Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality

 and
and
Abstract

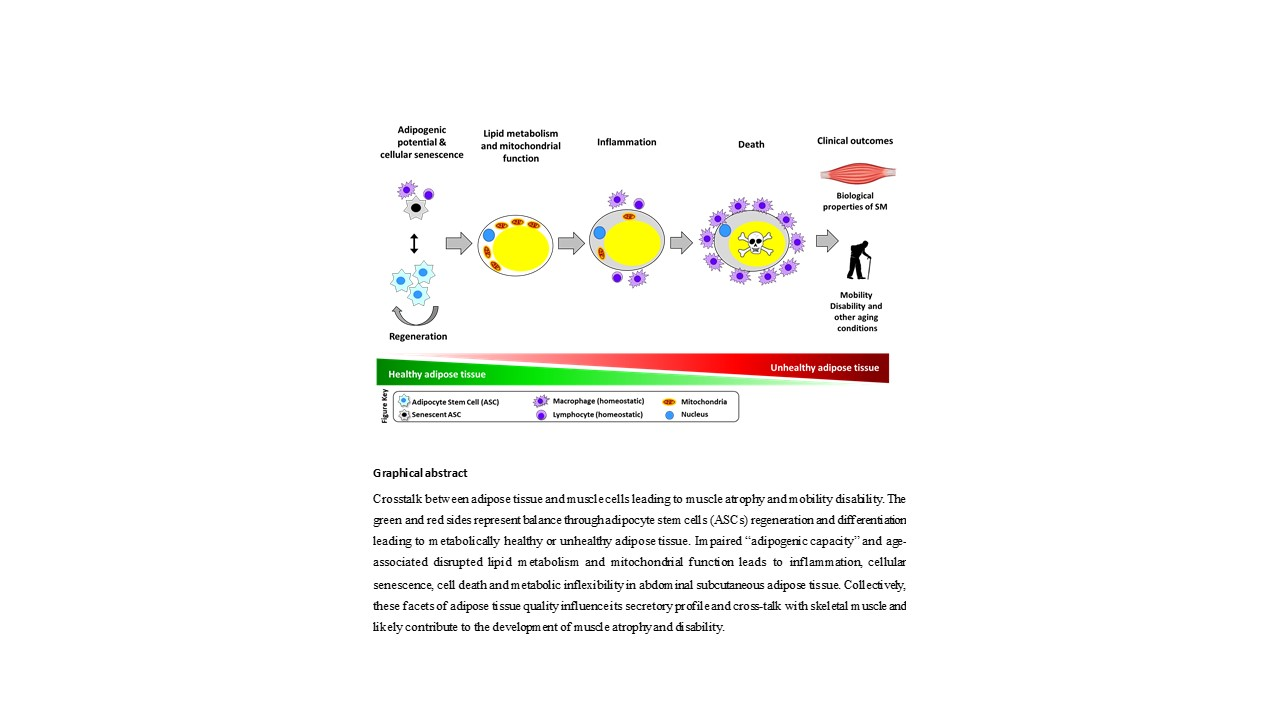{kind=link}
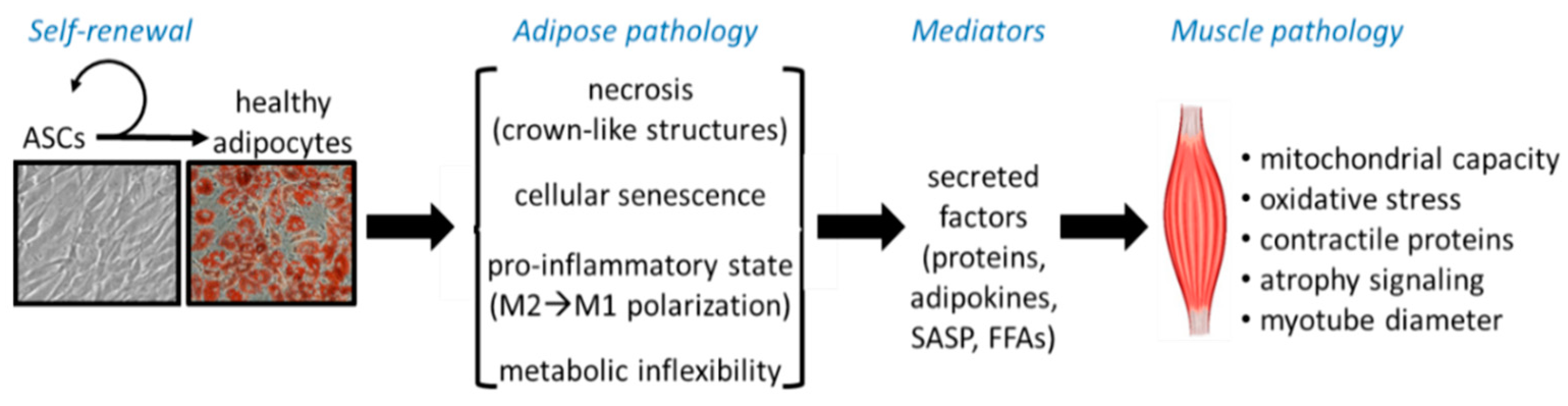{kind=link}
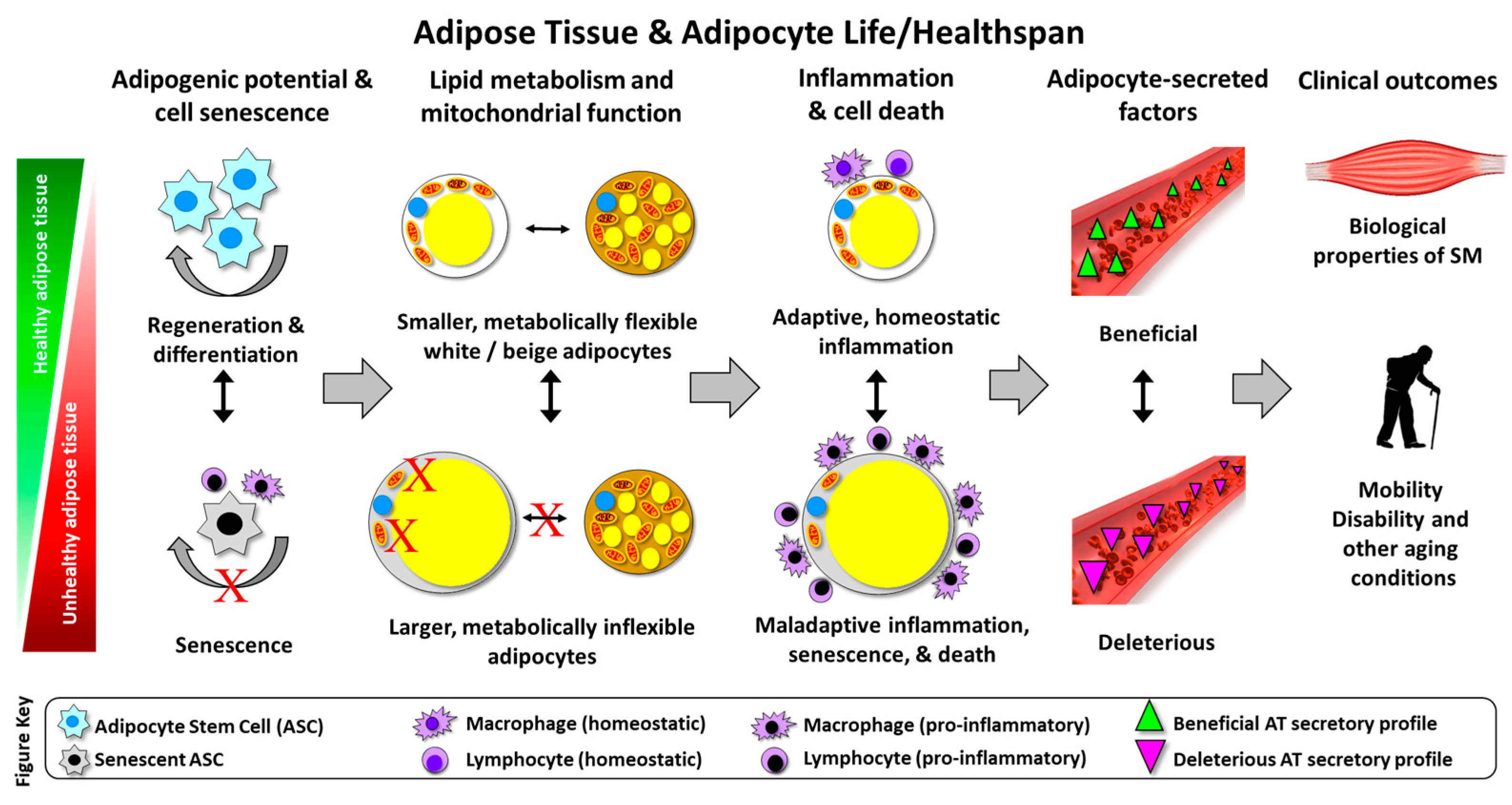{kind=link}
1. Introduction
2. Adipose Tissue and Lipid Distribution with Aging
3. Adipose Tissue Cell Death in Aging
4. Cellular Senescence in Adipose Tissue with Aging
5. Adipose Tissue Inflammation during Aging
6. Adipose Tissue Metabolic Flexibility and Aging
7. Adipose-Muscle Crosstalk, Mobility Disability, and Aging
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- U.S. Census Bureau. Older People Projected to Outnumber Children for First Time in U.S. History. Available online: https://census.gov/newsroom/press-releases/2018/cb18-41-population-projections.html (accessed on 22 October 2019).
- Centers for Disease Control and Prevention. Disability and Functioning (Noninstitutionalized Adults Aged 18 and Over). Available online: https://www.cdc.gov/nchs/fastats/disability.htm (accessed on 22 October 2019).
- Hardy, S.E.; Kang, Y.; Studenski, S.A.; Degenholtz, H.B. Ability to walk 1/4 mile predicts subsequent disability, mortality, and health care costs. J. Gen. Intern. Med. 2011, 26, 130–135. [Google Scholar] [CrossRef]
- Penninx, B.W.; Abbas, H.; Ambrosius, W.; Nicklas, B.J.; Davis, C.; Messier, S.P.; Pahor, M. Inflammatory markers and physical function among older adults with knee osteoarthritis. J. Rheumatol. 2004, 31, 2027–2031. [Google Scholar]
- Studenski, S.; Perera, S.; Patel, K.; Rosano, C.; Faulkner, K.; Inzitari, M.; Brach, J.; Chandler, J.; Cawthon, P.; Connor, E.B.; et al. Gait speed and survival in older adults. JAMA 2011, 305, 50–58. [Google Scholar] [CrossRef]
- Batsis, J.A. Obesity in the Older Adult: Special Issue. J. Nutr. Gerontol. Geriatr. 2019, 38, 1–5. [Google Scholar] [CrossRef]
- Newman, A.B.; Haggerty, C.L.; Goodpaster, B.; Harris, T.; Kritchevsky, S.; Nevitt, M.; Miles, T.P.; Visser, M.; Health, A. Body Composition Research Group. Strength and muscle quality in a well-functioning cohort of older adults: The Health, Aging and Body Composition Study. J. Am. Geriatr. Soc. 2003, 51, 323–330. [Google Scholar] [CrossRef]
- Newman, A.B.; Kupelian, V.; Visser, M.; Simonsick, E.; Goodpaster, B.; Nevitt, M.; Kritchevsky, S.B.; Tylavsky, F.A.; Rubin, S.M.; Harris, T.B.; et al. Sarcopenia: Alternative definitions and associations with lower extremity function. J. Am. Geriatr. Soc. 2003, 51, 1602–1609. [Google Scholar] [CrossRef]
- Bays, H.E. Adiposopathy is “sick fat” a cardiovascular disease? J. Am. Coll. Cardiol. 2011, 57, 2461–2473. [Google Scholar] [CrossRef]
- Berry, D.C.; Stenesen, D.; Zeve, D.; Graff, J.M. The developmental origins of adipose tissue. Development 2013, 140, 3939–3949. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Rouault, C.; Rodriguez-Cuenca, S.; Albert, V.; Edom-Vovard, F.; Vidal-Puig, A.; Clement, K.; Butler-Browne, G.S.; Lacasa, D. Human Adipocytes Induce Inflammation and Atrophy in Muscle Cells During Obesity. Diabetes 2015, 64, 3121–3134. [Google Scholar] [CrossRef]
- Trim, W.; Turner, J.E.; Thompson, D. Parallels in Immunometabolic Adipose Tissue Dysfunction with Ageing and Obesity. Front. Immunol. 2018, 9, 169. [Google Scholar] [CrossRef] [PubMed]
- Sachs, S.; Zarini, S.; Kahn, D.E.; Harrison, K.A.; Perreault, L.; Phang, T.; Newsom, S.A.; Strauss, A.; Kerege, A.; Schoen, J.A.; et al. Intermuscular adipose tissue directly modulates skeletal muscle insulin sensitivity in humans. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E866–E879. [Google Scholar] [CrossRef] [PubMed]
- Arner, P.; Bernard, S.; Salehpour, M.; Possnert, G.; Liebl, J.; Steier, P.; Buchholz, B.A.; Eriksson, M.; Arner, E.; Hauner, H.; et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011, 478, 110–113. [Google Scholar] [CrossRef]
- Sparks, L.M.; Pasarica, M.; Sereda, O.; deJonge, L.; Thomas, S.; Loggins, H.; Xie, H.; Miles, J.M.; Smith, S.R. Effect of adipose tissue on the sexual dimorphism in metabolic flexibility. Metabolism 2009, 58, 1564–1571. [Google Scholar] [CrossRef]
- Sparks, L.M.; Ukropcova, B.; Smith, J.; Pasarica, M.; Hymel, D.; Xie, H.; Bray, G.A.; Miles, J.M.; Smith, S.R. Relation of adipose tissue to metabolic flexibility. Diabetes Res. Clin. Pract. 2009, 83, 32–43. [Google Scholar] [CrossRef]
- Foryst-Ludwig, A.; Kreissl, M.C.; Benz, V.; Brix, S.; Smeir, E.; Ban, Z.; Januszewicz, E.; Salatzki, J.; Grune, J.; Schwanstecher, A.K.; et al. Adipose Tissue Lipolysis Promotes Exercise-induced Cardiac Hypertrophy Involving the Lipokine C16:1n7-Palmitoleate. J. Biol. Chem. 2015, 290, 23603–23615. [Google Scholar] [CrossRef]
- Stanford, K.I.; Goodyear, L.J. Exercise regulation of adipose tissue. Adipocyte 2016, 5, 153–162. [Google Scholar] [CrossRef]
- Pedersen, B.K. Muscles and their myokines. J. Exp. Biol. 2011, 214 Pt 2, 337–346. [Google Scholar] [CrossRef]
- Stanford, K.I.; Goodyear, L.J. Muscle-Adipose Tissue Cross Talk. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Kalinkovich, A.; Livshits, G. Sarcopenic obesity or obese sarcopenia: A cross talk between age-associated adipose tissue and skeletal muscle inflammation as a main mechanism of the pathogenesis. Ageing Res. Rev. 2017, 35, 200–221. [Google Scholar] [CrossRef]
- Mau, T.; Yung, R. Adipose tissue inflammation in aging. Exp. Gerontol. 2018, 105, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Meex, R.C.R.; Blaak, E.E.; van Loon, L.J.C. Lipotoxicity plays a key role in the development of both insulin resistance and muscle atrophy in patients with type 2 diabetes. Obes. Rev. 2019, 20, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Justice, J.N.; Thompson, L. Lipotoxicity, aging, and muscle contractility: Does fiber type matter? Geroscience 2019, 41, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Kuk, J.L.; Saunders, T.J.; Davidson, L.E.; Ross, R. Age-related changes in total and regional fat distribution. Ageing Res. Rev. 2009, 8, 339–348. [Google Scholar] [CrossRef]
- Chaurasia, B.; Summers, S.A. Ceramides—Lipotoxic Inducers of Metabolic Disorders. Trends Endocrinol. Metab. 2015, 26, 538–550. [Google Scholar] [CrossRef]
- Bandet, C.L.; Tan-Chen, S.; Bourron, O.; Le Stunff, H.; Hajduch, E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019, 20, 479. [Google Scholar] [CrossRef]
- St-Jean-Pelletier, F.; Pion, C.H.; Leduc-Gaudet, J.P.; Sgarioto, N.; Zovile, I.; Barbat-Artigas, S.; Reynaud, O.; Alkaterji, F.; Lemieux, F.C.; Grenon, A.; et al. The impact of ageing, physical activity, and pre-frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J. Cachexia Sarcopenia Muscle 2017, 8, 213–228. [Google Scholar] [CrossRef]
- Tumova, J.; Andel, M.; Trnka, J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar]
- Delmonico, M.J.; Harris, T.B.; Visser, M.; Park, S.W.; Conroy, M.B.; Velasquez-Mieyer, P.; Boudreau, R.; Manini, T.M.; Nevitt, M.; Newman, A.B.; et al. Body, Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 2009, 90, 1579–1585. [Google Scholar]
- Beavers, K.M.; Beavers, D.P.; Houston, D.K.; Harris, T.B.; Hue, T.F.; Koster, A.; Newman, A.B.; Simonsick, E.M.; Studenski, S.A.; Nicklas, B.J.; et al. Associations between body composition and gait-speed decline: Results from the Health, Aging, and Body Composition study. Am. J. Clin. Nutr. 2013, 97, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, R.; Collino, M.; Nigro, D.; Chiazza, F.; D’Antona, G.; Aragno, M.; Minetto, M.A. Accumulation of advanced glycation end-products and activation of the SCAP/SREBP Lipogenetic pathway occur in diet-induced obese mouse skeletal muscle. PLoS ONE 2015, 10, e0119587. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Chomentowski, P.; Ward, B.K.; Rossi, A.; Glynn, N.W.; Delmonico, M.J.; Kritchevsky, S.B.; Pahor, M.; Newman, A.B. Effects of physical activity on strength and skeletal muscle fat infiltration in older adults: A randomized controlled trial. J. Appl. Physiol. 2008, 105, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Giolo De Carvalho, F.; Sparks, L.M. Targeting White Adipose Tissue with Exercise or Bariatric Surgery as Therapeutic Strategies in Obesity. Biology 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Huang, T.L.; Tran, T.T.; Zhang, H.; Townsend, K.L.; Shadrach, J.L.; Cerletti, M.; McDougall, L.E.; Giorgadze, N.; Tchkonia, T.; et al. Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc. Natl. Acad. Sci. USA 2011, 108, 143–148. [Google Scholar] [CrossRef]
- Tsuji, W.; Rubin, J.P.; Marra, K.G. Adipose-derived stem cells: Implications in tissue regeneration. World J. Stem Cells 2014, 6, 312–321. [Google Scholar] [CrossRef]
- Raajendiran, A.; Ooi, G.; Bayliss, J.; O’Brien, P.E.; Schittenhelm, R.B.; Clark, A.K.; Taylor, R.A.; Rodeheffer, M.S.; Burton, P.R.; Watt, M.J. Identification of Metabolically Distinct Adipocyte Progenitor Cells in Human Adipose Tissues. Cell Rep. 2019, 27, 1528–1540. [Google Scholar] [CrossRef]
- Gao, H.; Volat, F.; Sandhow, L.; Galitzky, J.; Nguyen, T.; Esteve, D.; Astrom, G.; Mejhert, N.; Ledoux, S.; Thalamas, C.; et al. CD36 Is a Marker of Human Adipocyte Progenitors with Pronounced Adipogenic and Triglyceride Accumulation Potential. Stem Cells 2017, 35, 1799–1814. [Google Scholar] [CrossRef]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutr. 2012, 32, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Caso, G.; McNurlan, M.A.; Mileva, I.; Zemlyak, A.; Mynarcik, D.C.; Gelato, M.C. Peripheral fat loss and decline in adipogenesis in older humans. Metabolism 2013, 62, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Alt, E.U.; Senst, C.; Murthy, S.N.; Slakey, D.P.; Dupin, C.L.; Chaffin, A.E.; Kadowitz, P.J.; Izadpanah, R. Aging alters tissue resident mesenchymal stem cell properties. Stem Cell Res. 2012, 8, 215–225. [Google Scholar] [CrossRef]
- Palmer, A.K.; Kirkland, J.L. Aging and adipose tissue: Potential interventions for diabetes and regenerative medicine. Exp. Gerontol. 2016, 86, 97–105. [Google Scholar] [CrossRef]
- Campisi, J.; di Fagagna, F.D.A. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Childs, B.G.; Li, H.; van Deursen, J.M. Senescent cells: A therapeutic target for cardiovascular disease. J. Clin. Investig. 2018, 128, 1217–1228. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1233. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A senescent cell bystander effect: Senescence-induced senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [PubMed]
- LeBrasseur, N.K.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and the Biology of Aging, Disease, and Frailty. In Frailty: Pathophysiology, Phenotype and Patient Care; Karger Publishers: Basel, Switzerland, 2015; Volume 83, pp. 11–18. [Google Scholar]
- Tchkonia, T.; Morbeck, D.E.; Von Zglinicki, T.; Van Deursen, J.; Lustgarten, J.; Scrable, H.; Khosla, S.; Jensen, M.D.; Kirkland, J.L. Fat tissue, aging, and cellular senescence. Aging Cell 2010, 9, 667–684. [Google Scholar] [CrossRef]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological Aging: Links Among Adipose Tissue Dysfunction, Diabetes, and Frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef]
- Justice, J.N.; Gregory, H.; Tchkonia, T.; LeBrasseur, N.K.; Kirkland, J.L.; Kritchevsky, S.B.; Nicklas, B.J. Cellular Senescence Biomarker p16INK4a+ Cell Burden in Thigh Adipose is Associated With Poor Physical Function in Older Women. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 939–945. [Google Scholar] [CrossRef]
- Stout, M.B.; Tchkonia, T.; Pirtskhalava, T.; Palmer, A.K.; List, E.O.; Berryman, D.E.; Lubbers, E.R.; Escande, C.; Spong, A.; Masternak, M.M.; et al. Growth hormone action predicts age-related white adipose tissue dysfunction and senescent cell burden in mice. Aging (Albany NY) 2014, 6, 575–586. [Google Scholar] [CrossRef]
- Valli, A.; Harris, A.L.; Kessler, B.M. Hypoxia metabolism in ageing. Aging (Albany NY) 2015, 7, 465–466. [Google Scholar] [CrossRef][Green Version]
- Dalmas, E.; Clement, K.; Guerre-Millo, M. Defining macrophage phenotype and function in adipose tissue. Trends Immunol. 2011, 32, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ren, Z.; Pae, M.; Guo, W.; Cui, X.; Merrill, A.H.; Meydani, S.N. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J. Immunol. 2007, 179, 4829–4839. [Google Scholar] [CrossRef] [PubMed]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of Inflammation in Age-Related Sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef] [PubMed]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Cerk, I.K.; Wechselberger, L.; Oberer, M. Adipose Triglyceride Lipase Regulation: An Overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed]
- Danforth, E., Jr. Failure of adipocyte differentiation causes type II diabetes mellitus? Nat. Genet. 2000, 26, 13. [Google Scholar] [CrossRef]
- Gustafson, B.; Hedjazifar, S.; Gogg, S.; Hammarstedt, A.; Smith, U. Insulin resistance and impaired adipogenesis. Trends Endocrinol. Metab. 2015, 26, 193–200. [Google Scholar] [CrossRef]
- Unger, R.H.; Scherer, P.E. Gluttony, sloth and the metabolic syndrome: A roadmap to lipotoxicity. Trends Endocrinol. Metab. 2010, 21, 345–352. [Google Scholar] [CrossRef]
- Wang, M.Y.; Grayburn, P.; Chen, S.; Ravazzola, M.; Orci, L.; Unger, R.H. Adipogenic capacity and the susceptibility to type 2 diabetes and metabolic syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6139–6144. [Google Scholar] [CrossRef]
- Guo, W.; Pirtskhalava, T.; Tchkonia, T.; Xie, W.; Thomou, T.; Han, J.; Wang, T.; Wong, S.; Cartwright, A.; Hegardt, F.G.; et al. Aging results in paradoxical susceptibility of fat cell progenitors to lipotoxicity. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1041–E1051. [Google Scholar] [CrossRef] [PubMed]
- Berkes, C.A.; Tapscott, S.J. MyoD and the transcriptional control of myogenesis. Semin. Cell Dev. Biol. 2005, 16, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M. Cytoskeletal protein kinases: Titin and its relations in mechanosensing. Pflugers Arch. 2011, 462, 119–134. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J. Roles of titin in the structure and elasticity of the sarcomere. J. Biomed. Biotechnol. 2010, 2010, 612482. [Google Scholar] [CrossRef]
- Zhu, S.; Tian, Z.; Torigoe, D.; Zhao, J.; Xie, P.; Sugizaki, T.; Sato, M.; Horiguchi, H.; Terada, K.; Kadomatsu, T.; et al. Aging- and obesity-related peri-muscular adipose tissue accelerates muscle atrophy. PLoS ONE 2019, 14, e0221366. [Google Scholar] [CrossRef]
- Koster, A.; Patel, K.V.; Visser, M.; van Eijk, J.T.; Kanaya, A.M.; de Rekeneire, N.; Newman, A.B.; Tylavsky, F.A.; Kritchevsky, S.B.; Harris, T.B.; et al. Joint effects of adiposity and physical activity on incident mobility limitation in older adults. J. Am. Geriatr. Soc. 2008, 56, 636–643. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Carvalho, F.G.; Justice, J.N.; Freitas, E.C.d.; Kershaw, E.E.; Sparks, L.M. Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality. Nutrients 2019, 11, 2553. https://doi.org/10.3390/nu11112553
De Carvalho FG, Justice JN, Freitas ECd, Kershaw EE, Sparks LM. Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality. Nutrients. 2019; 11(11):2553. https://doi.org/10.3390/nu11112553
Chicago/Turabian StyleDe Carvalho, Flavia G., Jamie N. Justice, Ellen C. de Freitas, Erin E. Kershaw, and Lauren M. Sparks. 2019. "Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality" Nutrients 11, no. 11: 2553. https://doi.org/10.3390/nu11112553
APA StyleDe Carvalho, F. G., Justice, J. N., Freitas, E. C. d., Kershaw, E. E., & Sparks, L. M. (2019). Adipose Tissue Quality in Aging: How Structural and Functional Aspects of Adipose Tissue Impact Skeletal Muscle Quality. Nutrients, 11(11), 2553. https://doi.org/10.3390/nu11112553