Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides


Abstract
1. Introduction
2. Materials and Methods
2.1. Material
2.2. Enzyme Activity Test of TG2
2.3. Isopeptide Standard
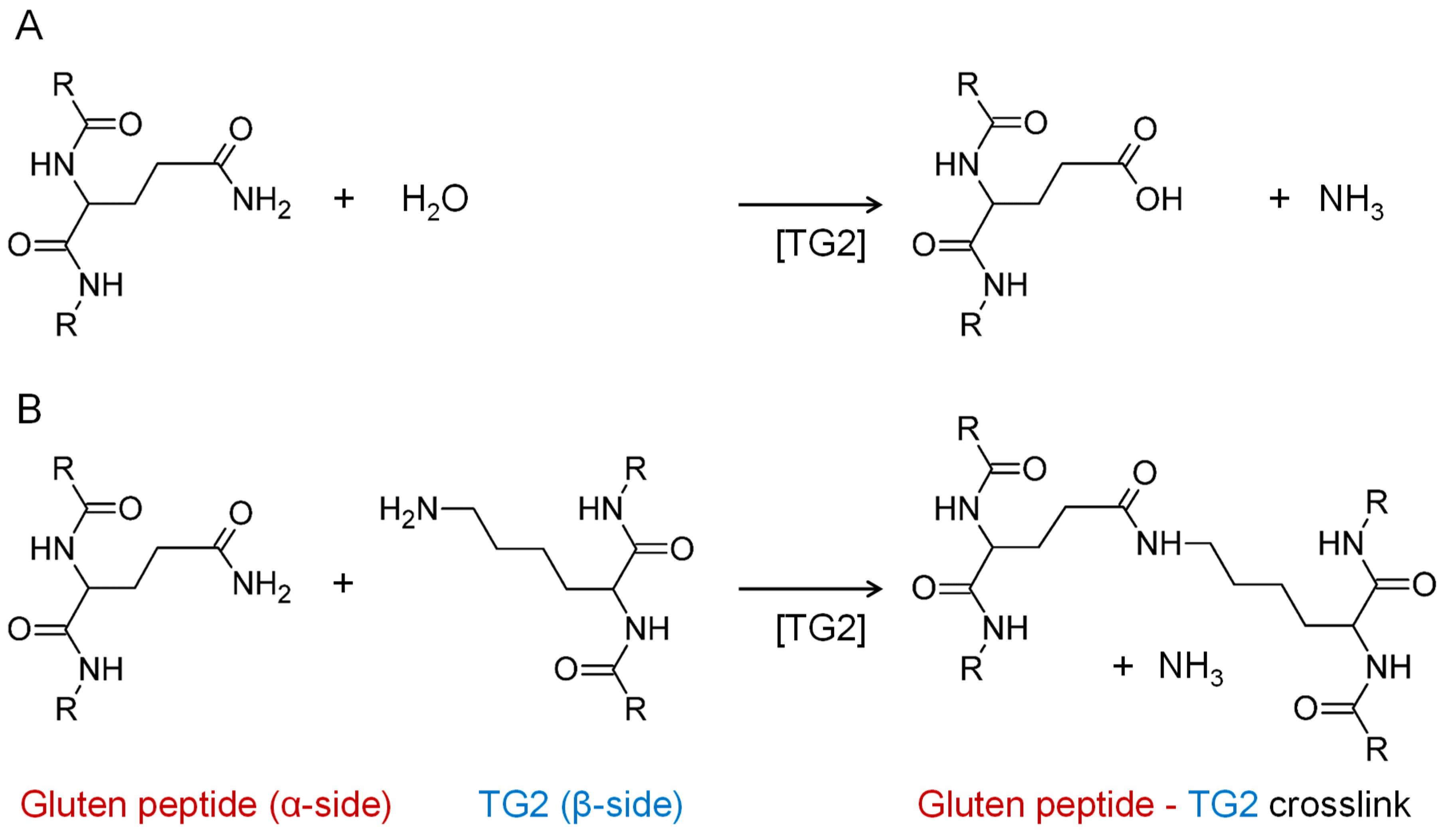
2.4. Model Reaction of TG2 and PepQ
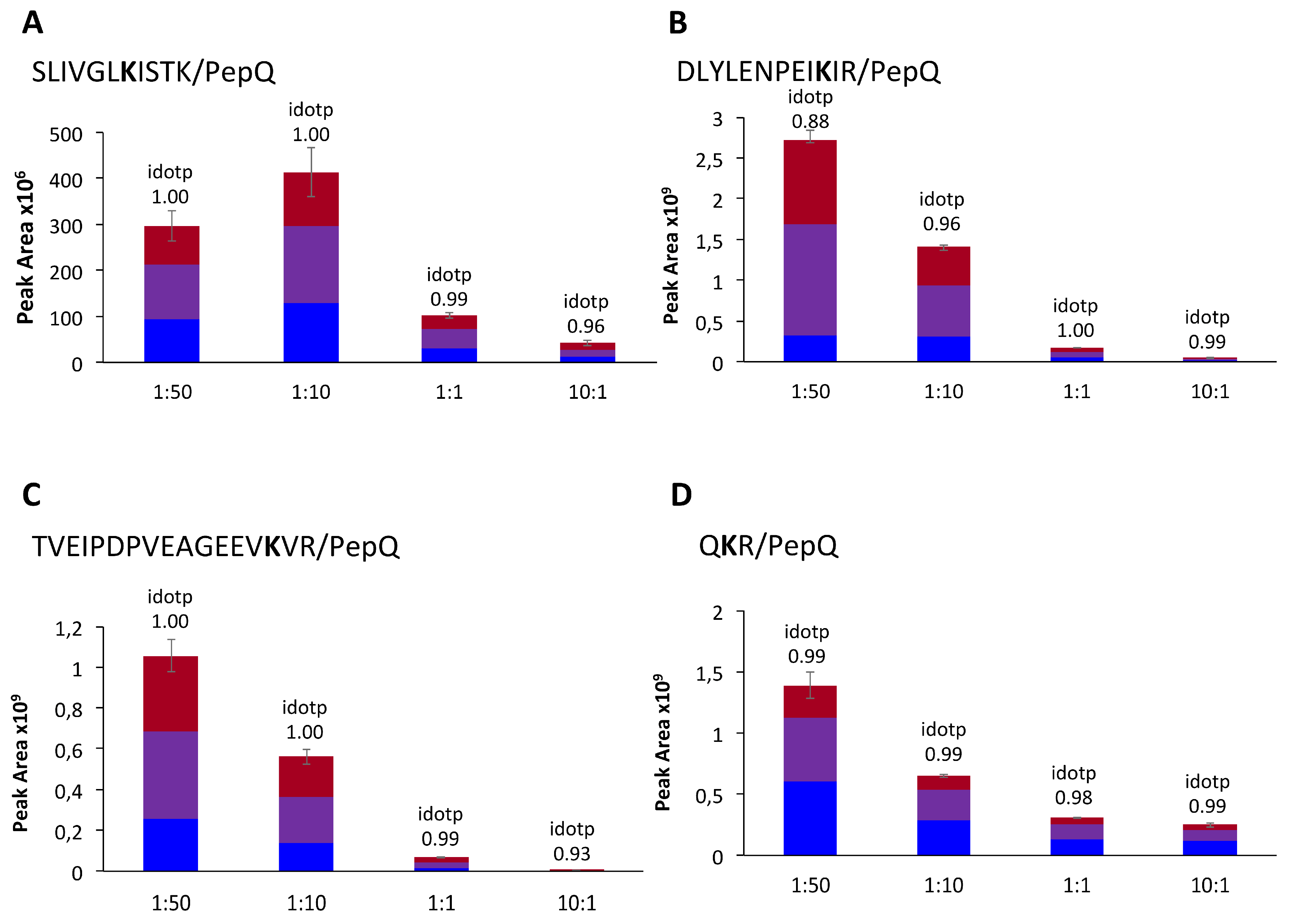
2.5. Model Reaction at Different Molar Ratios
2.6. Model Reaction with Three Different Model Peptides
2.7. Tryptic Digestion and Clean-Up by Solid Phase Extraction
2.8. Nanoscale Liquid Chromatography-Tandem Mass Spectrometry
2.9. Isopeptide Identification Using MaxQuant
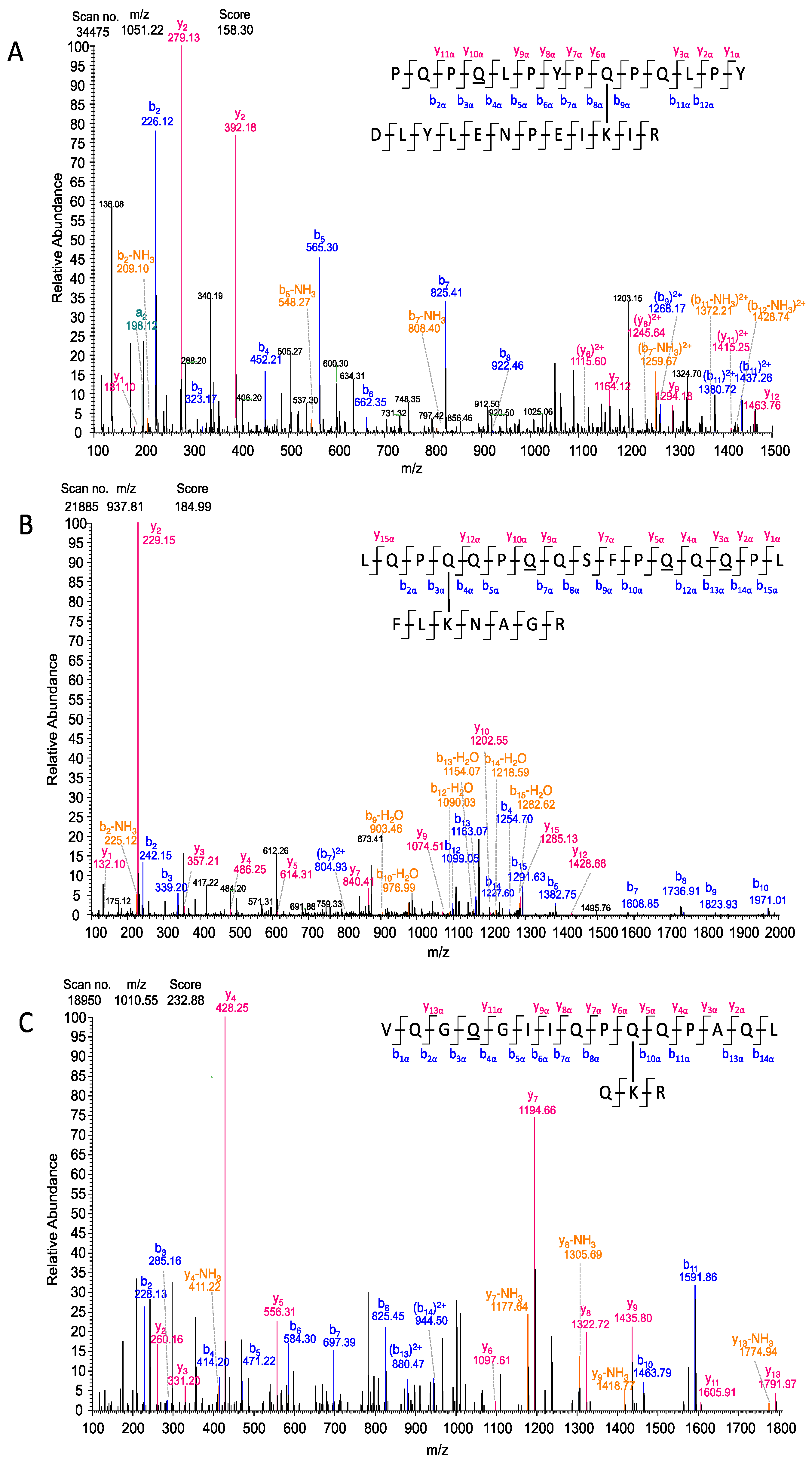
2.10. Assignment of MS/MS Fragments of the Isopeptide Sequences Using ProteinProspector
2.11. Isopeptide Confirmation Using Skyline
2.12. Isopeptide Identification Using pLink
2.13. 3D-Structure Model of TG2
3. Results
3.1. Determination of TG2 Enzyme Activity
3.2. Identification of the Isopeptide Standard
3.3. Identification of TG2-Peptides Involved in Isopeptide Formation
3.4. Identification of TG2-TG2 Crosslinks
3.5. Identification of Crosslinks and Deamidation Sites Within PepQ
3.6. Visualization of Isopeptides with Skyline
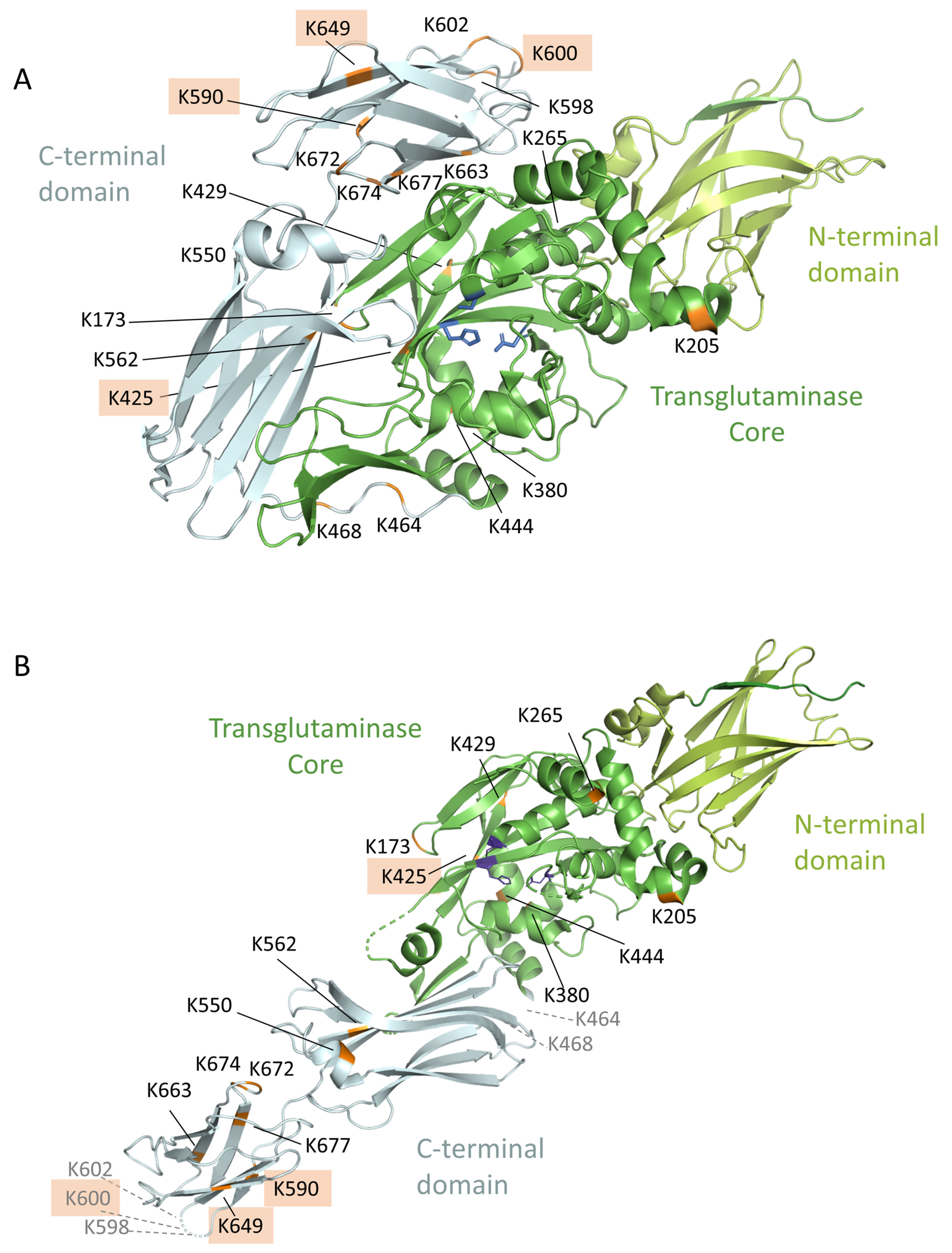
3.7. Estimation of Preferred Lysine Residues
3.8. Location of the Complex-Forming Lysine Residues in the 3D-Model of TG2
3.9. Identification of Isopeptides in the Extended Model System
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Singh, P.; Arora, A.; Strand, T.A.; Leffler, D.A.; Catassi, C.; Green, P.H.; Kelly, C.P.; Ahuja, V.; Makharia, G.K. Global prevalence of celiac disease: Systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 832e2–836e2. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.F.; Leffler, D.A.; Bai, J.; Biagi, F.; Fasano, A.; Green, P.H.; Hadjivassilou, M.; Kaukinen, K.; Kelly, C.P.; Leonard, J.N.; et al. The Oslo definitions for coeliac disease and related terms. Gut 2013, 62, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H.; Koehler, P.; Konitzer, K. Celiac Disease and Gluten—Multidisciplinary Challenges and Opportunities, 1st ed.; Academic Press: London, UK, 2014. [Google Scholar]
- Schuppan, D.; Junker, Y.; Barisani, D. Celiac disease: From pathogenesis to novel therapies. Gastroenterology 2009, 137, 1912–1933. [Google Scholar] [CrossRef] [PubMed]
- Koning, F. Celiac disease: Quantity matters. Semin. Immunopathol. 2012, 34, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Radek, J.T.; Jeong, J.M.; Murthy, S.N.; Ingham, K.C.; Lorand, L. Affinity of human erythrocyte transglutaminase for a 42-kDa gelatin-binding fragment of human plasma fibronectin. Proc. Natl. Acad. Sci. USA 1993, 90, 3152–3156. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, C.S.; Birckbichler, P.J.; Rice, R.H. Transglutaminases: Multifunctional cross-linking enzymes that stabilize tissues. FASEB J. 1991, 5, 3071–3077. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Esslinger, B.; Trapp, D.; Hahn, E.; Huff, T.; Seilmeier, W.; Wieser, H.; Schuppan, D. Cross linking to tissue transglutaminase and collagen favours gliadin toxicity in coeliac disease. Gut 2006, 55, 478–484. [Google Scholar] [CrossRef]
- Stamnaes, J.; Cardoso, I.; Iversen, R.; Sollid, L.M. Transglutaminase 2 strongly binds to an extracellular matrix component other than fibronectin via its second C-terminal beta-barrel domain. FEBS J. 2016, 283, 3994–4010. [Google Scholar] [CrossRef]
- IIsmaa, S.E.; Mearns, B.M.; Lorand, L.; Graham, R.M. Transglutaminases and disease: Lessons from genetically engineered mouse models and inherited disorders. Physiol. Rev. 2009, 89, 991–1023. [Google Scholar] [CrossRef]
- Sollid, L.M. Coeliac disease: Dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 2002, 2, 647–655. [Google Scholar] [CrossRef]
- Volta, U.; Molinaro, N.; Fusconi, M.; Cassani, F.; Biachi, F.B. IgA antiendomysial antibody test: A step forward in celiac disease screening. Dig. Dis. Sci. 1991, 36, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, W.; Ehins, T.; Bauer, M.; Donner, P.; Volta, U.; Riecken, E.O.; Schuppan, D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat. Med. 1997, 3, 797–801. [Google Scholar] [CrossRef] [PubMed]
- du Pré, M.F.; Sollid, L.M. T-cell and B-cell immunity in celiac disease. Best Pract. Res. Clin. Gastroenterol. 2015, 29, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Sollid, L.M.; Molberg, Ø.; McAdam, S.; Lundin, K.E.A. Autoantibodies in coliac disease: Tissue transglutaminase—Guilt by association? Gut 1997, 41, 851–852. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; du Pré, M.F.; Di Niro, R.; Sollid, L.M. Igs as substrates for transglutaminase 2: Implications for autoantibody production in celiac disease. J. Immunol. 2015, 195, 5159–5168. [Google Scholar] [CrossRef]
- Fleckenstein, B.; Qiao, S.-W.; Larsen, M.R.; Jung, G.; Roepstorff, P.; Sollid, L.M. Molecular characterization of covalent complexes between tissue transglutaminase and gliadin peptides. J. Biol. Chem. 2004, 279, 17607–17616. [Google Scholar] [CrossRef]
- Stamnaes, J.; Iversen, R.; du Pré, M.F.; Chen, X.; Sollid, L.M. Enhanced B cell receptor recognition of the autoantigen transglutaminase 2 by efficient catalytic self-mutimerization. PLoS ONE 2015, 10, e0134922. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.; Molberg, Ø.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef]
- Arentz-Hansen, H.; Körner, R.; Molberg, Ø.; Quarsten, H.; Vader, W.; Kooy, Y.M.C.; Knut, E.A.; Lundin, F.K.; Peter, R.; Ludvig, M.; et al. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 2000, 191, 603–661. [Google Scholar] [CrossRef]
- Dørum, S.; Arntzen, M.Ø.; Qiao, S.-W.; Holm, A.; Koehler, C.J.; Thiede, B.; Sollid, L.M.; Fleckenstein, B. The preferred substrates for transglutaminase 2 in a complex wheat gluten digest are peptide fragments harboring celiac disease T-cell epitopes. PLoS ONE 2010, 5, e14056. [Google Scholar] [CrossRef]
- Grossowicz, N.; Wainfan, E.; Borek, E.; Waelsch, H. The enzymatic formation of hydroxamic acids from glutamine and asparagine. J. Biol. Chem. 1950, 187, 111–125. [Google Scholar] [PubMed]
- Hahne, H.; Pachl, F.; Ruprecht, B.; Maier, S.K.; Klaeger, S.; Helm, D.; Médard, G.; Wilm, M.; Lemeer, S.; Kuster, B. DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods 2013, 10, 989–992. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Neuhauser, N.; Michalski, A.; Cox, J.; Mann, M. Expert system for computer-assisted annotation of MS/MS spectra. Mol. Cell. Proteom. 2012, 11, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Trnka, M.J.; Baker, P.R.; Robinson, P.J.J.; Burlingame, A.L.; Chalkley, R.J. Matching cross-linked peptide spectra: Only as good as the worse identification. Mol. Cell. Proteom. 2014, 13, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Rardin, M.J.; MacLean, B.X.; Zawadzka, A.M.; Frewen, B.E.; Cusack, M.P.; Sorensen, D.J.; Bereman, M.S.; Jing, E.; Wu, C.C.; et al. Platform independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in Skyline. Application to protein acetylation and phosphorylation. Mol. Cell. Proteom. 2012, 11, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Bing, Y.; Yan-Jie, W.; Ming, Z.; Sheng-Bo, F.; Jin-Zhong, L.; Kun, Z.; Shuang, L.; Hao, C.; Yu-Xin, L.; Hai-Feng, C.; et al. Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 4, 904–906. [Google Scholar] [CrossRef]
- Lu, S.; Fan, S.B.; Yang, B.; Li, Y.X.; Meng, J.M.; Wu, L.; Li, P.; Zhang, K.; Zhang, M.J.; Fu, Y.; et al. Mapping native disulfide bonds at a proteome scale. Nat. Methods 2015, 12, 329–331. [Google Scholar] [CrossRef]
- Vader, L.W.; de Ru, A.; van der Wal, Y.; Kooy, Y.M.C.; Benckhuijsen, W.; Mearin, M.L.; Drijfhout, J.W.; van Veelen, P.; Koning, F. Specificity of tissue transglutaminase explains cereal toxicity in Celiac Disease. J. Exp. Med. 2002, 195, 643–649. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Antonovici, M.; Ens WWilkins, J.A.; Standing, K.G. Deamidation of -Asn-Gly- sequences during sample preparation for proteomics: Consequences for MALDI and HPLC-MALDI analysis. Anal. Chem. 2006, 78, 6645–6650. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Iversen, R.; Mysling, S.; Hnida, K.; Jorgensen, T.J.D.; Sollid, L.M. Activity-regulating structural changes and autoantibody epitopes in transglutaminase 2 assessed by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. USA 2014, 111, 17146–17151. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) TG2-PepQ Isopeptides with Unambiguous Modification Sites (Localization Probability >75% for the Binding Site in PepQ) | ||||||||||
| Lysine Residue a | Sequence of TG2 peptide | Modification | Precursor | Charge State | MaxQuant Score | No. of Fragment Matches | Q6 Isopeptide Probabilities | Identification E-value pLink2 b | ||
| β-side c | α-side d,e | m/z | α f | β g | α | β | [%] | |||
| 173 | QEYVLTQQGFIYQGSAKFIK | PFPQPQLPY | 1139.59 | 3+ | 30.17 | 128.14 | 6 | 32 | 4.2 | 4.40 × 10−4 |
| 205 | FLKNAGR | PFPQPQLPY | 625.01 | 3+ | 135.77 | 35.63 | 18 | 10 | 100 | 2.24 × 10−11 |
| PFPEPQLPY | 625.34 | 3+ | 117.16 | 19.92 | 15 | 8 | 99.7 | - | ||
| 265 | WKNHGCQR | PFPQPQLPY | 699.36 | 3+ | 79.45 | 29.81 h | 12 | 4 | 100 | 6.36 × 10−6 |
| 380 | AIKEGDLSTK | PFPQPQLPY | 1065.07 | 2+ | 151.17 | 29.74 | 17 | 7 | 100 | 6.56 × 10−7 |
| PFPQPELPY | 710.71 | 3+ | 94.14 | 45.88 | 14 | 14 | 1.7 | - | ||
| 425 | SLIVGLKISTK | PFPQPQLPY | 742.77 | 3+ | 79.94 | 84.48 | 12 | 21 | 98.0 | 7.57 × 10−6 |
| 429 | ISTKSVGR | PFPQPQLPY | 639.02 | 3+ | 107.90 | 26.96 h | 17 | 5 | 100 | 4.57 × 10−6 |
| 429 | ISTKSVGRDER | PFPQPQLPY | 579.56 | 4+ | 168.23 | - i | 22 | - h | 100 | - |
| 444 | EDITHTYKYPEGSSEER j | PFPQPQLPY | 1036.82 | 3+ | 133.60 | 141.60 | 17 | 33 | 100 | 2.74 × 10−8 |
| PFPQPELPY | 1037.15 | 3+ | 119.29 | 96.45 | 14 | 29 | 0.3 | - | ||
| 444 | DEREDITHTYKYPEGSSEER | PFPQPQLPY | 877.91 | 4+ | 63.57 | 53.59 | 9 | 17 | 89.7 | - |
| 464 | ANHLNKLAEK | PFPQPQLPY | 735.73 | 3+ | 155.82 | 47.98 | 22 | 16 | 100 | - |
| 464 | ANHLNKLAEKEETGMAMR | PFPQPQLPY | 622.92 | 5+ | 110.31 | 92.31 | 17 | 25 | 100 | 1.07 × 10−6 |
| PFPEPQLPY | 623.11 | 5+ | 121.99 | 78.02 | 14 | 22 | 0.2 | - | ||
| 468 | LAEKEETGMAMR | PFPQPQLPY | 811.73 | 3+ | 116.73 | 95.09 | 17 | 21 | 100 | 6.50 × 10−11 |
| PFPEPQLPY | 812.06 | 3+ | 73.93 | 38.77 | 12 | 12 | 99.5 | - | ||
| 562 | DCLTESNLIKVR j | PFPQPQLPY | 820.10 | 3+ | 92.47 | 199.82 | 14 | 31 | 100 | 3.01 × 10−8 |
| 562 | YRDCLTESNLIKVR | PFPQPQLPY | 695.12 | 4+ | 157.86 | 111.74 | 19 | 26 | 100 | - |
| 590 | DLYLENPEIKIR j | PFPQPQLPY | 1285.67 | 2+ | 164.89 | 183.03 | 19 | 28 | 100 | 2.62 × 10−6 |
| PFPQPELPY | 1286.18 | 2+ | 59.21 | 108.56 | 8 | 21 | 13 | - | ||
| 598 | ILGEPKQK | PFPQPQLPY | 660.73 | 3+ | 118.01 | 57.18 h | 14 | 10 | 99.9 | 5.38 × 10−7 |
| 598 | ILGEPKQKR | PFPQPQLPY | 712.74 | 3+ | 108.64 | 51.95 | 15 | 12 | 100 | 3.56 × 10−7 |
| 600 | QKR k | PFPQPQLPY | 749.91 | 2+ | 195.98 | - i | 20 | -h | 100 | - |
| 649 | TVEIPDPVEAGEEVKVR i | PFPQPQLPY | 978.85 | 3+ | 29.16 | 122.10 | 10 | 24 | - | 1.04 × 10−2 |
| 663 | MDLLPLHMGLHKLVVNFESDKLK | PFPQPQLPY | 937.01 | 4+ | 93.20 | 103.02 | 15 | 32 | 98.5 | - |
| 677 | AVKGFR j | PFPQPQLPY | 872.98 | 2+ | 168.74 | 22.29 g | 19 | 7 | 100 | 1.04 × 10−6 |
| PFPQPELPY | 873.47 | 2+ | 151.51 | - i | 17 | -h | 0 | - | ||
| (B) TG2-PepQ Isopeptides with Ambiguous Modification Sites | ||||||||||
| 464 | ANHLNKLAEK | PFPEPQLPY | 552.30 | 4 | 53.17 | 48.28 | 9 | 14 | 50 | - |
| 550 | SVPLCILYEKYR | PFPQPQLPY | 851.12 | 3 | - i | 47.92 | - i | 11 | - | - |
| 598/600 | ILGEPKQK l | PFPQPELPY | 661.03 | 3+ | 98.30 | 65.25 | 12 | 10 | 6.8 | - |
| 600/602 | QKRKj k,l | PFPQPQLPY | 542.98 | 3+ | 121.99 | - i | 15 | - h | 100 | - |
| 672/674 | LVVNFESDKLK | PFPQPQLPY | 787.09 | 3+ | 104.75 | 103.91 | 17 | 20 | 100 | 8.51 × 10−7 |
| 672/674 | LVVNFESDKLKAVK h,l | PFPQPQLPY | 665.12 | 4+ | 50.70 | 52.27 | 9 | 13 | 97.2 | 1.57 × 10−2 b |
| Sequence of Isopeptide a | Precursor | Charge State | Residues in TG2 b | E-value pLink2 c | Number of MS2 Scans d | |
|---|---|---|---|---|---|---|
| m/z | K | Q | ||||
| VVSGMVNCNDDQGVLLGR/EKLVVR | 2601.35 | 4+ | 30 | 234 | 2.46 × 10−12 | 6 |
| VVSGMVNCNDDQGVLLGR/FLKNAGR | 2663.34 | 3+ | 205 | 234 | 1.77 × 10−17 | 10 |
| QEYVLTQQGFIYQGSAK/AIKEGDLSTK | 3003.53 | 3+ | 380 | 164 | 3.48 × 10−15 | 3 |
| VVSGMVNCNDDQGVLLGR/AIKEGDLSTK | 2919.45 | 3+ | 380 | 234 | 1.96 × 10−15 | 5 |
| AIKEGDLSTK/NHGCQR | 1757.86 | 4+ | 380 | 270 | 1.50 × 10−4 | 6 |
| NEFGEIQGDK/AIKEGDLSTK | 2180.07 | 3+ | 380 | 324 | 7.94 × 10−7 | 2 |
| VVTNYNSAHDQNSNLLIEYFR/AIKEGDLSTK e | 3540.76 | 4+ | 380 | 307 | 2.23 × 10−7 | 6 |
| VVSGMVNCNDDQGVLLGR/ISTKSVGRDER | 3105.54 | 5+ | 429 | 234 | 1.07 × 10−3 | 3 |
| EDITHTYKYPEGSSEER/VVSGMVNCNDDQGVLLGR | 3898.79 | 5+ | 444 | 234 | 1.62 × 10−8 | 6 |
| EDITHTYKYPEGSSEER/WKNHGCQR | 3051.36 | 5+ | 444 | 270 | 4.24 × 10−3 | 2 |
| VVTNYNSAHDQNSNLLIEYFR/EDITHTYKYPEGSSEER | 4520.09 | 6+ | 444 | 307 | 3.36 × 10−12 | 7 |
| EDITHTYKYPEGSSEER/NEFGEIQGDK | 3159.40 | 5+ | 444 | 324 | 1.94 × 10−3 | 4 |
| VGQSMNMGSDFDVFAHITNNTAEEYVCR/EDITHTYKYPEGSSEER e | 5158.24 | 5+ | 444 | 481 | 3.54 × 10−11 | 3 |
| VVSGMVNCNDDQGVLLGR/ANHLNKLAEK | 2995.51 | 4+ | 464 | 234 | 2.93 × 10−9 | 4 |
| VVTNYNSAHDQNSNLLIEYFR/ANHLNKLAEK e | 3616.81 | 5+ | 464 | 307 | 1.20 × 10−6 | 10 |
| VGQSMNMGSDFDVFAHITNNTAEEYVCR/ANHLNKLAEK | 4254.96 | 5+ | 464 | 481 | 9.92 × 10−8 | 5 |
| VGQSMNMGSDFDVFAHITNNTAEEYVCR/LEAKEETGMAMR e | 4482.98 | 5+ | 468 | 481 | 1.20 × 10−17 | 4 |
| QEYVLTQQGFIYQGSAK/DLYLENPEIKIR | 3444.77 | 4+ | 590 | 164 | 6.77 × 10−5 | 3 |
| VVSGMVNCNDDQGVLLGR/DLYLENPEIKIR | 3360.69 | 4+ | 590 | 234 | 7.07 × 10−11 | 7 |
| DLYLENPEIKIR/WKNHGCQR | 2513.27 | 4+ | 590 | 270 | 2.01 × 10−11 | 5 |
| VVTNYNSAHDQNSNLLIEYFR/DLYLENPEIKIR | 3981.99 | 5+ | 590 | 307 | 3.04 × 10−7 | 4 |
| ILGEPKQK/AVKGFR | 1571.92 | 4+ | 599 | 677 | 1.03 × 10−10 | 4 |
| ILGEPKQKR/AVKGFR | 1728.03 | 4+ | 599 | 677 | 1.21 × 10−3 | 3 |
| VVSGMVNCNDDQGVLLGR/TVEIPDPVEAGEEVKVR e | 3724.85 | 4+ | 649 | 234 | 6.78 × 10−13 | 6 |
| TVEIPDPVEAGEEVKVR/NHGCQR | 2563.26 | 4+ | 649 | 270 | 5.69 × 10−3 | 2 |
| VVTNYNSAHDQNSNLLIEYFR/TVEIPDPVEAGEEVKVR | 4346.16 | 5+ | 649 | 307 | 7.66 × 10−5 | 4 |
| LVVNFESDKLKAVK/NHGCQR | 2286.20 | 5+ | 672 | 270 | 8.80 × 10−6 | 2 |
| QEYVLTQQGFIYQGSAK/AVKGFR | 2619.36 | 4+ | 677 | 164 | 5.82 × 10−9 | 6 |
| QEYVLTQQGFIYQGSAK/FLKNAGR | 2747.42 | 3+ | 677 | 164 | 8.31 × 10−11 | 4 |
| QEYVLTQQGFIYQGSAK/TVEIPDPVEAGEEVKVR | 3808.93 | 6+ | 677 | 164 | 8.33 × 10−6 | 2 |
| QEYVLTQQGFIYQGSAK/AVKGFR | 2619.36 | 4+ | 677 | 169 | 5.15 × 10−7 | 10 |
| VVSGMVNCNDDQGVLLGR/AVKGFR e | 2535.28 | 4+ | 677 | 234 | 1.70 × 10−11 | 14 |
| WKNHGCQR/AVKGFR | 1687.86 | 4+ | 677 | 270 | 1.80 × 10−5 | 3 |
| VVTNYNSAHDQNSNLLIEYFR/AVKGFR e | 3156.59 | 4+ | 677 | 307 | 1.03 × 10−5 | 10 |
| NEFGEIQGDK/AVKGFR | 1795.90 | 3+ | 677 | 324 | 1.37 × 10−8 | 5 |
| VGQSMNMGSDFDVFAHITNNTAEEYVCR/AVKGFR e | 3794.74 | 4+ | 677 | 481 | 1.99 × 10−5 | 5 |
| (A) Isopeptides with Unambiguous Modification Sites (Localization Probability >75%) | |||||||||||||
| Position of K in TG2 a | m/z | z | Score b | Isopeptide Probability [%] | Deamidation Probability [%] | pLink2 Identification c | |||||||
| Q2 | Q4 | Q9 | Q11 | Q2 | Q4 | Q9 | Q11 | E-value | No. MS2 Spectra | ||||
| 205 | 818.77 | 3+ | 176.20 | - | - | 0.7 | 99.3 | 12.8 | 87.2 | - | - | 1.49 × 10−9 | 15 |
| 380 | 904.13 | 3+ | 153.63 | - | - | 0.4 | 99.6 | - | 100 | - | - | 1.37 × 10−9 | 8 |
| 464 | 929.48 | 3+ | 158.40 | - | - | 0.7 | 99.3 | - | 100 | - | - | 3.30 × 10−6 | 9 |
| 590 | 1051.22 | 3+ | 126.76 | 8.7 | 91.3 | - | - | - | - | 0.1 | 99.9 | 1.30 × 10−11 | 3 |
| 1051.55 | 3+ | 72.29 | 1.1 | 7.8 | 90.8 | 0.3 | 89.1 | 10.9 | 0.4 | 99.6 | 1.95 × 10−11 | 5 | |
| 598 | 854.46 | 3+ | 134.18 | - | - | 0.3 | 99.7 | 0.3 | 99.7 | - | - | 2.11 × 10−11 | 14 |
| 600c | 1104.59 | 2+ | 165.83 | - | - | - | 100 | - | 100 | - | - | - | - |
| 649 | 1172.60 | 3+ | 64.65 | - | 0.1 | 2.1 | 97.8 | 10.5 | 89.4 | - | 0.1 | 2.00 × 10−4 | 7 |
| 672 | 980.85 | 3+ | 138.94 | - | - | 6.2 | 93.8 | 6.4 | 93.6 | - | - | 1.36 × 10−9 | 3 |
| 677 | 776.08 | 3+ | 84.29 | 8.1 | 89.4 | 0.5 | 2.1 | 0.2 | 2.1 | 0.9 | 96.8 | 2.85 × 10−5 | 3 |
| (B) Isopeptides with Ambiguous Modification Sites | |||||||||||||
| 464 | 697.61 | 4+ | 61.64 | 0.9 | 0.8 | 91.7 | 6.5 | 49.9 | 50.1 | 6.8 | 93.2 | 1.46 × 10−3 | 6 |
| 468 | 1005.49 | 3+ | 43.99 | 0.9 | 4.6 | 50.6 | 43.9 | 10.0 | 84.3 | 1.0 | 4.6 | - | - |
| 1005.49 | 3+ | 40.73 | 0.2 | 2.2 | 19.3 | 78.3 | 41.8 | 55.2 | 0.8 | 2.2 | - | - | |
| 562 | 1013.85 | 3+ | 53.26 | - | 0.2 | 11.5 | 88.3 | 29.9 | 69.8 | 0.1 | 0.2 | 1.36 × 10−3 | 2 |
| 590 | 1051.22 | 3+ | 158.30 | - | - | 63.0 | 37.0 | - | 100 | - | - | - | - |
| 1051.22 | 3+ | 33.20 | 0.9 | 3.2 | 16.4 | 79.5 | 23.9 | 66.7 | 3.6 | 5.8 | 5.89 × 10−15 | 10 | |
| 600d | 694.03 | 3+ | 165.35 | - | - | 2.2 | 97.8 | 63.6 | 36.4 | - | - | - | - |
| 694.36 | 3+ | 44.62 | 10.2 | 28.8 | 23.8 | 37.3 | 76.3 | 62.1 | 52.0 | 9.6 | - | - | |
| (C) Isopeptides Identified with pLink2 d | |||||||||||||
| Crosslink | Deamidation | ||||||||||||
| 30 | 798.10 | 3+ | Q11 | Q4 | 1.20 × 10-4 | 3 | |||||||
| 797.77 | 3+ | Q4 | - | 7.62 × 10-5 | 3 | ||||||||
| 205 | 818.77 | 3+ | Q9 | Q4 | 7.67 × 10-3 | 6 | |||||||
| 429 | 832.77 | 3+ | Q11 | Q4 | 1.95 × 10-4 | 4 | |||||||
| (A) Isopeptides with Unambiguous Modification Sites (Localization Probability >75%) | |||||||||||||||||
| Position of K in TG2 a | m/z | z | Score b | Isopeptide Probability [%] | Deamidation Probability [%] | pLink2 Identification c | |||||||||||
| Q2 | Q4 | Q8 | Q10 | Q11 | Q14 | Q2 | Q4 | Q8 | Q10 | Q11 | Q14 | E-Value | No. MS2 Spectra | ||||
| 205 | 798.76 | 3+ | 142.10 | 0.6 | 1.0 | 0.4 | 87.4 | 10.6 | - | 99.4 | 99.0 | - | 0.6 | - | - | - | - |
| 798.43 | 3+ | 139.74 | - | - | - | 99.6 | 0.4 | - | 0.5 | 99.5 | - | - | - | - | 2.35 × 10−6 | 5 | |
| 380 | 884.13 | 3+ | 125.61 | 90.7 | 8.1 | 1.2 | - | - | - | 8.3 | 91.7 | 0.2 | 98.2 | 1.6 | - | 9.80 × 10−7 | 2 |
| 884.13 | 3+ | 128.46 | - | - | 0.1 | 99.1 | 0.8 | - | 0.4 | 99.6 | - | - | - | - | 8.31 × 10−7 | 3 | |
| 464 | 682.12 | 4+ | 149.69 | - | - | - | 99.8 | 0.2 | - | 1.4 | 98.6 | - | - | - | - | 1.06 × 10−6 | 7 |
| 682.36 | 4+ | 131.69 | 89.6 | 10.4 | - | - | - | - | 10.4 | 88.3 | 1.3 | 98.7 | 1.3 | - | - | - | |
| 590 | 1031.22 | 3+ | 157.56 | 93.6 | 6.4 | - | - | - | - | 6.4 | 93.6 | - | 98.3 | 1.7 | - | 6.60 × 10−11 | 8 |
| 1031.22 | 3+ | 126.32 | 10.5 | 89.5 | - | - | - | - | 89.5 | 10.5 | 0.1 | 96.9 | 3.1 | - | 1.61 × 10−8 | 8 | |
| 1031.54 | 3+ | 71.88 | - | - | - | - | 0.1 | 99.9 | 100 | 99.9 | 0.5 | 85.3 | 14.2 | - | 3.53 × 10−9 | 4 | |
| 598 | 834.13 | 3+ | 126.32 | - | 0.9 | 14.7 | 83.8 | 0.5 | - | 0.6 | 98.5 | - | 0.9 | - | - | 3.65 × 10−3 | 2 |
| 834.79 | 3+ | 75.46 | 98.9 | 1.1 | - | - | - | - | 1.1 | 97.4 | 2.5 | 87.8 | 11.6 | 99.6 | - | - | |
| 600 | 1010.05 | 2+ | 232.88 | - | - | - | 100 | - | - | 0.1 | 99.9 | - | - | - | - | - | - |
| 674.03 | 3+ | 139.19 | 99.2 | 0.8 | - | - | - | - | 0.8 | 99.2 | - | 93.3 | 6.7 | - | - | - | |
| 649 | 1151.95 | 3+ | 52.82 | - | - | - | 0.2 | 1.9 | 97.9 | - | - | - | - | - | - | - | - |
| 1152.60 | 3+ | 51.55 | 0.5 | 2.6 | 3.0 | 2.8 | 77.2 | 13.9 | 86.3 | 13.1 | 1.9 | 94.6 | 3.8 | 0.4 | - | - | |
| 672 | 960.85 | 3+ | 67.02 | 96.7 | 3.1 | 0.2 | - | - | - | 3.1 | 95.7 | 1.5 | 95.9 | 3.8 | - | - | - |
| 677 | 756.08 | 3+ | 120.60 | 93.0 | 7.0 | - | - | - | - | 7.0 | 92.9 | 0.1 | 96.3 | 3.7 | - | 2.12 × 10−5 | 5 |
| 756.08 | 3+ | 115.12 | 98.5 | 1.5 | - | - | - | - | 1.4 | 98.5 | 10.9 | 89.0 | 0.2 | - | 1.33 × 10−6 | 4 | |
| 755.75 | 3+ | 60.18 | - | - | 1.4 | 76.6 | 21.9 | 0.1 | 0.6 | 99.0 | 0.3 | 0.1 | - | - | 4.29 × 10−5 | 3 | |
| (B) Isopeptides with Ambiguous Modification Sites | |||||||||||||||||
| 649 | 1152.60 | 3+ | 78.53 | 58.2 | 14.9 | 26.0 | 0.8 | 0.1 | - | 35.0 | 62.1 | 3.0 | 77.2 | 22.7 | 0.1 | 9.03 × 10−4 | 3 |
| 1152.93 | 3+ | 60.40 | 3.3 | 30.0 | 5.0 | 60.3 | 0.4 | 0.9 | 96.1 | 66.5 | 2.2 | 35.9 | 8.7 | 90.5 | - | - | |
| (C) Isopeptides Identified with pLink2 d | |||||||||||||||||
| Crosslink | Deamidation | ||||||||||||||||
| 30 | 778.10 | 3+ | Q2 | Q4; Q10 | 8.12 × 10−5 | 3 | |||||||||||
| 778.10 | 3+ | Q4 | Q2; Q10 | 3.33 × 10−5 | 2 | ||||||||||||
| 380 | 884.46 | 3+ | Q2 | Q4; Q10; Q14 | 1.59 × 10−3 | 4 | |||||||||||
| 429 | 812.77 | 3+ | Q10 | Q2; Q4 | 4.79 × 10−5 | 6 | |||||||||||
| 464 | 909.15 | 3+ | Q11 | Q4 | 5.29 × 10−6 | 2 | |||||||||||
| 682.12 | 4+ | Q4 | Q10 | 4.42 × 10−7 | 5 | ||||||||||||
| 590 | 1031.22 | 3+ | Q8 | Q4; Q10 | 2.46 × 10−6 | 3 | |||||||||||
| 649 | 1152.60 | 3+ | Q11 | Q4; Q10 | 1.07 × 10−5 | 4 | |||||||||||
| 1152.60 | 3+ | Q8 | Q4; Q10 | 1.72 × 10−6 | 2 | ||||||||||||
| 1152.93 | 3+ | Q14 | Q2; Q10; Q14 | 2.12 × 10−6 | 2 | ||||||||||||
| 672 | 960.52 | 3+ | Q10 | Q4 | 2.81 × 10−4 | 2 | |||||||||||
| 960.52 | 3+ | Q4 | Q10 | 1.01 × 10−3 | 2 | ||||||||||||
| (A) Isopeptides with Unambiguous Modification Sites (Localization Probability >75%) | |||||||||||||||||||||||
| Position of K in TG2 a | m/z | z | Score b | Isopeptide Probability [%] | Deamidation Probability [%] | pLink2 Identification c | |||||||||||||||||
| Q2 | Q4 | Q5 | Q7 | Q8 | Q12 | Q13 | Q14 | Q15 | Q2 | Q4 | Q5 | Q7 | Q8 | Q12 | Q13 | Q14 | Q15 | E-value | No. MS2 Spectra | ||||
| 205 | 937.47 | 3+ | 184.99 | - | 80.3 | 11.9 | 7.8 | 0.1 | - | - | - | - | - | 7.4 | 3.7 | 88.6 | 0.2 | 80.2 | 28.7 | 91.0 | - | - | - |
| 590 | 1169.93 | 3+ | 133.23 | 4.9 | 90.1 | 4.9 | - | - | - | - | - | - | - | - | - | - | - | 99.3 | 16.4 | 82.1 | 2.2 | - | - |
| 600 | 812.74 | 3+ | 150.76 | - | 83.8 | 12.4 | 3.2 | 0.5 | - | - | - | - | - | 2.2 | 4.1 | 81.6 | 12.1 | 86.1 | 33.7 | 80.2 | - | ||
| 649 | 1291.31 | 3+ | 83.37 | 0.6 | 77.8 | 19.4 | 1.7 | 0.5 | - | - | - | - | 0.1 | 1.1 | 1.9 | 77.6 | 19.5 | 90.9 | 75.8 | 24.1 | 0.9 | - | - |
| 677 | 894.46 | 3+ | 140.22 | 0.3 | 87.4 | 11.9 | 0.3 | - | - | - | - | - | - | - | - | - | - | 98.4 | 16.8 | 84.6 | 0.2 | ||
| (B) Isopeptides with Ambiguous Modification Sites | |||||||||||||||||||||||
| 205 | 937.46 | 3+ | 169.37 | - | - | - | - | - | 76.3 | 22.8 | 0.9 | - | - | 92.8 | 7.6 | 24.3 | 75.2 | 1.0 | 2.2 | 96.8 | - | 1.49 × 10−5 | 7 |
| 937.46 | 3+ | 153.48 | 0.1 | 31.0 | 4.6 | 63.8 | 0.5 | - | - | - | - | - | 31.1 | 34.5 | 34.0 | 0.5 | 73.6 | 31.2 | 94.6 | 0.5 | 9.00 × 10−5 | 5 | |
| 380 | 1022.85 | 3+ | 153.23 | - | 21.0 | 2.6 | 76.2 | 0.2 | - | - | - | - | - | 32.4 | 48.5 | 19.0 | 0.1 | 82.7 | 29.5 | 85.8 | 2.1 | 1.82 × 10−4 | 6 |
| 464 | 1048.20 | 3+ | 104.94 | - | 23.9 | 18.4 | 27.4 | 27.2 | 2.5 | 0.3 | 0.3 | - | - | 2.6 | 20.3 | 29.0 | 28.9 | 94.4 | 53.1 | 48.2 | 0.1 | - | - |
| 786.64 | 4+ | 103.88 | 0.4 | 72.8 | 13.1 | 12.6 | 1.2 | - | - | - | - | 7.6 | 8.9 | 37.4 | 39.1 | 6.9 | 94.5 | 44.7 | 57.3 | 3.5 | - | - | |
| 786.64 | 4+ | 71.55 | - | 0.1 | 1.2 | 76.3 | 19.6 | 0.8 | 0.5 | 0.5 | 0.1 | 30.3 | 49.6 | 20.5 | 21.5 | 78.9 | 37.8 | 75.1 | 70.9 | 15.3 | 4.86 × 10−4 | 4 | |
| 590 | 1169.60 | 3+ | 147.56 | - | 12.4 | 12.4 | 67.5 | 7.7 | - | - | - | - | 0.2 | 53.2 | 21.9 | 21.1 | 3.6 | 60.7 | 84.4 | 53.8 | 1.2 | 4.70 × 10−9 | 2 |
| 598 | 973.17 | 3+ | 148.70 | - | 9.2 | 1.5 | 44.6 | 44.6 | - | - | - | - | - | 38.4 | 51.4 | 5.1 | 5.1 | 96.4 | 98.7 | 4.8 | - | - | - |
| 973.17 | 3+ | 140.77 | - | 30.6 | 6.2 | 60.6 | 2.5 | 0.1 | 0.1 | - | - | - | 32.9 | 32.4 | 33.4 | 1.5 | 87.0 | 95.0 | 17.8 | - | 1.15 × 10−3 | 4 | |
| 972.84 | 3+ | 125.50 | - | 45.6 | 45.6 | 8.6 | 0.2 | - | - | - | - | - | - | - | - | - | 48.7 | 76.9 | 70.7 | 3.6 | 8.06 × 10−4 | 2 | |
| 600 | 812.74 | 3+ | 124.50 | - | 14.4 | 81.1 | 3.3 | 1.2 | - | - | - | - | 0.1 | 78.7 | 11.2 | 7.9 | 2.1 | 89.7 | 55.1 | 55.1 | 0.1 | - | - |
| 649 | 1291.64 | 3+ | 78.33 | - | 0.4 | 3.4 | 21.1 | 70.9 | 0.4 | 0.2 | - | - | 0.6 | 49.0 | 54.1 | 72.3 | 23.5 | 82.3 | 50.7 | 52.7 | 14.8 | 2.85 × 10−5 | 2 |
| 1291.64 | 3+ | 78.33 | - | 0.4 | 3.4 | 21.1 | 70.9 | 0.4 | 0.2 | - | - | 0.6 | 49.0 | 54.1 | 72.3 | 23.5 | 82.3 | 50.7 | 52.7 | 14.8 | 1.39 × 10−5 | 2 | |
| 1291.64 | 3+ | 60.54 | 1.4 | 68.1 | 27.5 | 2.3 | 0.5 | 0.1 | - | - | - | - | 0.1 | 0.2 | 0.6 | 5.6 | 71.3 | 36.5 | 53.9 | 31.6 | 2.63 × 10−6 | 3 | |
| 677 | 894.79 | 3+ | 178.14 | - | 6.4 | 1.0 | 89.3 | 3.4 | - | - | - | - | - | 36.5 | 56.6 | 6.4 | 0.4 | 99.8 | 99.8 | 0.4 | - | - | - |
| 894.46 | 3+ | 120.59 | - | 5.1 | 70.1 | 23.3 | 1.2 | 0.1 | 0.1 | 0.1 | - | - | 0.1 | 0.1 | - | - | 98.8 | 49.4 | 49.4 | 2.1 | - | - | |
| 894.45 | 3+ | 51.70 | - | 0.1 | 0.1 | 0.4 | 0.4 | 24.7 | 24.7 | 24.7 | 24.8 | 0.3 | 47.1 | 52.9 | 49.0 | 54.9 | 74.0 | 74.0 | 74.0 | 73.8 | - | - | |
| (C) Isopeptides identified with pLink2 d | |||||||||||||||||||||||
| Crosslink | Deamidation | ||||||||||||||||||||||
| 30 | 916.48 | 3+ | Q4 | Q12; Q14 | 4.14 × 10−4 | 3 | |||||||||||||||||
| 380 | 1022.52 | 3+ | Q5 | Q12; Q13 | 6.95 × 10−5 | 3 | |||||||||||||||||
| 429 | 951.15 | 3+ | Q12 | Q4; Q14 | 6.77 × 10−5 | 2 | |||||||||||||||||
| 464 | 786.15 | 4+ | Q12 | Q4; Q13 | 3.16 × 10−4 | 6 | |||||||||||||||||
| 786.64 | 4+ | Q12 | Q2; Q10; Q14 | 3.39 × 10−4 | 3 | ||||||||||||||||||
| 590 | 1169.60 | 3+ | Q12 | Q4; Q7; Q13 | 1.21 × 10−6 | 2 | |||||||||||||||||
| 1169.60 | 3+ | Q7 | Q12; Q14 | 5.18 × 10−9 | 2 | ||||||||||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lexhaller, B.; Ludwig, C.; Scherf, K.A. Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides. Nutrients 2019, 11, 2263. https://doi.org/10.3390/nu11102263
Lexhaller B, Ludwig C, Scherf KA. Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides. Nutrients. 2019; 11(10):2263. https://doi.org/10.3390/nu11102263
Chicago/Turabian StyleLexhaller, Barbara, Christina Ludwig, and Katharina A. Scherf. 2019. "Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides" Nutrients 11, no. 10: 2263. https://doi.org/10.3390/nu11102263
APA StyleLexhaller, B., Ludwig, C., & Scherf, K. A. (2019). Comprehensive Detection of Isopeptides between Human Tissue Transglutaminase and Gluten Peptides. Nutrients, 11(10), 2263. https://doi.org/10.3390/nu11102263