Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation

 , , ,
, , ,  ,
,  ,
, 
 ,
,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Quantitative Real-Time PCR
2.3. Western Blotting
2.4. Immunofluorescence
2.5. Cell Death Analysis by Flow Cytometry
2.6. High-Performance Liquid Chromatography (HPLC)
2.7. Synthesis of SeChry
2.8. Synthesis of Folate-Targeted Polyurea Dendrimer Generation Four (PUREG4-FA) Nanoparticles
2.9. Preparation of SeChry@PUREG4-FA Nanoparticles
2.10. Inhibition of H2S-Synthesizing Enzymes by SeChry
2.11. Statistical Analysis
3. Results
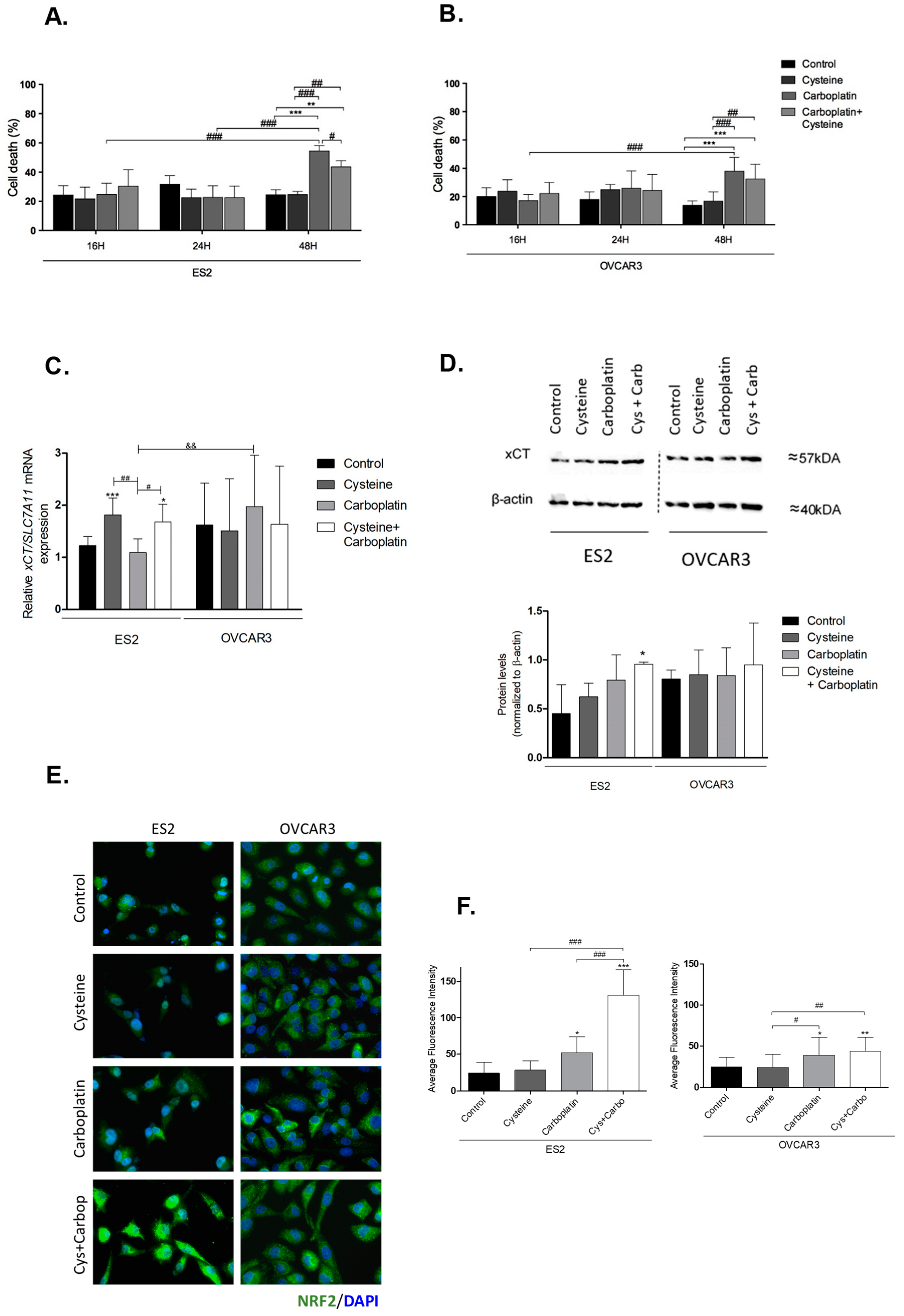
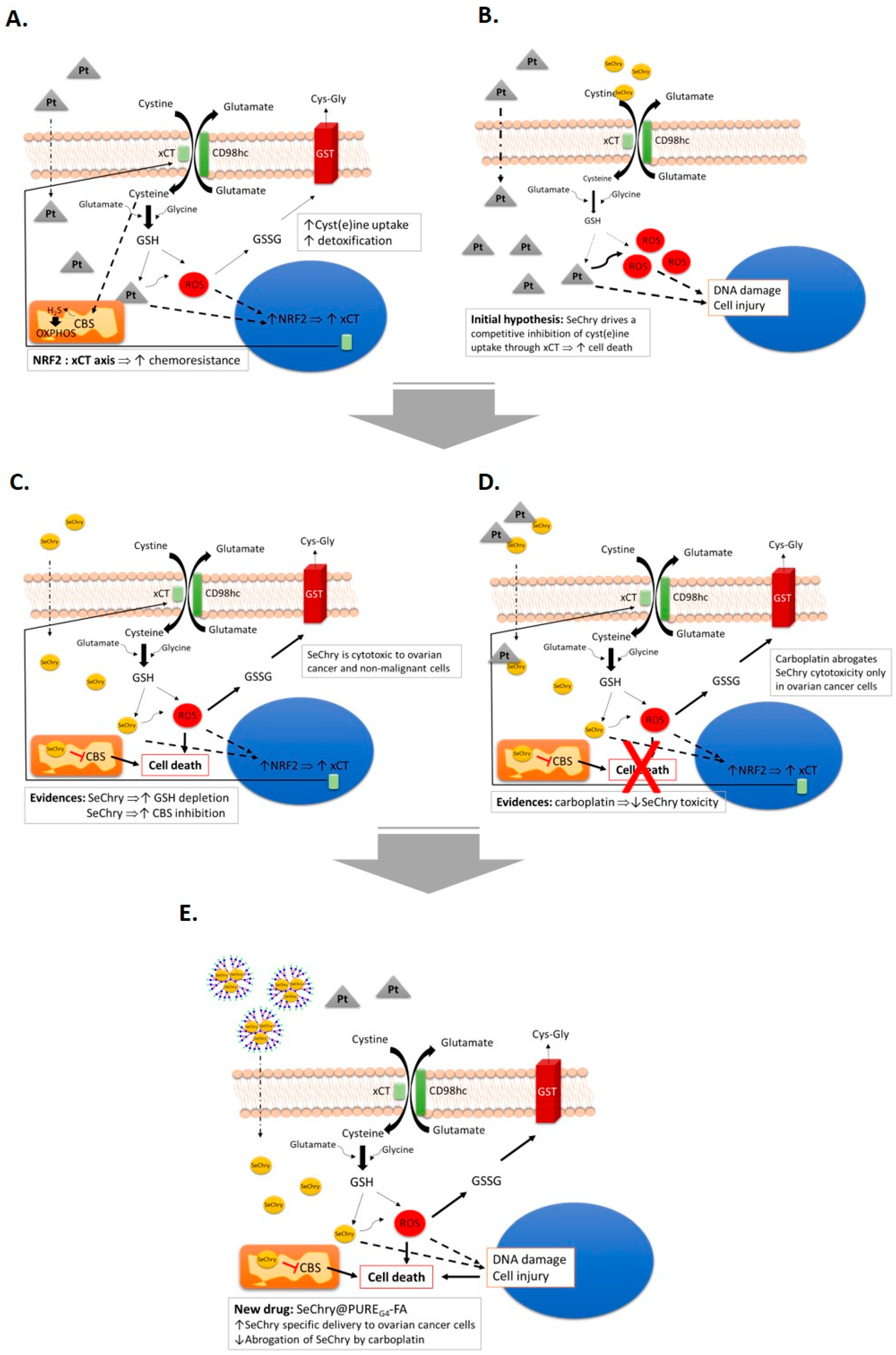
3.1. Cysteine Partially Abrogates Platinum-Induced Cell Death and Modulates xCT Expression
3.2. Cysteine and Carboplatin Modulate the Expression of Nrf2
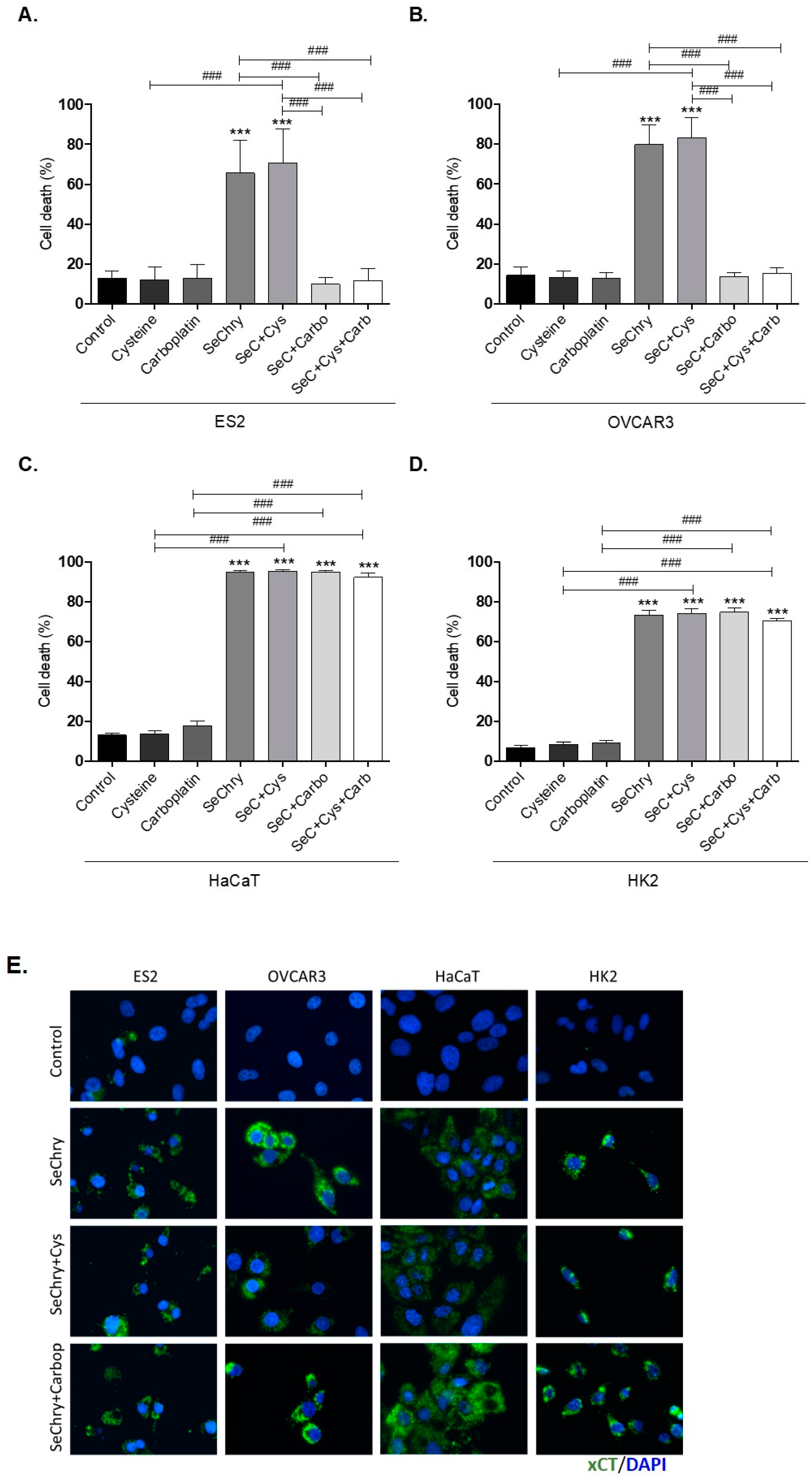
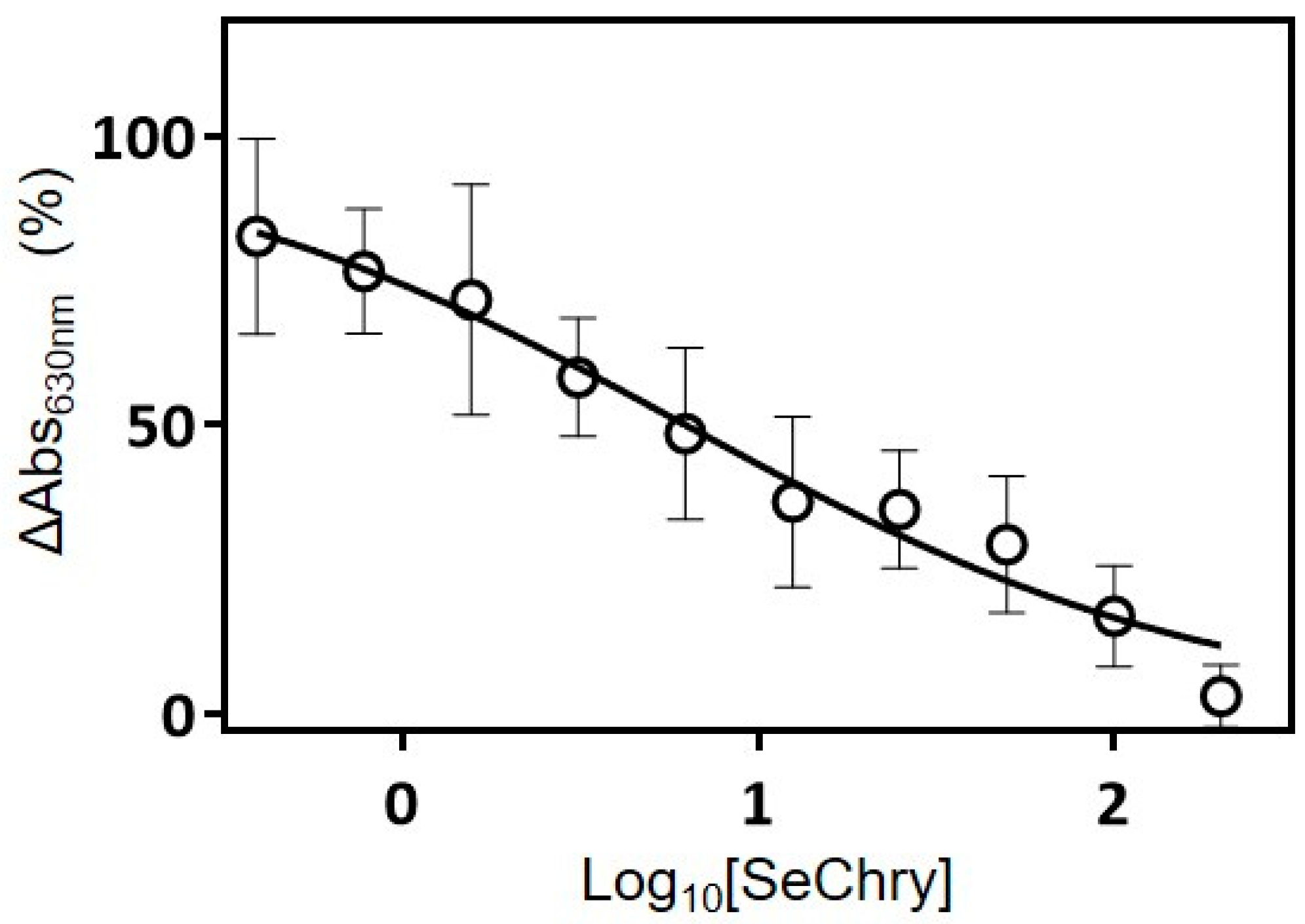
3.3. SeChry Induces Cell Death and xCT Expression in Ovarian Cancer Cells
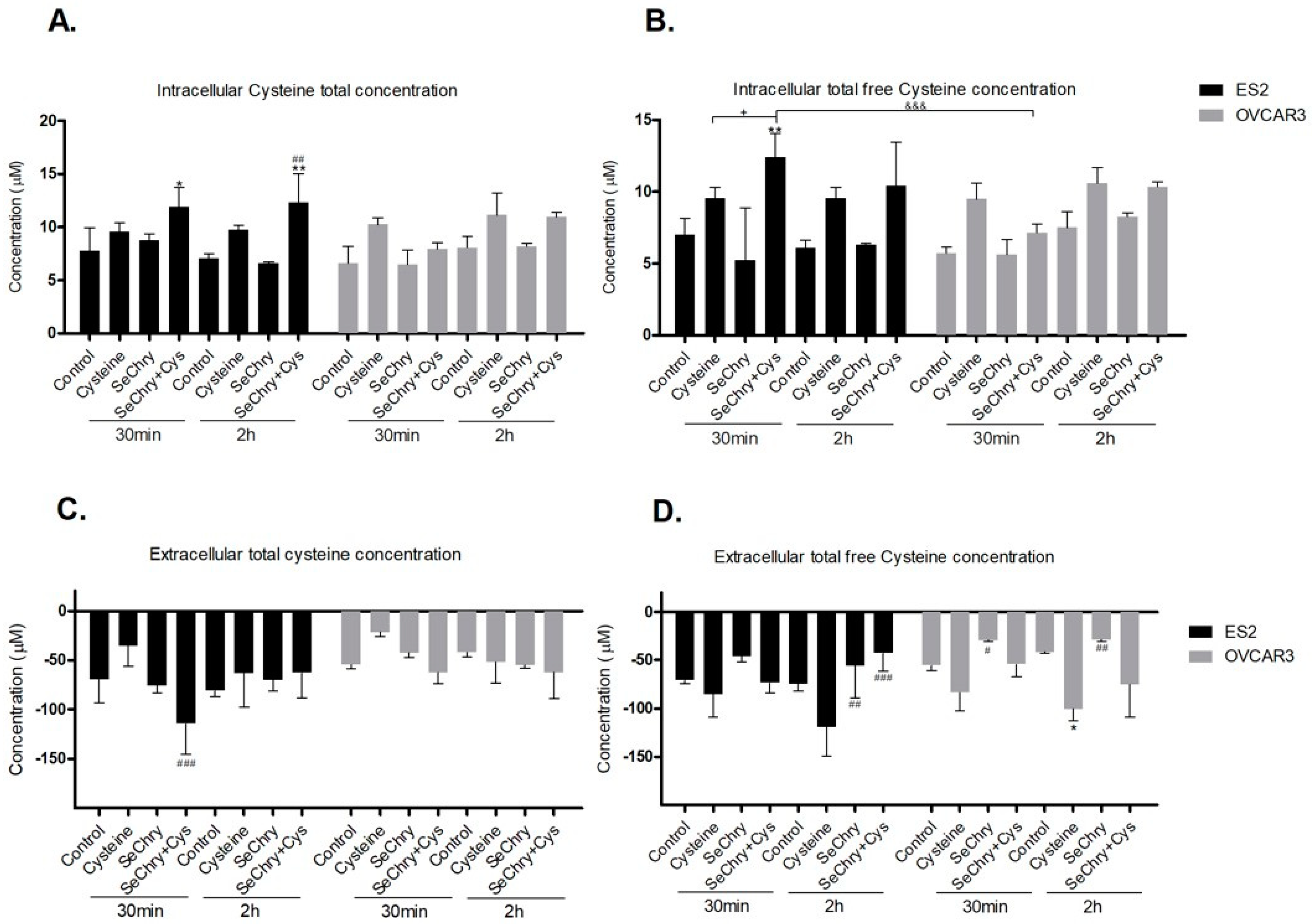
3.4. SeChry Does Not Impair But Enhances Cysteine Uptake
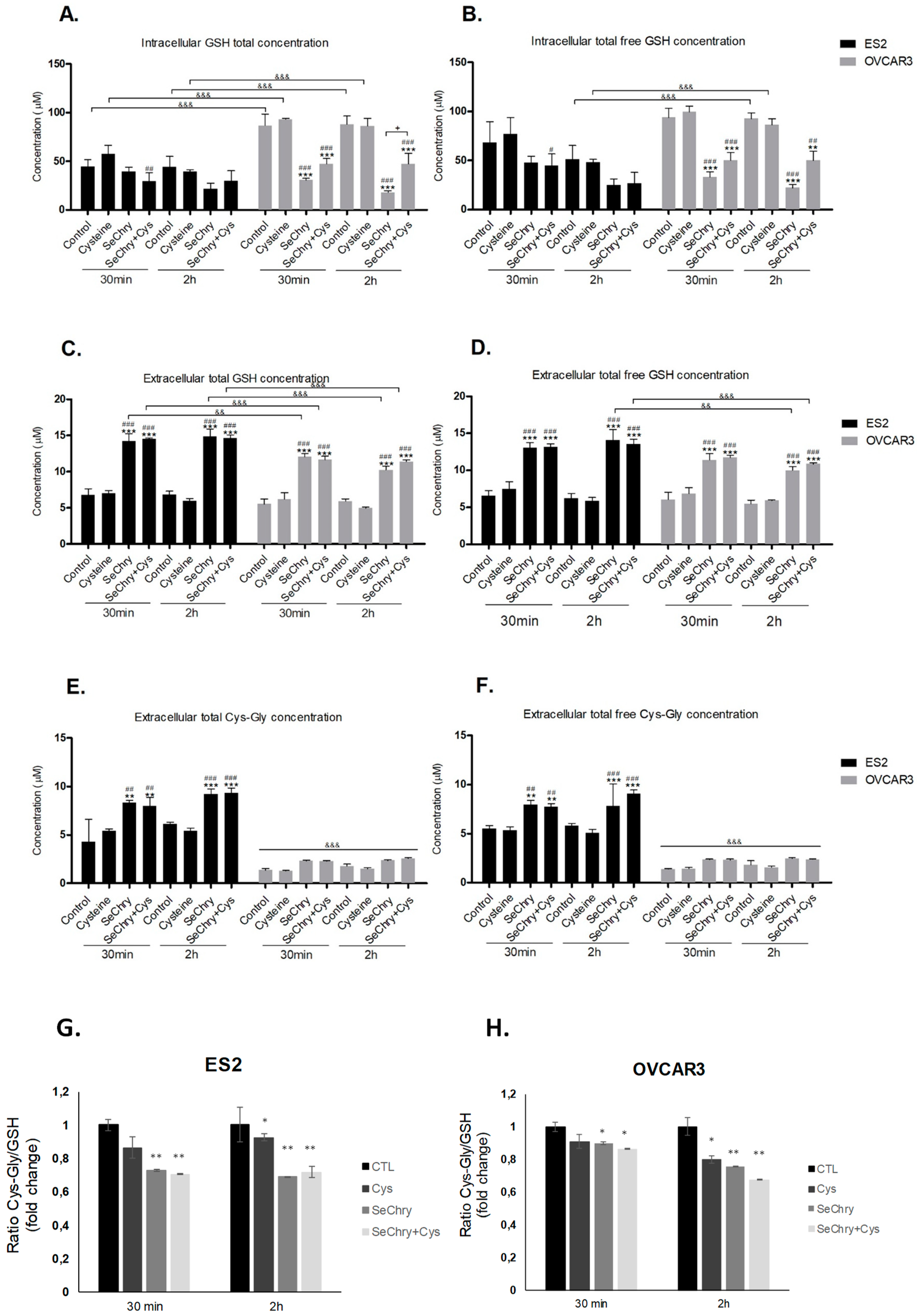
3.5. SeChry Enhances Glutathione (GSH) Turnover
3.6. SeChry Inhibits Cystathionine β-Synthase (CBS)
3.7. SeChry Delivery by PUREG4-FA Nanoparticles Allows the More Specific Targeting of Ovarian Cancer Cells, While Reducing Cytotoxicity in Non-Malignant Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Brett, M.R.; Jennifer, B.P.; Thomas, A.S.; Brett, M.R.; Jennifer, B.P.; Thomas, A.S. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef]
- Webb, P.M.; Jordan, S.J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef]
- Williams, T.I.; Toups, K.L.; Saggese, D.A.; Kalli, K.R.; Cliby, W.A.; Muddiman, D.C. Epithelial ovarian cancer: Disease etiology, treatment, detection, and investigational gene, metabolite, and protein biomarkers. J. Proteome Res. 2007, 6, 2936–2962. [Google Scholar] [CrossRef]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary ovarian cancer: Not only BRCA 1 and 2 genes. Biomed. Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef]
- Ho, G.Y.; Woodward, N.; Coward, J.I.G. Cispaltin versus carboplatin: Comparative review of therapeutic management in solid malignancies. Crit. Rev. Oncol. Hematol. 2016, 102, 37–46. [Google Scholar] [CrossRef]
- Rabik, C.A.; Dolan, M.E. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat. Rev. 2007, 33, 9–23. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef]
- He, P.-J.; Ge, R.-F.; Mao, W.J.; Chung, P.-S.; Ahn, J.-C.; Wu, H.-T. Oxidative stress induced by carboplatin promotes apoptosis and inhibits migration of HN‑3 cells. Oncol. Lett. 2018, 16, 7131–7138. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Cao, X.; Ding, L.; Xie, Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.S. A review of hydrogen sulfide synthesis, metabolism and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef]
- Johnson, S.; Ozols, R.; Hamilton, T. Mechanisms of drug resistance in ovarian cancer. Cancer 1993, 71, 644–649. [Google Scholar] [CrossRef]
- Ling, K.S.; Chen, G.D.; Tsai, H.J.; Lee, M.S.; Wang, P.H.; Liu, F.S. Mechanisms involved in chemoresistance in ovarian cancer. Taiwan J. Obstet. Gynecol. 2005, 44, 209–217. [Google Scholar] [CrossRef]
- Mytilineou, C.; Kramer, B.C.; Yabut, J.A. Glutathione depletion and oxidative stress. Parkinsonism Relat. Disord. 2002, 8, 385–387. [Google Scholar] [CrossRef]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef]
- Dhivya, H. Glutathione—A master antioxidant and an immune system modulator. J. Biol. Inf. Sci. 2012, 1, 28–30. [Google Scholar]
- Nunes, S.C.; Serpa, J. Glutathione in ovarian cancer: A double-edged sword. Int. J. Mol. Sci. 2018, 19, 1882. [Google Scholar] [CrossRef]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef]
- Prakash, M.; Shetty, M.S.; Tilak, P.; Anwar, N. Total thiols: Biomedical importance and their alteration in various disorders. Online J. Health Allied Sci. 2009, 8, 1–9. [Google Scholar]
- Burdo, J.; Dargusch, R.; Schubert, D. Distribution of the cystine/glutamate antiporter system xc-in the brain, kidney, and duodenum. J. Histochem. Cytochem. 2006, 54, 549–557. [Google Scholar] [CrossRef]
- Poisson, L.M.; Munkarah, A.; Madi, H.; Datta, I.; Hensley-Alford, S.; Tebbe, C.; Buekers, T.; Giri, S.; Rattan, R. A metabolomic approach to identifying platinum resistance in ovarian cancer. J. Ovarian Res. 2015, 8, 13. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc− in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Lopes-Coelho, F.; Gouveia-Fernandes, S.; Gonçalves, L.G.; Nunes, C.; Faustino, I.; Silva, F.; Félix, A.; Pereira, S.A.; Serpa, J. HNF1β drives glutathione (GSH) synthesis underlying intrinsic carboplatin resistance of ovarian clear cell carcinoma (OCCC). Tumor Biol. 2016, 37, 4813–4829. [Google Scholar] [CrossRef]
- Nunes, S.C.; Ramos, C.; Lopes-Coelho, F.; Sequeira, C.O.; Silva, F.; Gouveia-Fernandes, S.; Rodrigues, A.; Guimarães, A.; Silveira, M.; Abreu, S.; et al. Cysteine allows ovarian cancer cells to adapt to hypoxia and to escape from carboplatin cytotoxicity. Sci. Rep. 2018, 8, 8513. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Murphy, B.; Mustafi, S.B.; Dey, A.; Bhattacharya, R.; Mukherjee, P. Cystathionine b-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Roy-Chowdhuri, S.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine β-synthase in human breast Cancer. Free Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc(−) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Shin, C.S.; Mishra, P.; Watrous, J.D.; Carelli, V.; D’Aurelio, M.; Jain, M.; Chan, D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017, 8, 15074. [Google Scholar] [CrossRef]
- Wang, S.-F.; Chen, M.-S.; Chou, Y.-C.; Ueng, Y.-F.; Yin, P.-H.; Yeh, T.-S.; Lee, H.-C. Mitochondrial dysfunction enhances cisplatin resistance in human gastric cancer cells via the ROS-activated GCN2-eIF2α-ATF4-xCT pathway. Oncotarget 2016, 7, 74132. [Google Scholar] [CrossRef]
- Bannai, S. Induction of cystine and glutamate transport activity in human fibroblasts by diethylmaleate and other electrophilic agents. J. Biol. Chem. 1984, 259, 2435–2440. [Google Scholar]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Aboody, K.S. SLC7A11 overexpression in glioblastoma is associated with increased cancer stem cell-like properties. Stem. Cells Dev. 2017, 26, 1236–1246. [Google Scholar] [CrossRef]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadée, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef]
- Huang, Y. Pharmacogenetics/genomics of membrane transporters in cancer chemotherapy. Cancer Metastasis Rev. 2007, 26, 183–201. [Google Scholar] [CrossRef]
- Chen, L.; Li, X.; Liu, L.; Yu, B.; Xue, Y.; Liu, Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-γ-lyase function. Oncol. Rep. 2015, 33, 1465–1474. [Google Scholar] [CrossRef]
- Nagane, M.; Kanai, E.; Shibata, Y.; Shimizu, T.; Yoshioka, C. Sulfasalazine, an inhibitor of the cystine-glutamate antiporter, reduces DNA damage repair and enhances radiosensitivity in murine B16F10 melanoma. PLoS ONE 2018, 13, e0195151. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef]
- Yan, M.; Fan, J.; Wang, L.; Kuang, R. Sulfasalazine inhibits inflammation and fibrogenesis in pancreas via NF-κB signaling pathway in rats with oxidative stress-induced pancreatic injury. Drug Des. Dev. Ther. 2016, 10, 1743–1751. [Google Scholar] [CrossRef][Green Version]
- Tobe, T.; Ueda, K.; Aoki, A.; Okamoto, Y.; Kojima, N.; Jinno, H. Selenium uptake through cystine transporter mediated by glutathione conjugation. J. Toxicol. Sci. 2017, 42, 85–91. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Gundimeda, U.; Zhou, S.; Zung, K.; Forell, K.; Holmgren, A. Imbalance in protein thiol redox regulation and cancer-preventive efficacy of selenium. React. Oxyg. Species 2016, 2, 272–289. [Google Scholar] [CrossRef][Green Version]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef]
- Caffrey, P.B.; Frenkel, G.D. Prevention of carboplatin-induced resistance in human ovarian tumor xenografts by selenite. Anticancer Res. 2013, 33, 4249–4254. [Google Scholar]
- Lee, K.H.; Jeong, D. Bimodal actions of selenium essential for antioxidant and toxic pro-oxidant activities: The selenium paradox (Review). Mol. Med. Rep. 2012, 5, 299–304. [Google Scholar] [CrossRef]
- Björnstedt, M.; Fernandes, A.P. Selenium in the prevention of human cancers. EPMA J. 2010, 1, 389–395. [Google Scholar] [CrossRef]
- Olm, E.; Fernandes, A.P.; Hebert, C.; Rundlöf, A.-K.; Larsen, E.H.; Danielsson, O.; Björnstedt, M. Extracellular thiol-assisted selenium uptake dependent on the xc- cystine transporter explains the cancer-specific cytotoxicity of selenite. Proc. Natl. Acad. Sci. USA 2009, 106, 11400–11405. [Google Scholar] [CrossRef]
- Martins, I.L.; Charneira, C.; Gandin, V.; Ferreira da Silva, J.L.; Justino, G.C.; Telo, J.P.; Vieira, A.J.; Marzano, C.; Antunes, A.M. Selenium-containing chrysin and quercetin derivatives: Attractive scaffolds for cancer therapy. J. Med. Chem. 2015, 58, 4250–4265. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef]
- Rauf, A.; Khan, R.; Raza, M.; Khan, H.; Pervez, S.; De Feo, V.; Maione, F.; Mascolo, N. Suppression of inflammatory response by chrysin, a flavone isolated from Potentilla evestita Th. Wolf. In silico predictive study on its mechanistic effect. Fitoterapia 2015, 103, 129–135. [Google Scholar] [CrossRef]
- Gonçalves-Dias, C.; Morello, J.; Semedo, V.; Correia, M.J.; Coelho, N.R.; Monteiro, E.C.; Antunes, A.M.M.; Pereira, S.A. The mercapturomic profile of health and non-communicable diseases. High Throughput 2019, 8, 10. [Google Scholar] [CrossRef]
- Grilo, N.M.; João Correia, M.; Miranda, J.P.; Cipriano, M.; Serpa, J.; Marques, M.; Monteiro, E.C.; Antunes, A.M.M.; Diogo, L.N.; Pereira, S.A. Unmasking efavirenz neurotoxicity: Time matters to the underlying mechanisms. Eur. J. Pharm. Sci. 2017, 105, 47–54. [Google Scholar] [CrossRef]
- Restani, R.B.; Morgado, P.I.; Ribeiro, M.P.; Correia, I.J.; Aguiar-Ricardo, A.; Bonifácio, V.D.B. Biocompatible polyurea dendrimers with pH-dependent fluorescence. Angew. Chem. Int. Ed. 2012, 51, 5162–5165. [Google Scholar] [CrossRef]
- Yoon, K.; Harris, J.M.; Bentley, M.D.; Fang, Z.; Viegas, T. Multifunctional Forms of Polyoxazoline Copolymers and Drug Compositions Comprising the Same. U.S. Patent 8,501,899, 23 January 2012. [Google Scholar]
- Restani, R.B.; Conde, J.; Pires, R.F.; Martins, P.; Fernandes, A.R.; Baptista, P.V.; Bonifácio, V.D.B.; Aguiar-Ricardo, A. POxylated polyurea dendrimers: Smart core-shell vectors with IC50 lowering capacity. Macromol. Biosci. 2015, 15, 1045–1051. [Google Scholar] [CrossRef]
- Zuhra, K.; Sousa, P.M.F.; Paulini, G.; Lemos, A.R.; Kalme, Z.; Bisenieks, I.; Bisenieks, E.; Vigante, B.; Duburs, G.; Bandeiras, T.M.; et al. Screening pyridine derivatives against human hydrogen sulfide-synthesizing enzymes by orthogonal methods. Sci. Rep. 2019, 9, 684. [Google Scholar] [CrossRef]
- Nunes, S.C.; Lopes-Coelho, F.; Gouveia-Fernandes Sofia Ramos, C.; Pereira, S.A.; Serpa, J. Cysteine boosters the evolutionary adaptation to CoCl2 mimicked hypoxia conditions, favouring carboplatin resistance in ovarian cancer. BMC Evol. Biol. 2018, 18, 97. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; Calaza, K.C. Alterations in system expression in the retina of type 1 diabetic rats and the role of Nrf2. Mol. Neurobiol. 2018, 55, 7941–7948. [Google Scholar] [CrossRef]
- Khamari, R.; Trinh, A.; Gabert, P.E.; Corazao-Rozas, P.; Riveros-Cruz, S.; Balayssac, S.; Malet-Martino, M.; Dekiouk, S.; Joncquel Chevalier Curt, M.; Maboudou, P.; et al. Glucose metabolism and NRF2 coordinate the antioxidant response in melanoma resistant to MAPK inhibitors. Cell Death Dis. 2018, 9, 325. [Google Scholar] [CrossRef]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef]
- Wang, R. Phisiologycal implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Giuffrè, A.; Vicente, J.B. Hydrogen sulfide biochemistry and interplay with other gaseous. Oxidative Med. Cell. Longev. 2018, 2018, 6290931. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal. 2014, 20, 770–782. [Google Scholar] [CrossRef]
- Rizzo, A.; Napoli, A.; Roggiani, F.; Tomassetti, A.; Id, D.M. One-carbon metabolism: Biological players in epithelial ovarian cancer. Int. J. Mol. Sci. 2018, 19, 2092. [Google Scholar] [CrossRef]
- Wang, X.; Yang, R.; Yuan, C.; An, Y.; Tang, Q.; Chen, D. Preparation of folic acid-targeted temperature-sensitive magnetoliposomes and their antitumor effects in vitro and in vivo. Target. Oncol. 2018, 13, 481–494. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef]
- Stewart, D.J. Mechanisms of resistance to cisplatin and carboplatin. Crit. Rev. Oncol. Hematol. 2007, 63, 12–31. [Google Scholar] [CrossRef]
- Townsend, D.M.; Deng, M.; Zhang, L.; Lapus, M.G.; Hanigan, M.H. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003, 14, 1–10. [Google Scholar] [CrossRef]
- Ye, J.-L.; Gao, C.-Q.; Li, X.-G.; Jin, C.L.; Wang, D.; Shu, G.; Wang, W.C.; Kong, X.F.; Yao, K.; Yan, H.C.; et al. EAAT3 promotes amino acid transport and proliferation of porcine intestinal epithelial cells. Oncotarget 2016, 7, 38681. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, Z.; Song, R. Antisense lncRNA As-SLC7A11 suppresses epithelial ovarian cancer progression mainly by targeting SLC7A11. Pharmazie 2017, 72, 402–407. [Google Scholar] [CrossRef]
- O’Kane, R.L.; Martínez-López, I.; Dejoseph, M.R.; Viña, J.R.; Hawkins, R.A. Na(+)-dependent glutamate transporters (EAAT1, EAAT2, and EAAT3) of the blood-brain barrier. A mechanism for glutamate removal. J. Biol. Chem. 1999, 274, 31891–31895. [Google Scholar] [CrossRef] [PubMed]
- Furfaro, A.L.; Traverso, N.; Domenicotti, C.; Piras, S.; Moretta, L.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. The Nrf2/HO-1 axis in cancer cell growth and chemoresistance. Oxidative Med. Cell. Longev. 2016, 2016, 1958174. [Google Scholar] [CrossRef] [PubMed]
- Traverso, N.; Ricciarelli, R.; Nitti, M.; Marengo, B.; Furfaro, A.L.; Pronzato, M.A.; Marinari, U.M.; Domenicotti, C. Role of glutathione in cancer progression and chemoresisstance. Oxidative Med. Cell. Longev. 2013, 2013, 972913. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.M. Endometrial cancer. N. Engl. J. Med. 1980, 303, 522. [Google Scholar] [CrossRef]
- Hourihan, J.M.; Kenna, J.G.; Hayes, J.D. The gasotransmitter hydrogen sulfide induces Nrf2-target genes by inactivating the Keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between Cys-226 and Cys-613. Antioxid. Redox Signal. 2013, 19, 465–481. [Google Scholar] [CrossRef]
- Chakraborty, P.K.; Xiong, X.; Mustafi, S.B.; Bhattacharya, R.; Mukherjee, P. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget 2015, 6, 37367–37384. [Google Scholar] [CrossRef]
- Collery, P. Strategies for the development of selenium-based anticancer drugs. J. Trace Elem. Med. Biol. 2018, 50, 498–507. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Coletta, C.; Chao, C.; Szabo, C. The therapeutic potential of cystathionine-b-synthetase/hydrogen sulfide inhibition in cancer. Antioxid. Redox Signal. 2015, 22, 424–448. [Google Scholar] [CrossRef]
- Nichols, J.W.; Han, Y. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef]
- Palakurthi, S.; Yellepeddi, V.K.; Vangara, K.K. Recent trends in cancer drug resistance reversal strategies using nanoparticles. Expert Opin. Drug Deliv. 2012, 9, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Brannon-Peppas, L.; Blanchette, J.O. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Deliv. Rev. 2012, 64, 206–212. [Google Scholar] [CrossRef]
- Payne, N.C.; Geissler, A.; Button, A.; Sasuclark, A.R.; Schroll, A.L.; Ruggles, E.L.; Gladyshev, V.N.; Hondal, R.J. Comparison of the redox chemistry of sulfur- and selenium-containing analogs of uracil. Free Radic. Biol. Med. 2017, 104, 249–261. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, I.; Ramos, C.; Mendes, C.; Sequeira, C.O.; Tomé, C.S.; Fernandes, D.G.H.; Mota, P.; Pires, R.F.; Urso, D.; Hipólito, A.; et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation. Nutrients 2019, 11, 2523. https://doi.org/10.3390/nu11102523
Santos I, Ramos C, Mendes C, Sequeira CO, Tomé CS, Fernandes DGH, Mota P, Pires RF, Urso D, Hipólito A, et al. Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation. Nutrients. 2019; 11(10):2523. https://doi.org/10.3390/nu11102523
Chicago/Turabian StyleSantos, Inês, Cristiano Ramos, Cindy Mendes, Catarina O. Sequeira, Catarina S. Tomé, Dalila G.H. Fernandes, Pedro Mota, Rita F. Pires, Donato Urso, Ana Hipólito, and et al. 2019. "Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation" Nutrients 11, no. 10: 2523. https://doi.org/10.3390/nu11102523
APA StyleSantos, I., Ramos, C., Mendes, C., Sequeira, C. O., Tomé, C. S., Fernandes, D. G. H., Mota, P., Pires, R. F., Urso, D., Hipólito, A., Antunes, A. M. M., Vicente, J. B., Pereira, S. A., Bonifácio, V. D. B., Nunes, S. C., & Serpa, J. (2019). Targeting Glutathione and Cystathionine β-Synthase in Ovarian Cancer Treatment by Selenium–Chrysin Polyurea Dendrimer Nanoformulation. Nutrients, 11(10), 2523. https://doi.org/10.3390/nu11102523