Effects of Nutrient Intake during Pregnancy and Lactation on the Endocrine Pancreas of the Offspring

{kind=link}
Abstract
:1. Introduction
2. Pancreas
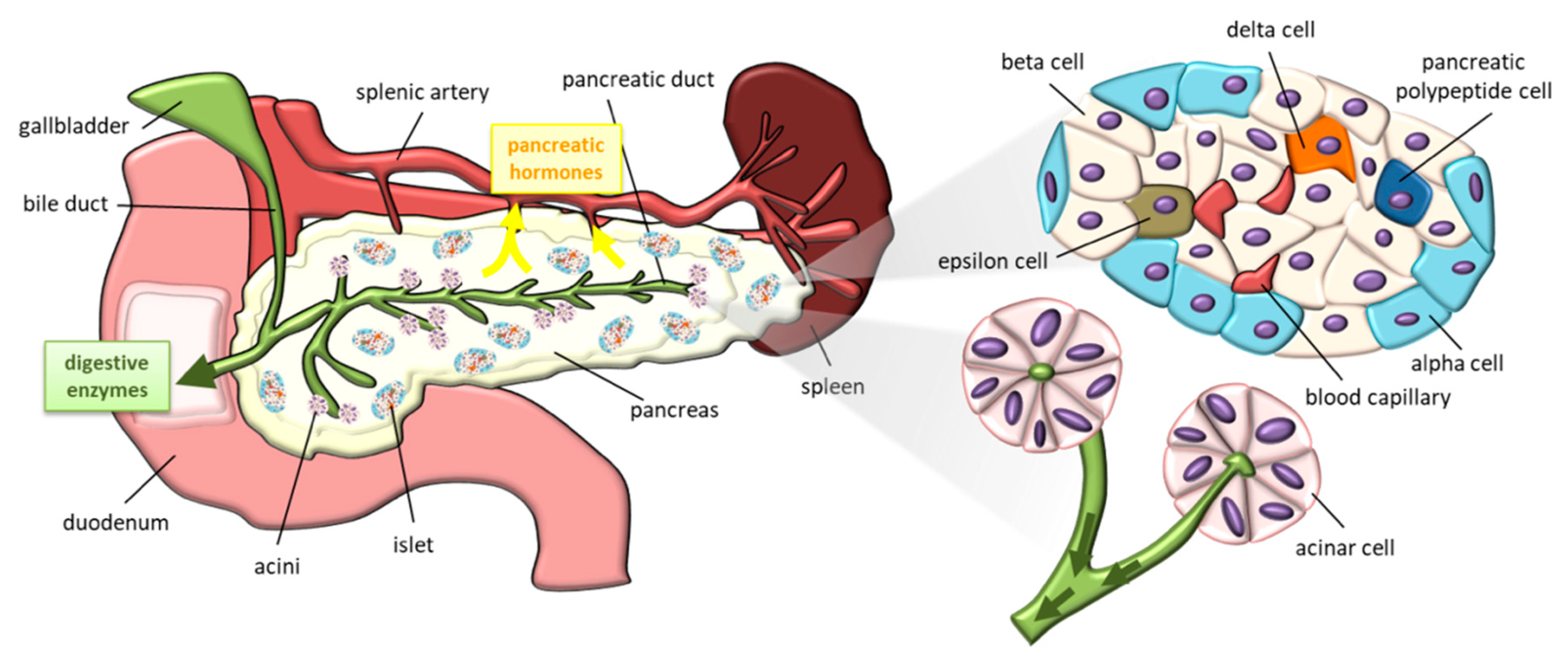
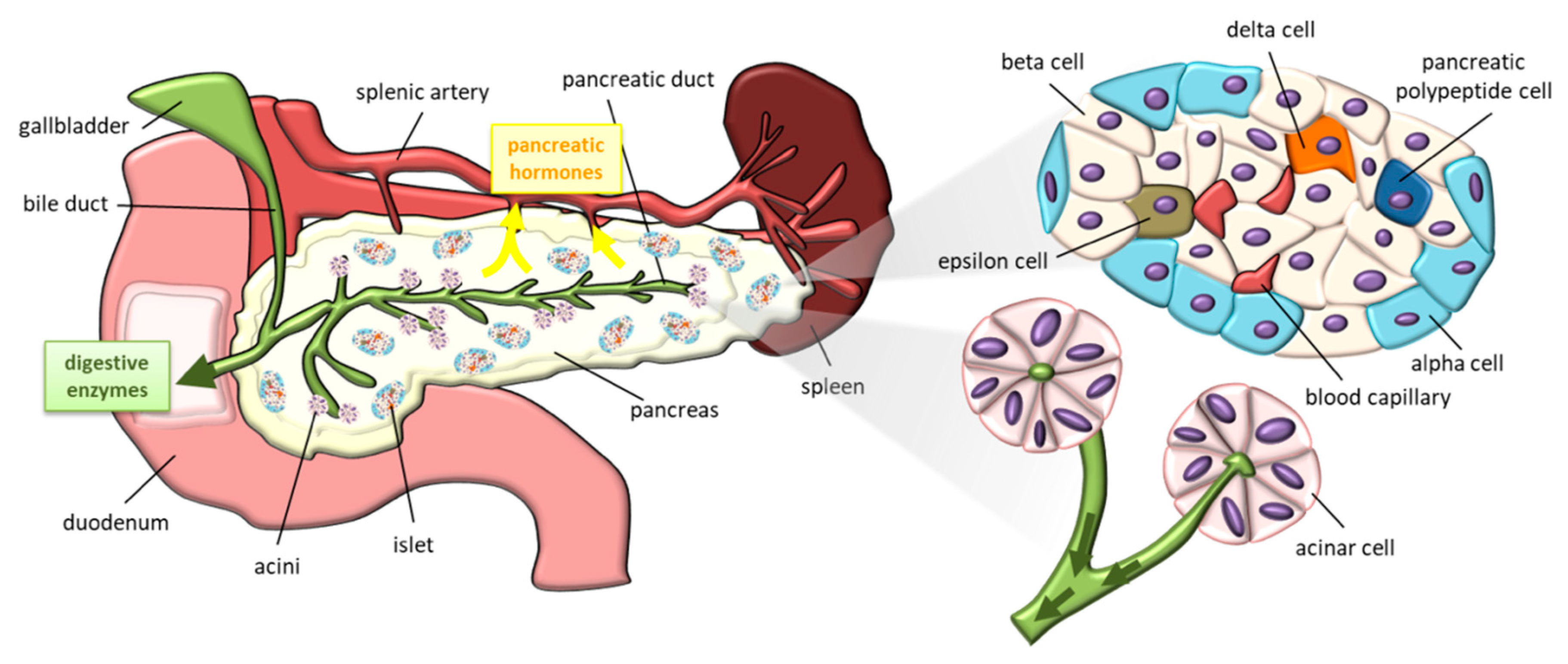
2.1. Pancreas Anatomy
2.2. Development and Plasticity of the Pancreas
2.3. Regulation of Insulin Secretion
2.3.1. Glucose-Stimulated Insulin Secretion
2.3.2. Other Contributors to Insulin Secretion
3. Influence of Dietary Intakes during Gestation and or Lactation
3.1. Excess of Nutrients
3.1.1. Carbohydrate Excess
3.1.2. Lipid Excess
3.1.3. Protein Excess
3.2. Nutrient Restriction
3.2.1. Carbohydrate and Lipid Restriction
3.2.2. Protein Restriction
4. Impact of Ultra-Processed Food Consumption
5. Other Molecules that Can Influence Pancreas Development and Function: The Case of Xenobiotics
6. Importance of Lactation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunter, D.S.; Hazel, S.J.; Kind, K.L.; Owens, J.A.; Pitcher, J.B.; Gatford, K.L. Programming the brain: Common outcomes and gaps in knowledge from animal studies of IUGR. Physiol. Behav. 2016, 164, 233–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Bonner-Weir, S. Life and death of the pancreatic beta cells. Trends Endocrinol. Metab. 2000, 11, 375–378. [Google Scholar] [CrossRef]
- Bonner-Weir, S.; Aguayo-Mazzucato, C.; Weir, G.C. Dynamic development of the pancreas from birth to adulthood. Ups. J. Med. Sci. 2016, 121, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.C.; Brissova, M. Pancreas development in humans. Curr. Opin. Endocrinol. Diabetes Obes. 2014, 21, 77–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Hales, C.N.; Fall, C.H.; Osmond, C.; Phipps, K.; Clark, P.M. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): Relation to reduced fetal growth. Diabetologia 1993, 36, 62–67. [Google Scholar] [CrossRef]
- Zheng, M.; Lamb, K.E.; Grimes, C.; Laws, R.; Bolton, K.; Ong, K.K.; Campbell, K. Rapid weight gain during infancy and subsequent adiposity: A systematic review and meta-analysis of evidence. Obes. Rev. 2018, 19, 321–332. [Google Scholar] [CrossRef]
- McMillen, I.C.; Adam, C.L.; Muhlhausler, B.S. Early origins of obesity: Programming the appetite regulatory system. J. Physiol. 2005, 565, 9–17. [Google Scholar] [CrossRef]
- Jorgensen, M.C.; Ahnfelt-Ronne, J.; Hald, J.; Madsen, O.D.; Serup, P.; Hecksher-Sorensen, J. An illustrated review of early pancreas development in the mouse. Endocr. Rev. 2007, 28, 685–705. [Google Scholar] [CrossRef]
- Abdulreda, M.H.; Caicedo, A.; Berggren, P.O. A Natural Body Window to Study Human Pancreatic Islet Cell Function and Survival. CellR4 Repair Replace. Regen. Reprogram. 2013, 1, 111–122. [Google Scholar]
- Babic, T.; Travagli, R.A. Neural Control of the Pancreas. Available online: https://www.pancreapedia.org/reviews/neural-control-of-pancreas (accessed on 22 September 2016).
- Golson, M.L.; Kaestner, K.H. Epigenetics in formation, function, and failure of the endocrine pancreas. Mol. Metab. 2017, 6, 1066–1076. [Google Scholar] [CrossRef] [PubMed]
- Rankin, M.M.; Kushner, J.A. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009, 58, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Moullé, V.S.; Vivot, K.; Tremblay, C.; Zarrouki, B.; Ghislain, J.; Poitout, V. Glucose and fatty acids synergistically and reversibly promote beta cell proliferation in rats. Diabetologia 2017, 60, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Hellerstrom, C.; Swenne, I. Functional maturation and proliferation of fetal pancreatic beta-cells. Diabetes 1991, 40, 89–93. [Google Scholar] [CrossRef]
- Aguayo-Mazzucato, C.; Koh, A.; El Khattabi, I.; Li, W.C.; Toschi, E.; Jermendy, A.; Juhl, K.; Mao, K.; Weir, G.C.; Sharma, A.; et al. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia 2011, 54, 583–593. [Google Scholar] [CrossRef]
- Henquin, J.C.; Nenquin, M. Immaturity of insulin secretion by pancreatic islets isolated from one human neonate. J. Diabetes Investig. 2018, 9, 270–273. [Google Scholar] [CrossRef]
- Henquin, J.C.; Nenquin, M. Dynamics and Regulation of Insulin Secretion in Pancreatic Islets from Normal Young Children. PLoS ONE 2016, 11, e0165961. [Google Scholar] [CrossRef]
- Bernard-Kargar, C.; Ktorza, A. Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 2001, 50, S30–S35. [Google Scholar] [CrossRef]
- Nielsen, J.H. Beta cell adaptation in pregnancy: A tribute to Claes Hellerstrom. Ups J. Med. Sci. 2016, 121, 151–154. [Google Scholar] [CrossRef]
- Scaglia, L.; Smith, F.E.; Bonner-Weir, S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 1995, 136, 5461–5468. [Google Scholar] [CrossRef]
- Kloppel, G.; Lohr, M.; Habich, K.; Oberholzer, M.; Heitz, P.U. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv. Synth. Pathol. Res. 1985, 4, 110–125. [Google Scholar] [PubMed]
- Assmann, A.; Ueki, K.; Winnay, J.N.; Kadowaki, T.; Kulkarni, R.N. Glucose effects on beta-cell growth and survival require activation of insulin receptors and insulin receptor substrate 2. Mol. Cell. Biol. 2009, 29, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Okada, T.; Liew, C.W.; Hu, J.; Hinault, C.; Michael, M.D.; Krtzfeldt, J.; Yin, C.; Holzenberger, M.; Stoffel, M.; Kulkarni, R.N. Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 8977–8982. [Google Scholar] [CrossRef] [PubMed]
- Stamateris, R.E.; Sharma, R.B.; Kong, Y.; Ebrahimpour, P.; Panday, D.; Ranganath, P.; Zou, B.; Levitt, H.; Parambil, N.A.; O’Donnell, C.P.; et al. Glucose Induces Mouse beta-Cell Proliferation via IRS2, MTOR, and Cyclin D2 but Not the Insulin Receptor. Diabetes 2016, 65, 981–995. [Google Scholar] [CrossRef]
- Curry, D.L.; Bennett, L.L.; Grodsky, G.M. Dynamics of insulin secretion by the perfused rat pancreas. Endocrinology 1968, 83, 572–584. [Google Scholar] [CrossRef]
- Moullé, V.S.; Ghislain, J.; Poitout, V. Nutrient regulation of pancreatic beta-cell proliferation. Biochimie 2017, 143, 10–17. [Google Scholar] [CrossRef]
- Prentki, M.; Matschinsky, F.M.; Madiraju, S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013, 18, 162–185. [Google Scholar] [CrossRef]
- Ahren, B.; Holst, J.J. The cephalic insulin response to meal ingestion in humans is dependent on both cholinergic and noncholinergic mechanisms and is important for postprandial glycemia. Diabetes 2001, 50, 1030–1038. [Google Scholar] [CrossRef]
- Guemes, A.; Herrero, P.; Bondia, J.; Georgiou, P. Modeling the effect of the cephalic phase of insulin secretion on glucose metabolism. Med. Biol. Eng. Comput. 2019, 57, 1173–1186. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic Beta-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef]
- Hill, J.O.; Wyatt, H.R.; Peters, J.C. The Importance of Energy Balance. Eur. Endocrinol. 2013, 9, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Farabi, S.S.; Hernandez, T.L. Low-Carbohydrate Diets for Gestational Diabetes. Nutrients 2019, 11, 1737. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Gardea, M.O.; Gonzales-Pacheco, D.M.; Reader, D.M.; Thomas, A.M.; Wang, S.R.; Gregory, R.P.; Piemonte, T.A.; Thompson, K.L.; Moloney, L. Academy of Nutrition and Dietetics Gestational Diabetes Evidence-Based Nutrition Practice Guideline. J. Acad. Nutr. Diet. 2018, 118, 1719–1742. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; McIntyre, H.D.; Cruickshank, J.K.; McCance, D.R.; Dyer, A.R.; Metzger, B.E.; Lowe, L.P.; Trimble, E.R.; Coustan, D.R.; Hadden, D.R.; et al. The hyperglycemia and adverse pregnancy outcome study: Associations of GDM and obesity with pregnancy outcomes. Diabetes Care 2012, 35, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, A.; Harder, T.; Brunn, M.; Harder, A.; Roepke, K.; Wittrock-Staar, M.; Ziska, T.; Schellong, K.; Rodekamp, E.; Melchior, K.; et al. Hypothalamic proopiomelanocortin promoter methylation becomes altered by early overfeeding: An epigenetic model of obesity and the metabolic syndrome. J. Physiol. 2009, 587, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, R.N.; Warrington, N.M.; Cavadino, A.; Tyrrell, J.; Nodzenski, M.; Horikoshi, M.; Geller, F.; Myhre, R.; Richmond, R.C.; Paternoster, L.; et al. Genome-wide association study of offspring birth weight in 86 577 women identifies five novel loci and highlights maternal genetic effects that are independent of fetal genetics. Hum. Mol. Genet. 2018, 27, 742–756. [Google Scholar] [CrossRef]
- Zhang, P.; Zhu, D.; Zhang, Y.; Li, L.; Chen, X.; Zhang, W.; Shi, R.; Tao, J.; Han, B.; Xu, Z. Synergetic Effects of Prenatal and Postnatal High Sucrose Intake on Glucose Tolerance and Hepatic Insulin Resistance in Rat Offspring. Mol. Nutr. Food Res. 2018, 62. [Google Scholar] [CrossRef]
- Ozkan, H.; Topsakal, S.; Ozmen, O. Investigation of the diabetic effects of maternal high-glucose diet on rats. Biomed. Pharm. 2019, 110, 609–617. [Google Scholar] [CrossRef]
- Ishikawa, K.; Tsunekawa, S.; Ikeniwa, M.; Izumoto, T.; Iida, A.; Ogata, H.; Uenishi, E.; Seino, Y.; Ozaki, N.; Sugimura, Y.; et al. Long-term pancreatic beta cell exposure to high levels of glucose but not palmitate induces DNA methylation within the insulin gene promoter and represses transcriptional activity. PLoS ONE 2015, 10, e0115350. [Google Scholar] [CrossRef]
- Siemelink, M.; Verhoef, A.; Dormans, J.A.; Span, P.N.; Piersma, A.H. Dietary fatty acid composition during pregnancy and lactation in the rat programs growth and glucose metabolism in the offspring. Diabetologia 2002, 45, 1397–1403. [Google Scholar] [CrossRef] [Green Version]
- Bringhenti, I.; Moraes-Teixeira, J.A.; Cunha, M.R.; Ornellas, F.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Maternal obesity during the preconception and early life periods alters pancreatic development in early and adult life in male mouse offspring. PLoS ONE 2013, 8, e55711. [Google Scholar] [CrossRef] [PubMed]
- Graus-Nunes, F.; Dalla Corte Frantz, E.; Lannes, W.R.; da Silva Menezes, M.C.; Mandarim-de-Lacerda, C.A.; Souza-Mello, V. Pregestational maternal obesity impairs endocrine pancreas in male F1 and F2 progeny. Nutrition 2015, 31, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Ailhaud, G.; Massiera, F.; Weill, P.; Legrand, P.; Alessandri, J.M.; Guesnet, P. Temporal changes in dietary fats: Role of n-6 polyunsaturated fatty acids in excessive adipose tissue development and relationship to obesity. Prog. Lipid Res. 2006, 45, 203–236. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Cetin, I.; Brenna, J.T.; Perinatal Lipid Intake Working, Group. Dietary fat intakes for pregnant and lactating women. Br. J. Nutr. 2007, 98, 873–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, P.A.; DiPatrizio, N.V. Impact of maternal western diet-induced obesity on offspring mortality and peripheral endocannabinoid system in mice. PLoS ONE 2018, 13, e0205021. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, B.M.; Souza-Mello, V.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Maternal high-fat diet is associated with altered pancreatic remodelling in mice offspring. Eur. J. Nutr. 2013, 52, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, E.; Sosa-Larios, T.; Calzada, L.; Ibanez, C.A.; Mendoza-Rodriguez, C.A.; Morales, A.; Morimoto, S. Decreased basal insulin secretion from pancreatic islets of pups in a rat model of maternal obesity. J. Endocrinol. 2016, 231, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Dyrskog, S.E.; Gregersen, S.; Hermansen, K. High-fat feeding during gestation and nursing period have differential effects on the insulin secretory capacity in offspring from normal Wistar rats. Rev. Diabet Stud. 2005, 2, 136–145. [Google Scholar] [CrossRef]
- Taylor, P.D.; McConnell, J.; Khan, I.Y.; Holemans, K.; Lawrence, K.M.; Asare-Anane, H.; Persaud, S.J.; Jones, P.M.; Petrie, L.; Hanson, M.A.; et al. Impaired glucose homeostasis and mitochondrial abnormalities in offspring of rats fed a fat-rich diet in pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R134–R139. [Google Scholar] [CrossRef]
- Ong, Z.Y.; Muhlhausler, B.S. Maternal “junk-food” feeding of rat dams alters food choices and development of the mesolimbic reward pathway in the offspring. FASEB J. 2011, 25, 2167–2179. [Google Scholar] [CrossRef]
- Haumaitre, C.; Lenoir, O.; Scharfmann, R. Histone deacetylase inhibitors modify pancreatic cell fate determination and amplify endocrine progenitors. Mol. Cell. Biol. 2008, 28, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine beta- and delta-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [PubMed]
- Julvez, J.; Fernandez-Barres, S.; Gignac, F.; Lopez-Vicente, M.; Bustamante, M.; Garcia-Esteban, R.; Vioque, J.; Llop, S.; Ballester, F.; Fernandez-Somoano, A.; et al. Maternal seafood consumption during pregnancy and child attention outcomes: A cohort study with gene effect modification by PUFA-related genes. Int. J. Epidemiol. 2019. Available online: https://academic.oup.com/ije/advance-article-abstract/doi/10.1093/ije/dyz197/5579829 (accessed on 2 October 2019). [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Johnson, G.A.; Herring, C.; Seo, H.; Dai, Z.; Wang, J.; Wu, Z.; Wang, X. Functional amino acids in the development of the pig placenta. Mol. Reprod. Dev. 2017, 84, 870–882. [Google Scholar] [CrossRef]
- Herring, C.M.; Bazer, F.W.; Johnson, G.A.; Wu, G. Impacts of maternal dietary protein intake on fetal survival, growth, and development. Exp. Biol. Med. (Maywood) 2018, 243, 525–533. [Google Scholar] [CrossRef]
- Kucia, M.; Langhammer, M.; Gors, S.; Albrecht, E.; Hammon, H.M.; Nurnberg, G.; Metges, C.C. High-protein diet during gestation and lactation affects mammary gland mRNA abundance, milk composition and pre-weaning litter growth in mice. Animal 2011, 5, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Coupe, B.; Delamaire, E.; Hoebler, C.; Grit, I.; Even, P.; Fromentin, G.; Darmaun, D.; Parnet, P. Hypothalamus integrity and appetite regulation in low birth weight rats reared artificially on a high-protein milk formula. J. Nutr. Biochem. 2011, 22, 956–963. [Google Scholar] [CrossRef]
- Delamaire, E.; Parnet, P.; Coupe, B.; Hoebler, C.; Blat, S.; Poupeau, G.; Boquien, C.Y.; Champ, M.; Darmaun, D. Long term metabolic impact of high protein neonatal feeding: A preliminary study in male rat pups born with a low birth weight. Clin. Nutr. 2012, 31, 741–748. [Google Scholar] [CrossRef]
- Switkowski, K.M.; Jacques, P.F.; Must, A.; Hivert, M.F.; Fleisch, A.; Gillman, M.W.; Rifas-Shiman, S.; Oken, E. Higher Maternal Protein Intake during Pregnancy Is Associated with Lower Cord Blood Concentrations of Insulin-like Growth Factor (IGF)-II, IGF Binding Protein 3, and Insulin, but Not IGF-I, in a Cohort of Women with High Protein Intake. J. Nutr. 2017, 147, 1392–1400. [Google Scholar] [CrossRef] [Green Version]
- Closa-Monasterolo, R.; Ferre, N.; Luque, V.; Zaragoza-Jordana, M.; Grote, V.; Weber, M.; Koletzko, B.; Socha, P.; Gruszfeld, D.; Janas, R.; et al. Sex differences in the endocrine system in response to protein intake early in life. Am. J. Clin. Nutr. 2011, 94, 1920S–1927S. [Google Scholar] [CrossRef]
- Oken, E.; Morton-Eggleston, E.; Rifas-Shiman, S.L.; Switkowski, K.M.; Hivert, M.F.; Fleisch, A.F.; Mantzoros, C.; Gillman, M.W. Sex-Specific Associations of Maternal Gestational Glycemia with Hormones in Umbilical Cord Blood at Delivery. Am. J. Perinatol. 2016, 33, 1273–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, K.M.; Barker, D.J. Fetal nutrition and adult disease. Am. J. Clin. Nutr. 2000, 71, 1344S–1352S. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J.; Clark, P.M.; Cox, L.J.; Fall, C.; Osmond, C.; Winter, P.D. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 1991, 303, 1019–1022. [Google Scholar] [CrossRef]
- Van Assche, F.A.; Aerts, L.; De Prins, F.A. The fetal endocrine pancreas. Eur. J. Obstet. Gynecol. Reprod. Biol. 1984, 18, 267–272. [Google Scholar] [CrossRef]
- Snoeck, A.; Remacle, C.; Reusens, B.; Hoet, J.J. Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol. Neonate 1990, 57, 107–118. [Google Scholar] [CrossRef]
- Calzada, L.; Morales, A.; Sosa-Larios, T.C.; Reyes-Castro, L.A.; Rodriguez-Gonzalez, G.L.; Rodriguez-Mata, V.; Zambrano, E.; Morimoto, S. Maternal protein restriction during gestation impairs female offspring pancreas development in the rat. Nutr. Res. 2016, 36, 855–862. [Google Scholar] [CrossRef]
- Park, J.H.; Stoffers, D.A.; Nicholls, R.D.; Simmons, R.A. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J. Clin. Investig. 2008, 118, 2316–2324. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, T.L.; Mande, A.; Barbour, L.A. Nutrition therapy within and beyond gestational diabetes. Diabetes Res. Clin. Pract. 2018, 145, 39–50. [Google Scholar] [CrossRef]
- Barbour, L.A.; Farabi, S.S.; Friedman, J.E.; Hirsch, N.M.; Reece, M.S.; Van Pelt, R.E.; Hernandez, T.L. Postprandial Triglycerides Predict Newborn Fat More Strongly than Glucose in Women with Obesity in Early Pregnancy. Obesity 2018, 26, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Moore, D.; Subramanian, A.; Cheng, K.K.; Toulis, K.A.; Qiu, X.; Saravanan, P.; Price, M.J.; Nirantharakumar, K. Gestational dyslipidaemia and adverse birthweight outcomes: A systematic review and meta-analysis. Obes. Rev. 2018, 19, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Barbour, L.A.; Hernandez, T.L. Maternal Lipids and Fetal Overgrowth: Making Fat from Fat. Clin. Ther. 2018, 40, 1638–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumfield, M.L.; Hure, A.J.; MacDonald-Wicks, L.K.; Smith, R.; Simpson, S.J.; Giles, W.B.; Raubenheimer, D.; Collins, C.E. Dietary balance during pregnancy is associated with fetal adiposity and fat distribution. Am. J. Clin. Nutr. 2012, 96, 1032–1041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Imhoff-Kunsch, B.; Girard, A.W. Biological mechanisms for nutritional regulation of maternal health and fetal development. Paediatr. Perinat. Epidemiol. 2012, 26, 4–26. [Google Scholar] [CrossRef]
- Coupe, B.; Amarger, V.; Grit, I.; Benani, A.; Parnet, P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology 2010, 151, 702–713. [Google Scholar] [CrossRef]
- Coupe, B.; Grit, I.; Darmaun, D.; Parnet, P. The timing of “catch-up growth” affects metabolism and appetite regulation in male rats born with intrauterine growth restriction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R813–R824. [Google Scholar] [CrossRef]
- Martin Agnoux, A.; Alexandre-Gouabau, M.C.; Le Drean, G.; Antignac, J.P.; Parnet, P. Relative contribution of foetal and post-natal nutritional periods on feeding regulation in adult rats. Acta Physiol. (Oxf.) 2014, 210, 188–201. [Google Scholar] [CrossRef]
- Agnoux, A.M.; Antignac, J.P.; Simard, G.; Poupeau, G.; Darmaun, D.; Parnet, P.; Alexandre-Gouabau, M.C. Time window-dependent effect of perinatal maternal protein restriction on insulin sensitivity and energy substrate oxidation in adult male offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R184–R197. [Google Scholar] [CrossRef] [Green Version]
- Frantz, E.D.; Aguila, M.B.; Pinheiro-Mulder Ada, R.; Mandarim-de-Lacerda, C.A. Transgenerational endocrine pancreatic adaptation in mice from maternal protein restriction in utero. Mech. Ageing Dev. 2011, 132, 110–116. [Google Scholar] [CrossRef]
- Bertin, E.; Gangnerau, M.N.; Bailbe, D.; Portha, B. Glucose metabolism and beta-cell mass in adult offspring of rats protein and/or energy restricted during the last week of pregnancy. Am. J. Physiol. 1999, 277, E11–E17. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, S.; Calzada, L.; Sosa, T.C.; Reyes-Castro, L.A.; Rodriguez-Gonzalez, G.L.; Morales, A.; Nathanielsz, P.W.; Zambrano, E. Emergence of ageing-related changes in insulin secretion by pancreatic islets of male rat offspring of mothers fed a low-protein diet. Br. J. Nutr. 2012, 107, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.P.; Phillips, A.R.; Harris, P.M.; Cooper, G.J. Impaired insulin secretion in perfused pancreases isolated from offspring of female rats fed a low protein whey-based diet. JOP 2008, 9, 477–488. [Google Scholar] [PubMed]
- Monteiro, C.A.; Cannon, G.; Levy, R.B.; Moubarac, J.C.; Louzada, M.L.; Rauber, F.; Khandpur, N.; Cediel, G.; Neri, D.; Martinez-Steele, E.; et al. Ultra-processed foods: What they are and how to identify them. Public Health Nutr. 2019, 22, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Poti, J.M.; Braga, B.; Qin, B. Ultra-processed Food Intake and Obesity: What Really Matters for Health-Processing or Nutrient Content? Curr. Obes. Rep. 2017, 6, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Juul, F.; Martinez-Steele, E.; Parekh, N.; Monteiro, C.A.; Chang, V.W. Ultra-processed food consumption and excess weight among US adults. Br. J. Nutr. 2018, 120, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Srour, B.; Fezeu, L.K.; Kesse-Guyot, E.; Alles, B.; Mejean, C.; Andrianasolo, R.M.; Chazelas, E.; Deschasaux, M.; Hercberg, S.; Galan, P.; et al. Ultra-processed food intake and risk of cardiovascular disease: Prospective cohort study (NutriNet-Sante). BMJ 2019, 365, l1451. [Google Scholar] [CrossRef]
- Rohatgi, K.W.; Tinius, R.A.; Cade, W.T.; Steele, E.M.; Cahill, A.G.; Parra, D.C. Relationships between consumption of ultra-processed foods, gestational weight gain and neonatal outcomes in a sample of US pregnant women. PeerJ 2017, 5, e4091. [Google Scholar] [CrossRef]
- Sartorelli, D.S.; Crivellenti, L.C.; Zuccolotto, D.C.C.; Franco, L.J. Relationship between minimally and ultra-processed food intake during pregnancy with obesity and gestational diabetes mellitus. Cad. Saude Publica 2019, 35, e00049318. [Google Scholar] [CrossRef]
- Gomes, C.B.; Malta, M.B.; Papini, S.J.; Benicio, M.H.D.; Corrente, J.E.; Carvalhaes, M. Adherence to dietary patterns during pregnancy and association with maternal characteristics in pregnant Brazilian women. Nutrition 2019, 62, 85–92. [Google Scholar] [CrossRef]
- Gomes, C.B.; Malta, M.B.; Louzada, M.; Benicio, M.H.D.; Barros, A.J.D.; Carvalhaes, M. Ultra-processed Food Consumption by Pregnant Women: The Effect of an Educational Intervention with Health Professionals. Matern. Child Health J. 2019, 23, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Staud, F.; Karahoda, R. Trophoblast: The central unit of fetal growth, protection and programming. Int. J. Biochem. Cell. Biol. 2018, 105, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Fontes, K.N.; Reginatto, M.W.; Silva, N.L.; Andrade, C.B.V.; Bloise, F.F.; Monteiro, V.R.S.; Silva-Filho, J.L.; Imperio, G.E.; Pimentel-Coelho, P.M.; Pinheiro, A.A.S.; et al. Dysregulation of placental ABC transporters in a murine model of malaria-induced preterm labor. Sci. Rep. 2019, 9, 11488. [Google Scholar] [CrossRef] [PubMed]
- Bordoni, L.; Nasuti, C.; Di Stefano, A.; Marinelli, L.; Gabbianelli, R. Epigenetic Memory of Early-Life Parental Perturbation: Dopamine Decrease and DNA Methylation Changes in Offspring. Oxid. Med. Cell. Longev. 2019, 2019, 1472623. [Google Scholar] [CrossRef]
- Nasuti, C.; Fattoretti, P.; Carloni, M.; Fedeli, D.; Ubaldi, M.; Ciccocioppo, R.; Gabbianelli, R. Neonatal exposure to permethrin pesticide causes lifelong fear and spatial learning deficits and alters hippocampal morphology of synapses. J. Neurodev. Disord. 2014, 6, 7. [Google Scholar] [CrossRef]
- Vrijheid, M.; Casas, M.; Gascon, M.; Valvi, D.; Nieuwenhuijsen, M. Environmental pollutants and child health-A review of recent concerns. Int J. Hyg. Environ. Health 2016, 219, 331–342. [Google Scholar] [CrossRef]
- Garcia-Arevalo, M.; Alonso-Magdalena, P.; Servitja, J.M.; Boronat-Belda, T.; Merino, B.; Villar-Pazos, S.; Medina-Gomez, G.; Novials, A.; Quesada, I.; Nadal, A. Maternal Exposure to Bisphenol-A During Pregnancy Increases Pancreatic beta-Cell Growth During Early Life in Male Mice Offspring. Endocrinology 2016, 157, 4158–4171. [Google Scholar] [CrossRef]
- Roze, J.C.; Darmaun, D.; Boquien, C.Y.; Flamant, C.; Picaud, J.C.; Savagner, C.; Claris, O.; Lapillonne, A.; Mitanchez, D.; Branger, B.; et al. The apparent breastfeeding paradox in very preterm infants: Relationship between breast feeding, early weight gain and neurodevelopment based on results from two cohorts, EPIPAGE and LIFT. BMJ Open 2012, 2, e000834. [Google Scholar] [CrossRef]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. N. Am. 2013, 60, 49–74. [Google Scholar] [CrossRef]
- Koletzko, B.; Agostoni, C.; Bergmann, R.; Ritzenthaler, K.; Shamir, R. Physiological aspects of human milk lipids and implications for infant feeding: A workshop report. Acta Paediatr. 2011, 100, 1405–1415. [Google Scholar] [CrossRef]
- Andreas, N.J.; Kampmann, B.; Mehring Le-Doare, K. Human breast milk: A review on its composition and bioactivity. Early Hum. Dev. 2015, 91, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Makela, J.; Linderborg, K.; Niinikoski, H.; Yang, B.; Lagstrom, H. Breast milk fatty acid composition differs between overweight and normal weight women: The STEPS Study. Eur. J. Nutr. 2013, 52, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Martin Agnoux, A.; Antignac, J.P.; Boquien, C.Y.; David, A.; Desnots, E.; Ferchaud-Roucher, V.; Darmaun, D.; Parnet, P.; Alexandre-Gouabau, M.C. Perinatal protein restriction affects milk free amino acid and fatty acid profile in lactating rats: Potential role on pup growth and metabolic status. J. Nutr. Biochem. 2015, 26, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Human milk and formula fatty acids. J. Pediatr. 1992, 120, S56–S61. [Google Scholar] [CrossRef]
- Ranade, P.S.; Rao, S.S. Maternal long-chain PUFA supplementation during protein deficiency improves brain fatty acid accretion in rat pups by altering the milk fatty acid composition of the dam. J. Nutr. Sci. 2013, 2, e5. [Google Scholar] [CrossRef] [Green Version]
- Padberg, S.; Buhrer, C.; Menzel, J.; Weikert, C.; Schaefer, C.; Abraham, K. Xenobiotics and pathogens in breast milk: A risk for the child. Bundesgesundheitsblatt Gesundh. Gesundh. 2018, 61, 960–970. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moullé, V.S.; Parnet, P. Effects of Nutrient Intake during Pregnancy and Lactation on the Endocrine Pancreas of the Offspring. Nutrients 2019, 11, 2708. https://doi.org/10.3390/nu11112708
Moullé VS, Parnet P. Effects of Nutrient Intake during Pregnancy and Lactation on the Endocrine Pancreas of the Offspring. Nutrients. 2019; 11(11):2708. https://doi.org/10.3390/nu11112708
Chicago/Turabian StyleMoullé, Valentine Suzanne, and Patricia Parnet. 2019. "Effects of Nutrient Intake during Pregnancy and Lactation on the Endocrine Pancreas of the Offspring" Nutrients 11, no. 11: 2708. https://doi.org/10.3390/nu11112708