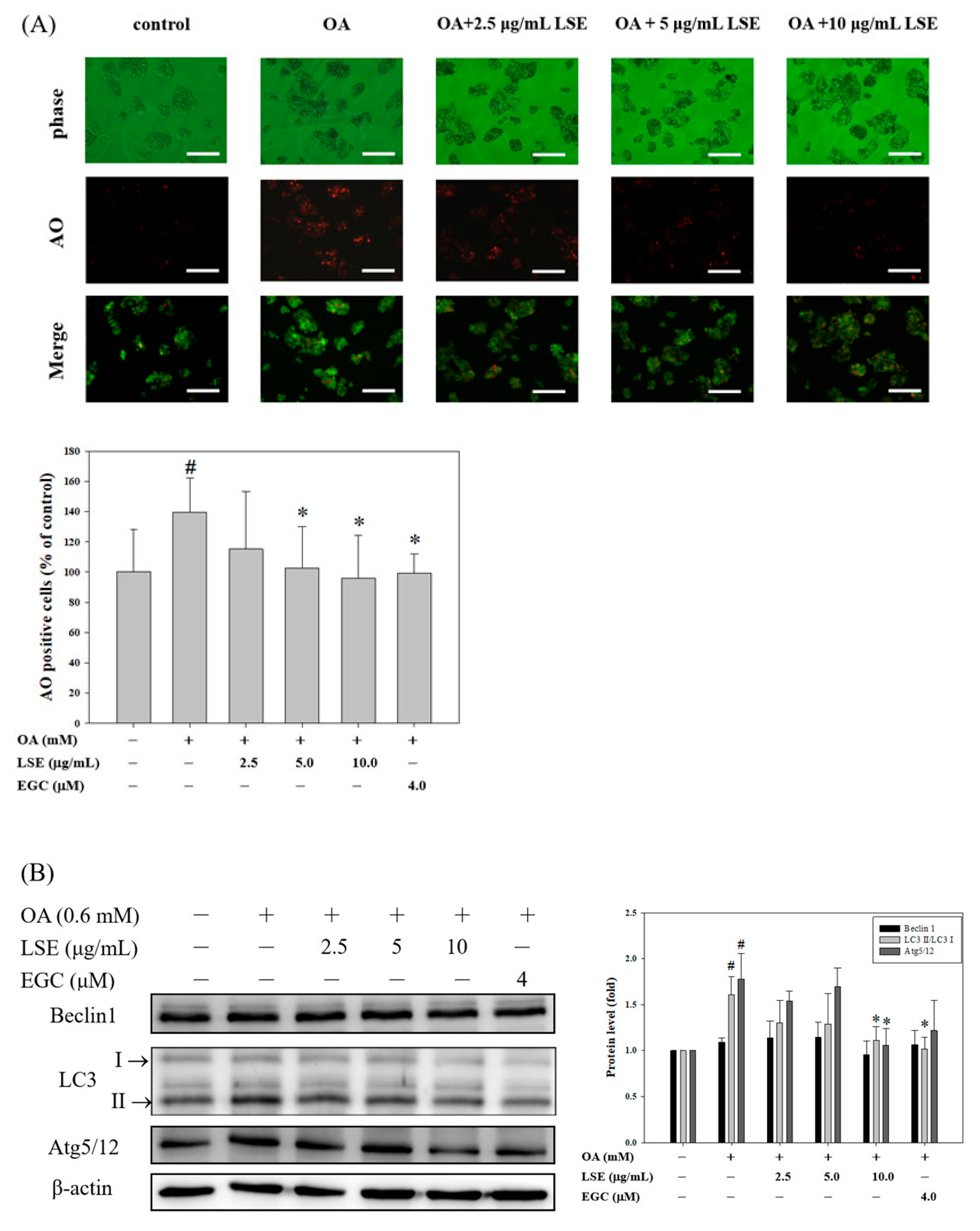
3.4. Effects of LSE on OA-Induced HepG2 Cell Autophagy
3.5. Effects of LSE on OA-Induced HepG2 Cell Apoptosis
4. Discussion
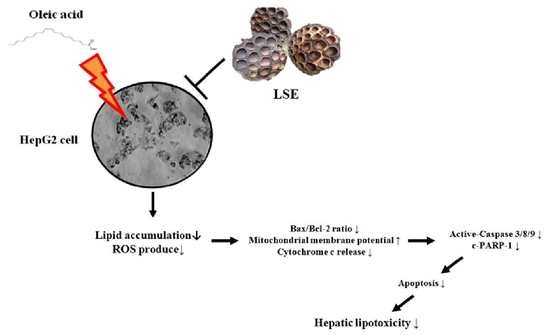
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jin, X.; Yang, Y.D.; Chen, K.; Lv, Z.Y.; Zheng, L.; Liu, Y.P.; Chen, S.H.; Yu, C.H.; Jiang, X.Y.; Zhang, C.Y.; et al. HDMCP uncouples yeast mitochondrial respiration and alleviates steatosis in L02 and hepG2 cells by decreasing ATP and H2O2 levels: A novel mechanism for NAFLD. J. Hepatol. 2009, 50, 1019–1028. [Google Scholar] [CrossRef]
- Zhou, Y.; Ding, Y.L.; Zhang, J.L.; Zhang, P.; Wang, J.Q.; Li, Z.H. Alpinetin improved high fat diet-induced non-alcoholic fatty liver disease (NAFLD) through improving oxidative stress, inflammatory response and lipid metabolism. Biomed. Pharm. 2018, 97, 1397–1408. [Google Scholar] [CrossRef]
- Marchisello, S.; Di Pino, A.; Scicali, R.; Urbano, F.; Piro, S.; Purrello, F.; Rabuazzo, A.M. Pathophysiological, Molecular and Therapeutic Issues of Nonalcoholic Fatty Liver Disease: An Overview. Int. J. Mol. Sci. 2019, 20, 1948. [Google Scholar] [CrossRef] [PubMed]
- Calanna, S.; Scicali, R.; Di Pino, A.; Knop, F.K.; Piro, S.; Rabuazzo, A.M.; Purrello, F. Lipid and liver abnormalities in haemoglobin A1c-defined prediabetes and type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Chen, X.; Cheng, S.; Shu, L.; Yan, M.; Yao, L.; Wang, B.; Huang, S.; Zhou, L.; Yang, Z.; et al. FTO promotes SREBP1c maturation and enhances CIDEC transcription during lipid accumulation in HepG2 cells. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 538–548. [Google Scholar] [CrossRef]
- Xie, C.; Chen, Z.; Zhang, C.; Xu, X.; Jin, J.; Zhan, W.; Han, T.; Wang, J. Dihydromyricetin ameliorates oleic acid-induced lipid accumulation in L02 and HepG2 cells by inhibiting lipogenesis and oxidative stress. Life Sci. 2016, 157, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Jin, X.L.; Wang, K.; Li, Q.Q.; Tian, W.L.; Xue, X.F.; Wu, L.M.; Hu, F.L. Antioxidant and anti-inflammatory effects of Chinese propolis during palmitic acid-induced lipotoxicity in cultured hepatocytes. J. Funct. Foods. 2017, 34, 216–223. [Google Scholar] [CrossRef]
- Aray, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n-6/n-3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef]
- Zhang, D.D.; Zhang, J.G.; Wu, X.; Liu, Y.; Gu, S.Y.; Zhu, G.H.; Wang, Y.Z.; Liu, G.L.; Li, X.Y. Nuciferine downregulates Per-Arnt-Sim kinase expression during its alleviation of lipogenesis and inflammation on oleic acid-induced hepatic steatosis in HepG2 cells. Front. Pharm. 2015, 6, 238. [Google Scholar] [CrossRef]
- Sica, V.; Galluzzi, L.; Bravo-San Pedro, J.M.; Izzo, V.; Maiuri, M.C.; Kroemer, G. Organelle-specific initiation of autophagy. Mol. Cell. 2015, 59, 522–539. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.A.; Tavallai, S.; Hamed, H.A.; Cruickshanks, N.; Dent, P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cell. Signal. 2014, 26, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Xingfeng, G.; Daijie, W.; Wenjuan, D.; Jinhua, D.; Xiao, W. Preparative isolation and purification of four flavonoids from the petals of Nelumbo nucifera by high-speed counter-current chromatography. Phytochem. Anal. 2010, 21, 268–272. [Google Scholar]
- Chaudhuri, P.K.; Singh, D. A new lipid and other constituents from the rhizomes of Nelumbo nucifera. J. Asian Nat. Prod. Res. 2009, 11, 583–587. [Google Scholar] [CrossRef]
- Lu, Y.; Ma, W.; Hu, R.; Dai, X.; Pan, Y. Ionic liquid-based microwave-assisted extraction of phenolic alkaloids from the medicinal plant Nelumbo nucifera Gaertn. J. Chromatogr. A 2008, 1208, 42–46. [Google Scholar] [CrossRef]
- Park, Y.S.; Towantakavanit, K.; Kowalska, T.; Jung, S.T.; Ham, K.S.; Heo, B.G.; Cho, J.Y.; Yun, J.G.; Kim, H.J.; Gorinstein, S. Bioactive compounds and antioxidant and antiproliferative activities of Korean white lotus cultivars. J. Med. Food 2009, 12, 1057–1064. [Google Scholar] [CrossRef]
- Lin, H.Y.; Kuo, Y.H.; Lin, Y.L.; Chiang, W. Antioxidative effect and active components from leaves of Lotus (Nelumbo nucifera). J. Agric. Food Chem. 2009, 57, 6623–6629. [Google Scholar] [CrossRef]
- Ling, Z.Q.; Xie, B.J.; Yang, E.L. Isolation, characterization, and determination of antioxidative activity of oligomeric procyanidins from the seedpod of Nelumbo nucifera Gaertn. J. Agric. Food Chem. 2005, 53, 2441–2445. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, H.; Xie, B.; Yan, Y.; Li, J.; Xu, F.; Qin, Y. Whole body radioprotective activity of an acetone-water extract from the seedpod of Nelumbo nucifera Gaertn. Seedpod. Food Chem. Toxicol. 2010, 48, 3374–3384. [Google Scholar] [CrossRef]
- Gong, Y.; Liu, L.; Xie, B.; Liao, Y.; Yang, E.; Sun, Z. Ameliorative effects of lotus seedpod proanthocyanidins on cognitive de fi cits and oxidative damage in senescence-accelerated mice. Behav. Brain Res. 2008, 194, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Rong, S.; Xie, B.; Sun, Z.; Zhang, L.; Wu, H.; Yao, P.; Zhang, Y.; Liu, L. Procyanidins extracted from the lotus seedpod ameliorate scopolamine-induced memory impairment in mice. Phytother. Res. 2009, 23, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, H.; Lv, Z.; Li, S.; Hu, B.; Guan, Y.; Xie, B.; Sun, Z. Oligomeric procyanidins of lotus seedpod inhibits the formation of advanced glycation end-products by scavenging reactive carbonyls. Food Chem. 2013, 138, 1493–1502. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sui, Y.; Wu, Q.; Xie, B.; Sun, Z. Attenuated mTOR signaling and enhanced glucose homeostasis by dietary supplementation with lotus seedpod oligomeric procyanidins in streptozotocin (STZ)-induced diabetic mice. J. Agric. Food Chem. 2017, 65, 3801–3810. [Google Scholar] [CrossRef]
- Tseng, H.C.; Tsai, P.M.; Chou, Y.H.; Lee, Y.C.; Lin, H.H.; Chen, J.H. In Vitro and In Vivo Protective Effects of Flavonoid-Enriched Lotus Seedpod Extract on Lipopolysaccharide-Induced Hepatic Inflammation. Am. J. Chin. Med. 2019, 47, 153–176. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.E. A human hepatocellular in vitro model to investigate steatosis. Chem. Biol. Interact. 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Jin, Y.; Tan, Y.; Chen, L.; Liu, Y.; Ren, Z. Reactive Oxygen Species Induces Lipid Droplet Accumulation in HepG2 Cells by Increasing Perilipin 2 Expression. Int. J. Mol. Sci. 2018, 19, 3445. [Google Scholar] [CrossRef]
- Yan, C.; Sun, W.; Wang, X.; Long, J.; Liu, X.; Feng, Z.; Liu, J. Punicalagin attenuates palmitate-induced lipotoxicity in HepG2 cells by activating the Keap1-Nrf2 antioxidant defense system. Mol. Nutr. Food Res. 2016, 60, 1139–1149. [Google Scholar] [CrossRef]
- Lee, M.S.; Tsai, C.W.; Wang, C.P.; Chen, J.H.; Lin, H.H. Anti-prostate cancer potential of gossypetin via inducing apoptotic and autophagic cell death. Mol. Carcinog. 2017, 56, 2578–2592. [Google Scholar] [CrossRef]
- Chen, C.T.; Wang, Z.H.; Hsu, C.C.; Lin, H.H.; Chen, J.H. In Vivo Protective Effects of Diosgenin against Doxorubicin-Induced Cardiotoxicity. Nutrients 2015, 7, 4938–4954. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, A.Y.; Chang, S.H.; Yu, K.N.; Kim, J.H.; Cho, M.H. Role of p53 in the cellular response following oleic acid accumulation in Chang liver cells. Toxicol. Lett. 2014, 224, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Lipotoxic diseases. Annu. Rev. Med. 2002, 53, 319–336. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Sedlak, T.W.; Oltvai, Z.N.; Yang, E.; Wang, K.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA 1995, 92, 7834–7838. [Google Scholar] [CrossRef]
- Moravcová, A.; Červinková, Z.; Kučera, O.; Mezera, V.; Rychtrmoc, D.; Lotková, H. The effect of oleic and palmitic acid on induction of steatosis and cytotoxicity on rat hepatocytes in primary culture. Physiol. Res. 2015, 64, 627–636. [Google Scholar]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef]
- Pan, Z.G.; An, X.S. SARM1 deletion restrains NAFLD induced by high fat diet (HFD) through reducing inflammation, oxidative stress and lipid accumulation. Biochem. Biophys. Res. Commun. 2018, 498, 416–423. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, L.; Wang, D.; Huo, Y.; Ji, B. Anthocyanin-rich extracts from blackberry, wild blueberry, strawberry, and chokeberry: Antioxidant activity and inhibitory effect on oleic acid-induced hepatic steatosis in vitro. J. Sci. Food Agric. 2016, 96, 2494–2503. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH:causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Li, J.; Cordero, P.; Nguyen, V.; Oben, J.A. The role of vitamins in the pathogenesis of non-alcoholic fatty liver disease. Integr. Med. Insights 2016, 11, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Ota, T. Micronutrient antioxidants and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2016, 17, 1379. [Google Scholar] [CrossRef] [PubMed]
- Bae, M.; Park, Y.K.; Lee, J.Y. Food components with anti-fibrotic activity and implications in prevention of liver disease. J. Nutr. Biochem. 2017, 55, 11–12. [Google Scholar]
- Abenavoli, L.; Milic, N.; Luzza, F.; Boccuto, L.; Lorenzo, A.D. Polyphenols treatment in patients with nonalcoholic fatty liver disease. J. Transl. Int. Med. 2017, 5, 144–147. [Google Scholar] [CrossRef]
- Yang, J.P.; Shin, J.H.; Seo, S.H.; Kim, S.G.; Lee, S.H.; Shin, E.H. Effects of Antioxidants in Reducing Accumulation of Fat in Hepatocyte. Int. J. Mol. Sci. 2018, 19, 2563. [Google Scholar] [CrossRef]
- Khambu, B.; Yan, S.; Huda, N.; Liu, G.; Yin, X.M. Autophagy in non-alcoholic fatty liver disease and alcoholic liver disease. Liver Res. 2018, 2, 112–119. [Google Scholar] [CrossRef]
- Rautou, P.E.; Mansouri, A.; Lebrec, D.; Durand, F.; Valla, D.; Moreau, R. Autophagy in liver diseases. J. Hepatol. 2010, 53, 1123–1134. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef]
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Eccleston, H.B.; Bailey, S.M. Mitochondrial dysfunction and oxidative stress in the pathogenesis of alcohol- and obesity-induced fatty liver diseases. Free Radical. Biol. Med. 2008, 44, 1259–1272. [Google Scholar] [CrossRef]
- Xiong, S.; Mu, T.; Wang, G.; Jiang, X. Mitochondria-mediated apoptosis in mammals. Protein Cell 2014, 5, 737–749. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.-T.; Lai, Y.-H.; Lin, H.-H.; Chen, J.-H. Lotus Seedpod Extracts Reduced Lipid Accumulation and Lipotoxicity in Hepatocytes. Nutrients 2019, 11, 2895. https://doi.org/10.3390/nu11122895
Liu Y-T, Lai Y-H, Lin H-H, Chen J-H. Lotus Seedpod Extracts Reduced Lipid Accumulation and Lipotoxicity in Hepatocytes. Nutrients. 2019; 11(12):2895. https://doi.org/10.3390/nu11122895
Chicago/Turabian StyleLiu, Yen-Tze, Yen-Hsun Lai, Hui-Hsuan Lin, and Jing-Hsien Chen. 2019. "Lotus Seedpod Extracts Reduced Lipid Accumulation and Lipotoxicity in Hepatocytes" Nutrients 11, no. 12: 2895. https://doi.org/10.3390/nu11122895
APA StyleLiu, Y.-T., Lai, Y.-H., Lin, H.-H., & Chen, J.-H. (2019). Lotus Seedpod Extracts Reduced Lipid Accumulation and Lipotoxicity in Hepatocytes. Nutrients, 11(12), 2895. https://doi.org/10.3390/nu11122895