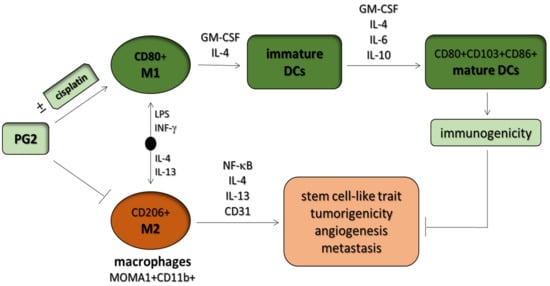
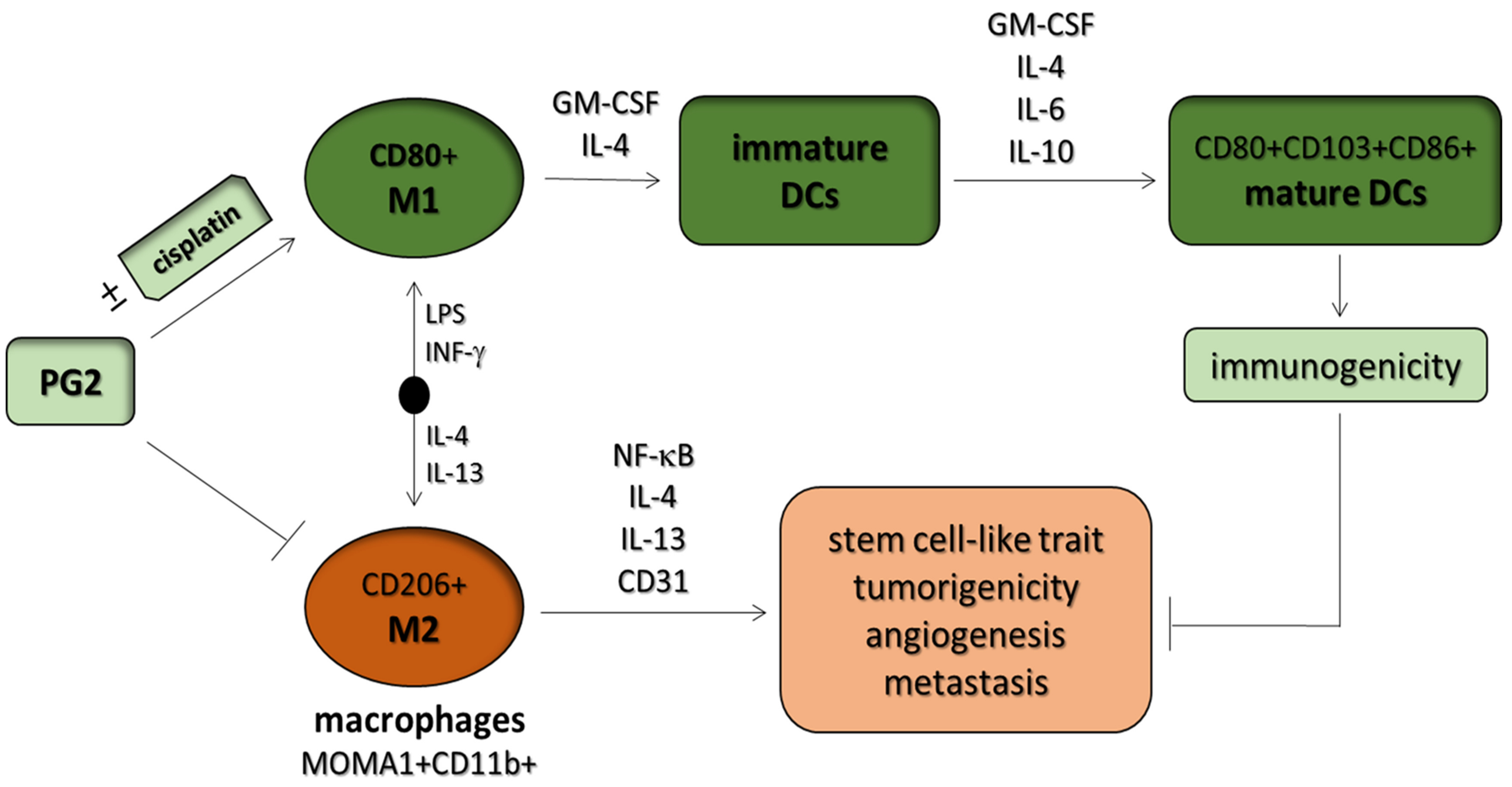
Astragalus polysaccharides (PG2) Enhances the M1 Polarization of Macrophages, Functional Maturation of Dendritic Cells, and T Cell-Mediated Anticancer Immune Responses in Patients with Lung Cancer

 ,
,  ,
,  and
and
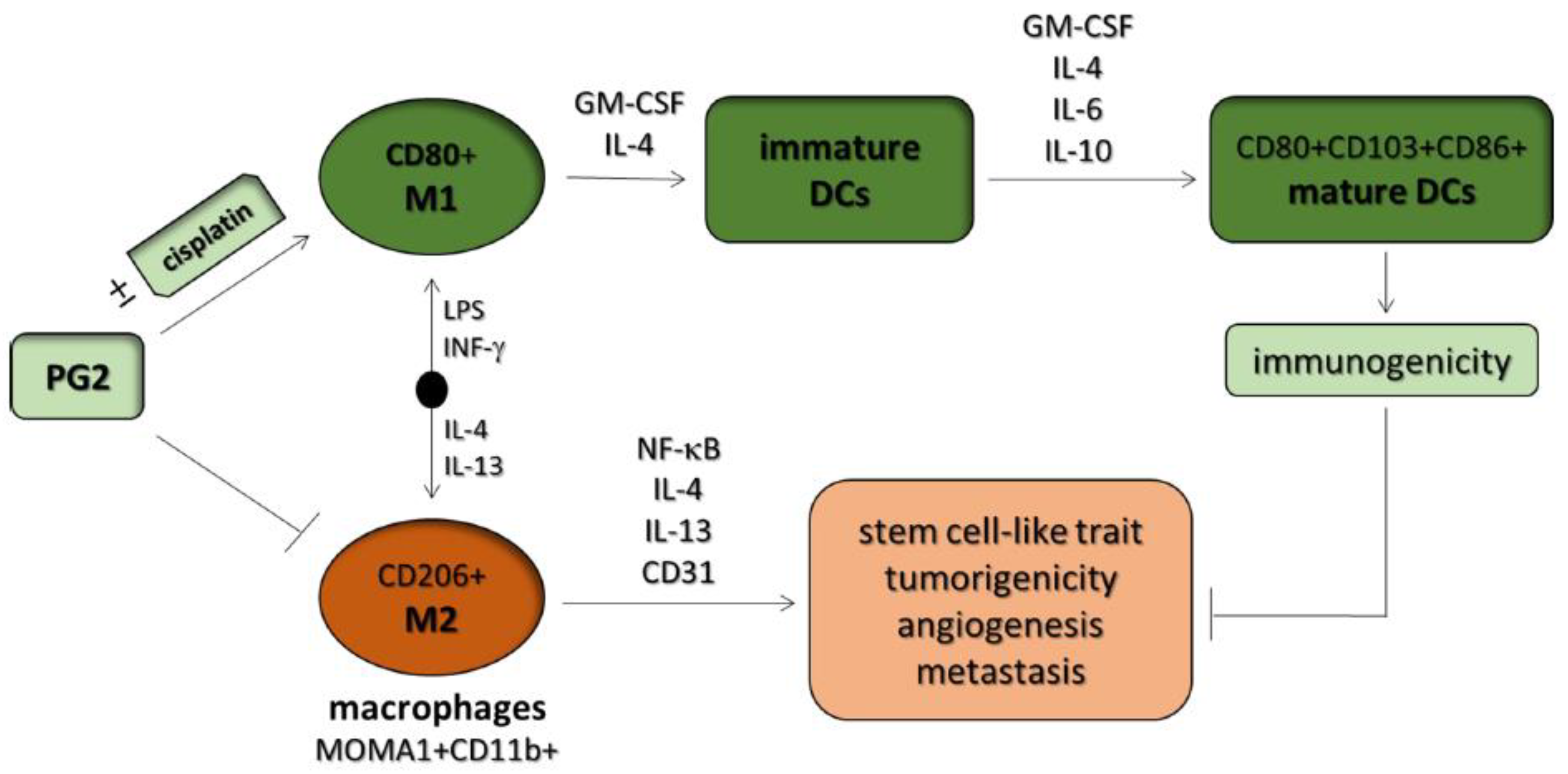
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Peripheral Blood Mononuclear Cells (PBMCs) Culture and Isolation of Dendritic Cells
2.3. Cell Lines and Culture
2.4. Western Blot
2.5. Flow Cytometry Analyses
2.6. Methylthiazolyldiphenyl-Tetrazolium Bromide (MTT) Viability Assay
2.7. Cell Migration and Invasion Assays
2.8. Colony Formation Assay
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Tumor Implantation and Growth in Syngeneic Mice Models
2.11. Ethics Approval and Consent to Participate
2.12. Statistical Analysis
3. Results
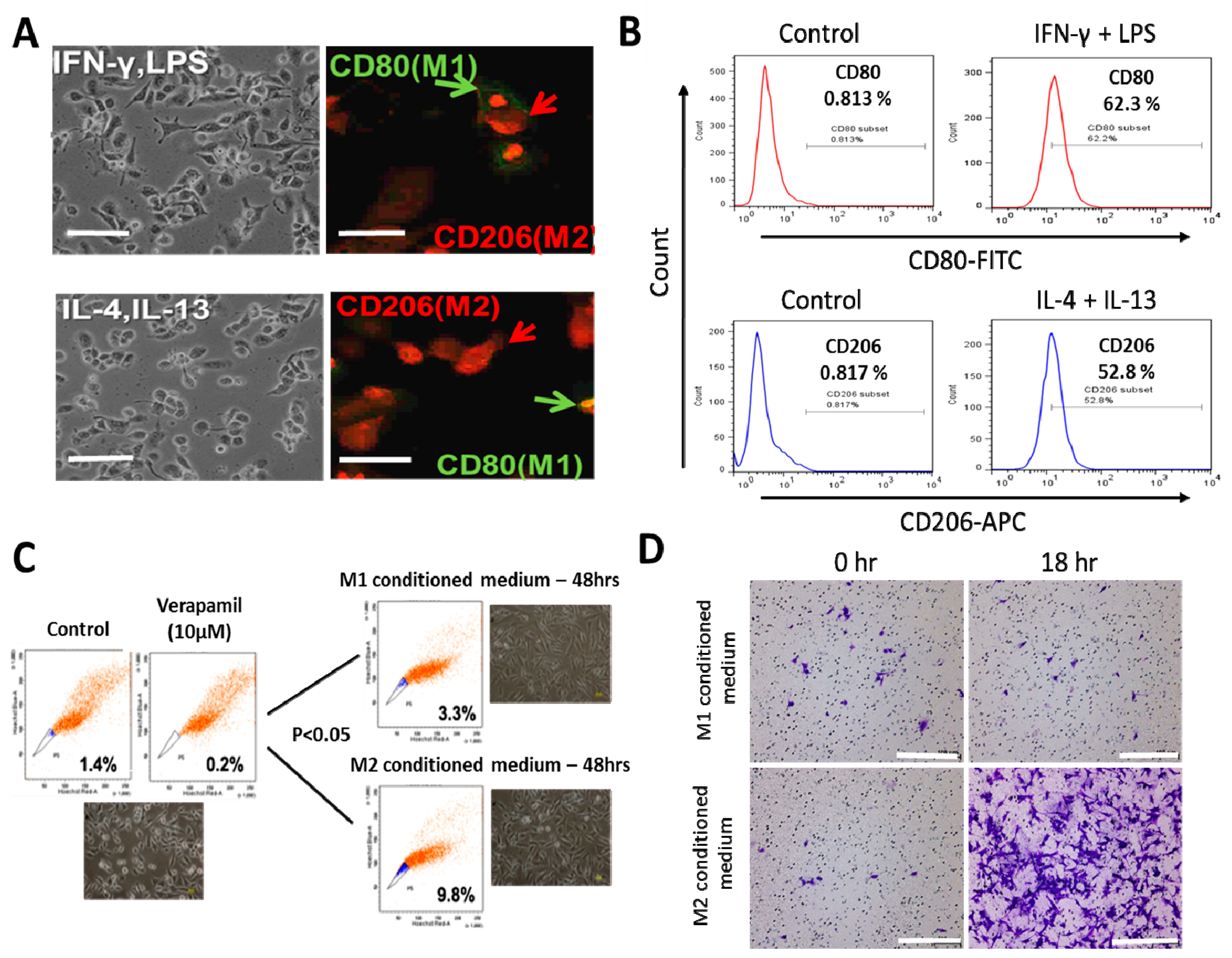
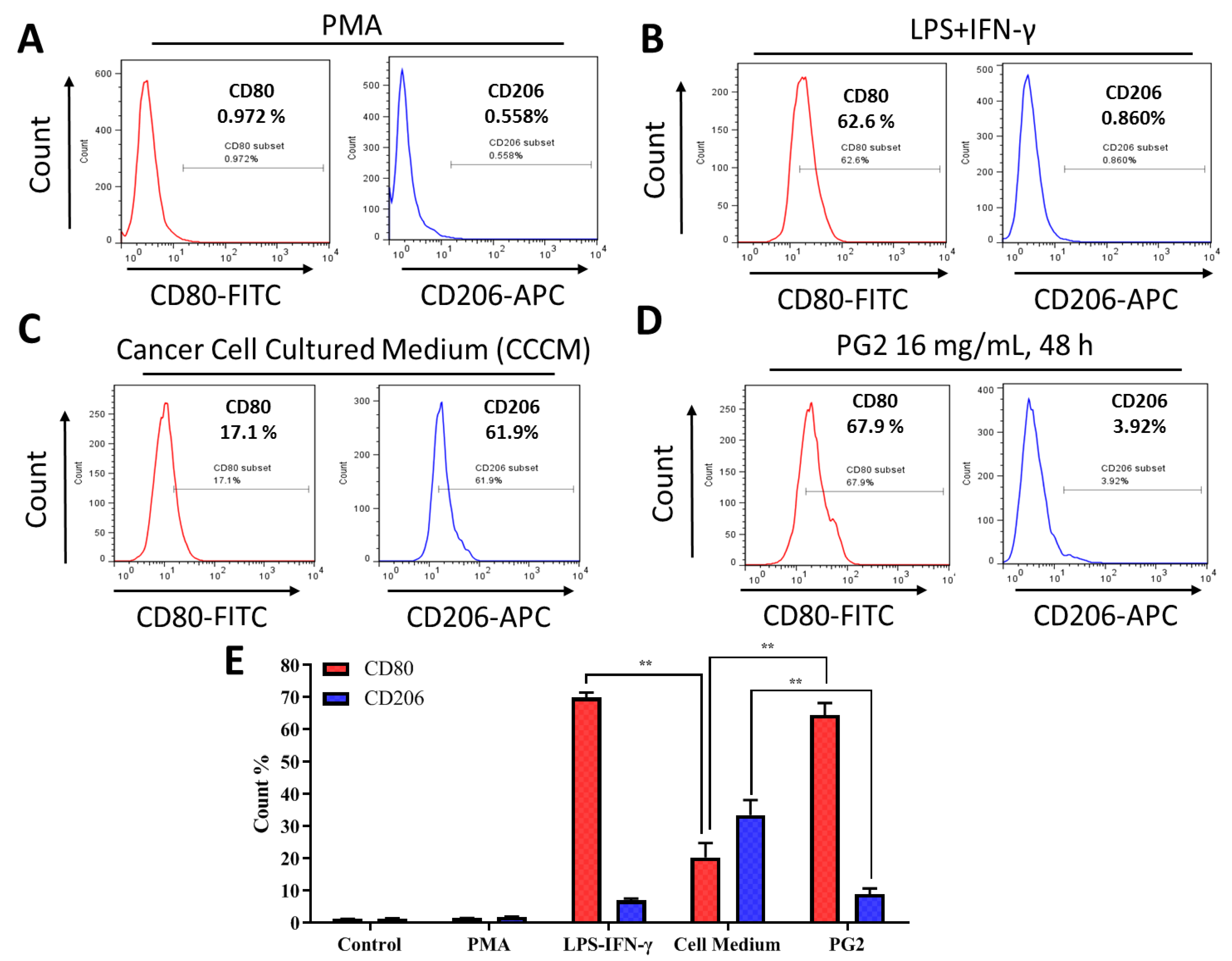
3.1. Macrophages Respond Differentially to Different Inflammatory Cytokine Stimuli
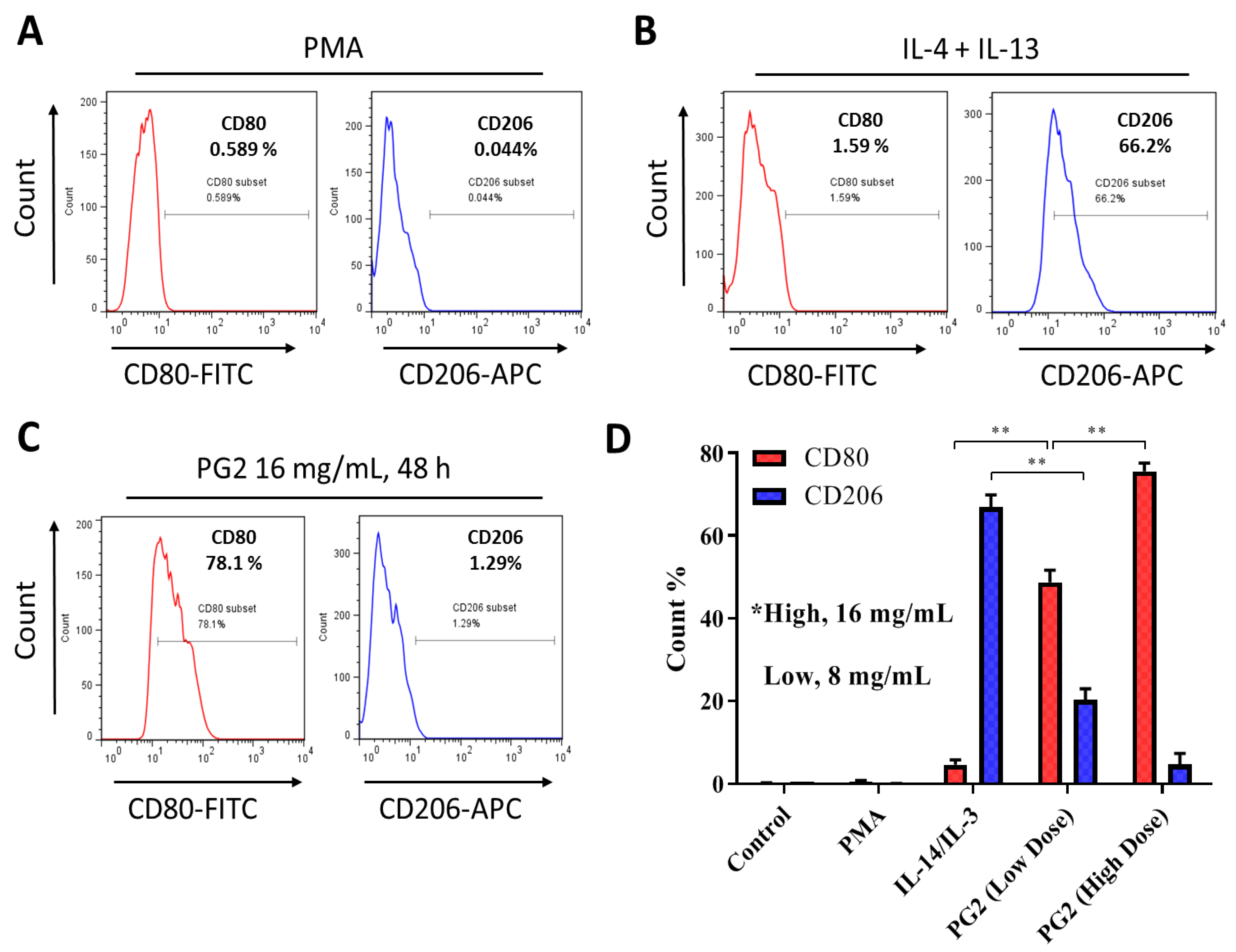
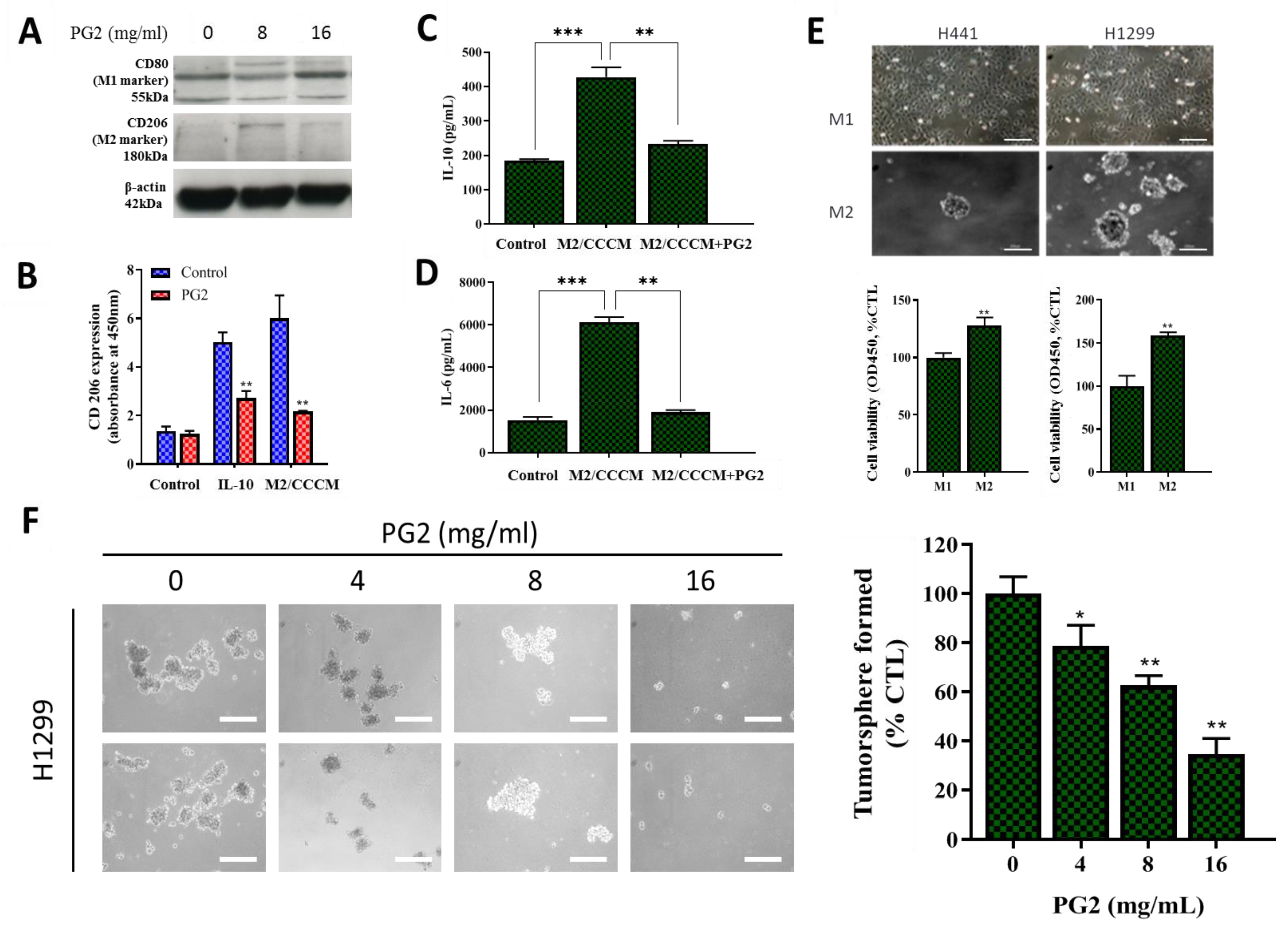
3.2. PG2 Enhances M1 Polarization While Down-Regulating IL-4/IL-13-Induced M2 Polarization Dose-Dependently
3.3. The Enhancement of M1 Macrophage Polarization by PG2 Is Akin to the Effect of LPS/IFN-γ Stimulation of MDMs
3.4. PG2 Represses the Tumor-Promoting Effects of Anti-Inflammatory Cytokines and Inhibits the NSCLC Stem Cell-Like Phenotypes Induced by M2-Conditioned Medium
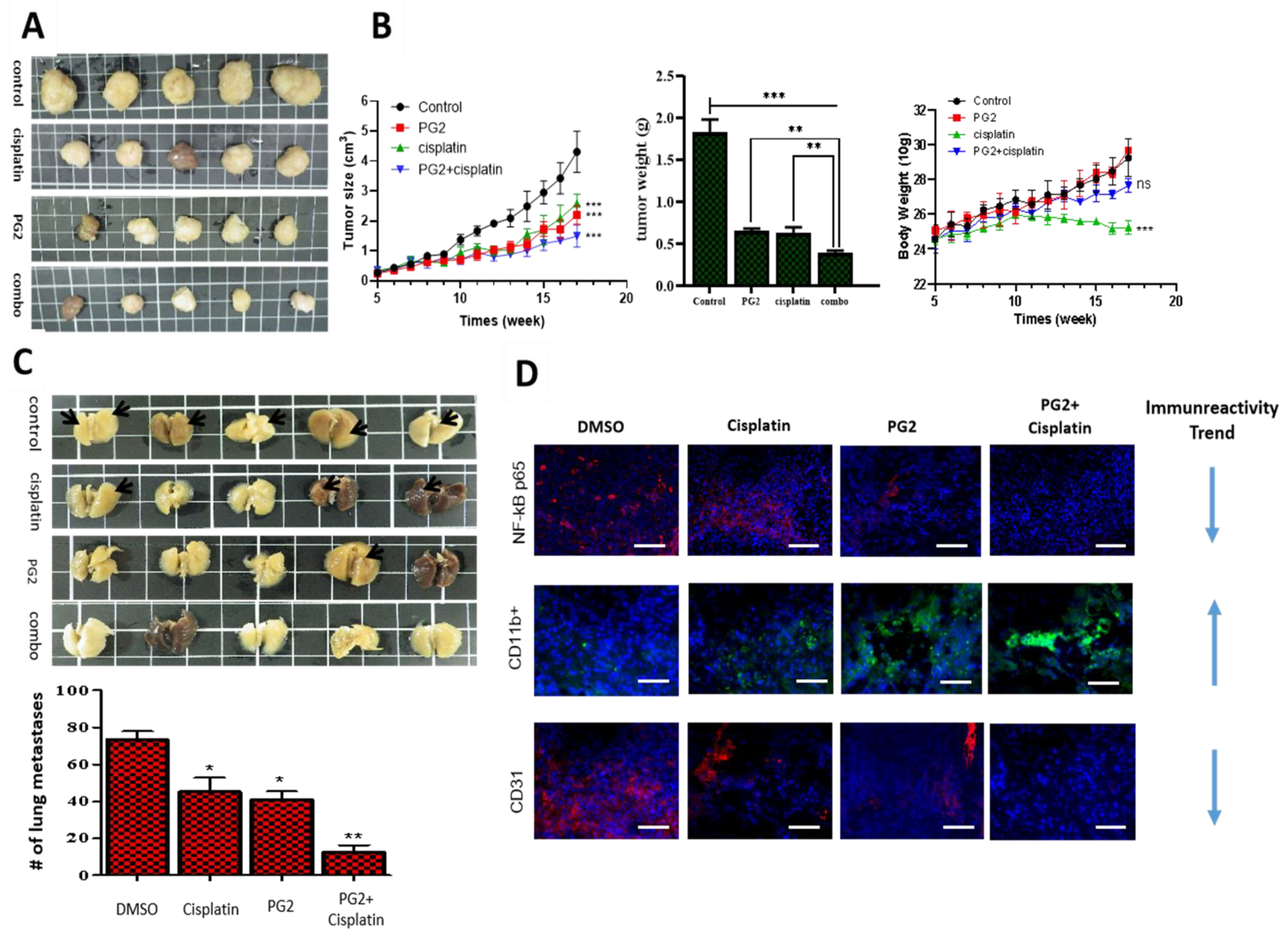
3.5. PG2 Suppresses Tumorigenicity and Metastasis in Syngeneic C57BL/6 Mice, and Potentiates Anticancer Effect of Cisplatin In Vivo by Modulating Inflammation-Associated Macrophage Activity and Angiogenesis
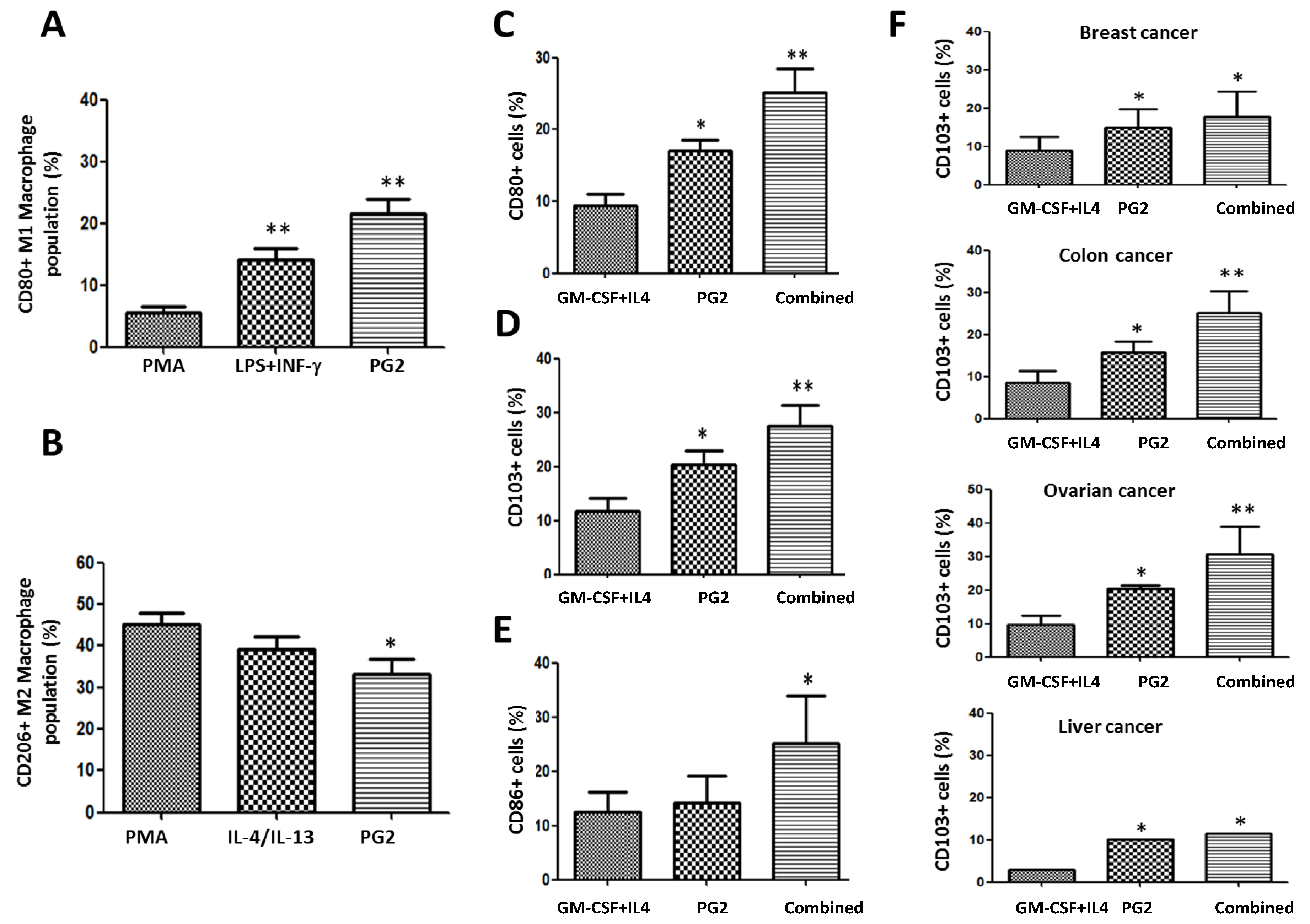
3.6. PG2 Up-Modulates the CD80+ M1/CD206+ M2 Macrophage Ratio and Increases the Population of CD80+, CD103+, and CD86+ Functionally Matured Dendritic Cells Ex Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Data and Materials
Abbreviations
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Yang, J.J.; Lam, K.C. Treating patients with EGFR-sensitizing mutations: First line or second line—Is there a difference? J. Clin. Oncol. 2013, 31, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Non Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Haslam, A.; Prasad, V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Franco, F.; Ho, P.C. Metabolic Regulation of Tregs in Cancers: Opportunities for Immunotherapy. Trends Cancer 2017, 3, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009, 206, 1327–1337. [Google Scholar] [CrossRef]
- Finn, O.J. Immuno-oncology: Understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. 2012, 23 (Suppl. 8), viii6–viii9. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Gocheva, V.; Wang, H.W.; Gadea, B.B.; Shree, T.; Hunter, K.E.; Garfall, A.L.; Berman, T.; Joyce, J.A. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010, 24, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Takeya, M.; Komohara, Y. Role of tumor-associated macrophages in human malignancies: Friend or foe? Pathol. Int. 2016, 66, 491–505. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Ohtaki, Y.; Ishii, G.; Nagai, K.; Ashimine, S.; Kuwata, T.; Hishida, T.; Nishimura, M.; Yoshida, J.; Takeyoshi, I.; Ochiai, A. Stromal macrophage expressing CD204 is associated with tumor aggressiveness in lung adenocarcinoma. J. Thorac. Oncol. 2010, 5, 1507–1515. [Google Scholar] [CrossRef]
- Cao, W.; Peters, J.H.; Nieman, D.; Sharma, M.; Watson, T.; Yu, J. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br. J. Cancer 2015, 113, 738–746. [Google Scholar] [CrossRef]
- Feng, Z.; Wang, Z.; Yang, M.; Zhou, L.; Bao, Y. Polysaccharopeptide exerts immunoregulatory effects via MyD88-dependent signaling pathway. Int. J. Biol. Macromol. 2016, 82, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Dong, B.; Tan, Y.; Yu, S.; Bao, Y.X. A study on the immunomodulation of polysaccharopeptide through the TLR4-TIRAP/MAL-MyD88 signaling pathway in PBMCs from breast cancer patients. Immunopharmacol. Immunotoxicol. 2013, 35, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Lin, C.Y.; Chen, J.S.; Ho, C.L.; Rau, K.M.; Tsai, J.T.; Chang, C.S.; Yeh, S.P.; Cheng, C.F.; Lai, Y.L. Karnofsky Performance Status as a Predictive Factor for Cancer-Related Fatigue Treatment with Astragalus Polysaccharides (PG2) Injection—A Double Blind, Multi-Center, Randomized Phase IV Study. Cancers 2019, 11, 128. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Kuo, K.T.; Bamodu, O.A.; Lin, Y.K.; Wang, C.H.; Lee, K.Y.; Wang, L.S.; Yeh, C.T.; Tsai, J.T. Astragalus polysaccharide (PG2) Ameliorates Cancer Symptom Clusters, as well as Improves Quality of Life in Patients with Metastatic Disease, through Modulation of the Inflammatory Cascade. Cancers 2019, 11, 1054. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Yao, Y.M.; Zhang, S.W.; Sheng, Z.Y. Astragalus polysaccharides regulate T cell-mediated immunity via CD11c(high)CD45RB(low) DCs in vitro. J. Ethnopharmacol. 2011, 136, 457–464. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed.; The National Academies Collection: Reports funded by National Institutes of Health; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Sainz, B.; Carron, E.; Vallespinós, M.; Machado, H.L.; Machado, H.L. Cancer Stem Cells and Macrophages: Implications in Tumor Biology and Therapeutic Strategies. Mediat. Inflamm. 2016, 2016, 9012369. [Google Scholar] [CrossRef]
- Reck, M.; Heigener, D.F.; Mok, T.; Soria, J.C.; Rabe, K.F. Management of non-small-cell lung cancer: Recent developments. Lancet 2013, 382, 709–719. [Google Scholar] [CrossRef]
- Fennell, D.A.; Summers, Y.; Cadranel, J.; Benepal, T.; Christoph, D.C.; Lal, R.; Das, M.; Maxwell, F.; Visseren-Grul, C.; Ferry, D. Cisplatin in the modern era: The backbone of first-line chemotherapy for non-small cell lung cancer. Cancer Treat. Rev. 2016, 44, 42–50. [Google Scholar] [CrossRef]
- Shigdar, S.; Li, Y.; Bhattacharya, S.; O’Connor, M.; Pu, C.; Lin, J.; Wang, T.; Xiang, D.; Kong, L.; Wei, M.Q.; et al. Inflammation and cancer stem cells. Cancer Lett. 2014, 345, 271–278. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef]
- Hiasa, M.; Abe, M.; Nakano, A.; Oda, A.; Amou, H.; Kido, S.; Takeuchi, K.; Kagawa, K.; Yata, K.; Hashimoto, T.; et al. GM-CSF and IL-4 induce dendritic cell differentiation and disrupt osteoclastogenesis through M-CSF receptor shedding by up-regulation of TNF-alpha converting enzyme (TACE). Blood 2009, 114, 4517–4526. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Wu, J.M.; Wang, M.Y.; Wu, M.H.; Chen, K.Y.; Yeh, S.L.; Lin, M.T. Modulatory Effects of Astragalus Polysaccharides on T-Cell Polarization in Mice with Polymicrobial Sepsis. Mediat. Inflamm. 2015, 2015, 826319. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Qiu, S.D.; Chen, H.X.; Tian, H.; Wang, H.X. The immunotherapeutic effects of Astragalus polysaccharide in type 1 diabetic mice. Biol. Pharm. Bull. 2007, 30, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, X.; Zhang, Y.; Xu, J.; Zhang, L.; Li, Z.; Liu, W.; Ouyang, J.; Han, S.; He, X. Astragalus polysaccharide induces anti-inflammatory effects dependent on AMPK activity in palmitate-treated RAW264.7 cells. Int. J. Mol. Med. 2013, 31, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.H.; Ma, Z.X.; Zhu, J.; Yu, X.H.; Weng, D.P. Characterization of polysaccharide from Astragalus radix as the macrophage stimulator. Cell Immunol. 2011, 271, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Hsiao, Y.J.; Chen, H.Y.; Chen, H.W.; Ho, C.C.; Chen, Y.Y.; Liu, Y.C.; Hong, T.H.; Yu, S.L.; Chen, J.J.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef]
- Gao, L.; Zhou, Y.; Zhou, S.X.; Yu, X.J.; Xu, J.M.; Zuo, L.; Luo, Y.H.; Li, X.A. PLD4 promotes M1 macrophages to perform antitumor effects in colon cancer cells. Oncol. Rep. 2016, 37, 408–416. [Google Scholar] [CrossRef]
- Qu, Z.; Sun, F.; Zhou, J.; Li, L.; Shapiro, S.D.; Xiao, G. Interleukin-6 Prevents the Initiation but Enhances the Progression of Lung Cancer. Cancer Res. 2015, 75, 3209–3215. [Google Scholar] [CrossRef]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial–mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Kuang, C.M.; Fu, X.; Hua, Y.J.; Ye, Z.H.; Li, Y.; Peng, Q.H.; Li, Y.Z.; Chen, S.; Qian, C.N.; Huang, W.; et al. BST2 confers cisplatin resistance via NF-κB signaling in nasopharyngeal cancer. Cell Death Dis. 2017, 8, e2874. [Google Scholar] [CrossRef] [PubMed]
- Privratsky, J.R.; Tourdot, B.E.; Newman, D.K.; Newman, P.J. The Anti-Inflammatory Actions of Platelet Endothelial Cell Adhesion Molecule-1 Do Not Involve Regulation of Endothelial Cell NF-kB. J. Immunol. 2010, 184, 3157–3163. [Google Scholar] [CrossRef]
- Privratsky, J.R.; Newman, P.J. PECAM-1: Regulator of endothelial junctional integrity. Cell Tissue Res. 2014, 355, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Biswas, S.K.; Lawrence, T.; Sica, A.; Lewis, C.E. Regulation of macrophage function in tumors: The multifaceted role of NF-κB. Blood 2009, 113, 3139–3146. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Rothe, M.; Schnorfeil, F.M.; Deiser, K.; Krupka, C.; Augsberger, C.; Schlüter, M.; Neitz, J.; Subklewe, M. Targeting LAG-3 and PD-1 to Enhance T Cell Activation by Antigen-Presenting Cells. Front. Immunol. 2018, 9, 385. [Google Scholar] [CrossRef]
- Han, T.H.; Jin, P.; Ren, J.Q.; Slezak, S.; Marincola, F.M.; Stroncek, D.F. Evaluation of 3 clinical dendritic cell maturation protocols containing lipopolysaccharide and interferon-gamma. J. Immunother. 2009, 32, 399–407. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamodu, O.A.; Kuo, K.-T.; Wang, C.-H.; Huang, W.-C.; Wu, A.T.H.; Tsai, J.-T.; Lee, K.-Y.; Yeh, C.-T.; Wang, L.-S. Astragalus polysaccharides (PG2) Enhances the M1 Polarization of Macrophages, Functional Maturation of Dendritic Cells, and T Cell-Mediated Anticancer Immune Responses in Patients with Lung Cancer. Nutrients 2019, 11, 2264. https://doi.org/10.3390/nu11102264
Bamodu OA, Kuo K-T, Wang C-H, Huang W-C, Wu ATH, Tsai J-T, Lee K-Y, Yeh C-T, Wang L-S. Astragalus polysaccharides (PG2) Enhances the M1 Polarization of Macrophages, Functional Maturation of Dendritic Cells, and T Cell-Mediated Anticancer Immune Responses in Patients with Lung Cancer. Nutrients. 2019; 11(10):2264. https://doi.org/10.3390/nu11102264
Chicago/Turabian StyleBamodu, Oluwaseun Adebayo, Kuang-Tai Kuo, Chun-Hua Wang, Wen-Chien Huang, Alexander T.H. Wu, Jo-Ting Tsai, Kang-Yun Lee, Chi-Tai Yeh, and Liang-Shun Wang. 2019. "Astragalus polysaccharides (PG2) Enhances the M1 Polarization of Macrophages, Functional Maturation of Dendritic Cells, and T Cell-Mediated Anticancer Immune Responses in Patients with Lung Cancer" Nutrients 11, no. 10: 2264. https://doi.org/10.3390/nu11102264
APA StyleBamodu, O. A., Kuo, K.-T., Wang, C.-H., Huang, W.-C., Wu, A. T. H., Tsai, J.-T., Lee, K.-Y., Yeh, C.-T., & Wang, L.-S. (2019). Astragalus polysaccharides (PG2) Enhances the M1 Polarization of Macrophages, Functional Maturation of Dendritic Cells, and T Cell-Mediated Anticancer Immune Responses in Patients with Lung Cancer. Nutrients, 11(10), 2264. https://doi.org/10.3390/nu11102264