A Designer Synbiotic Attenuates Chronic-Binge Ethanol-Induced Gut-Liver Injury in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Ethanol Exposure Model and Supplementations
2.3. Biochemical Assays
2.4. Immunohistochemistry
2.5. Oil Red O Staining
2.6. Western Blot
2.7. Real Time Polymerase Chain Reaction
2.8. Detection of Short-Chain Fatty Acids in Fecal Samples
2.9. Statistical Analysis
3. Results
3.1. Synbiotic Supplementation Altered SCFA Levels in Cecum
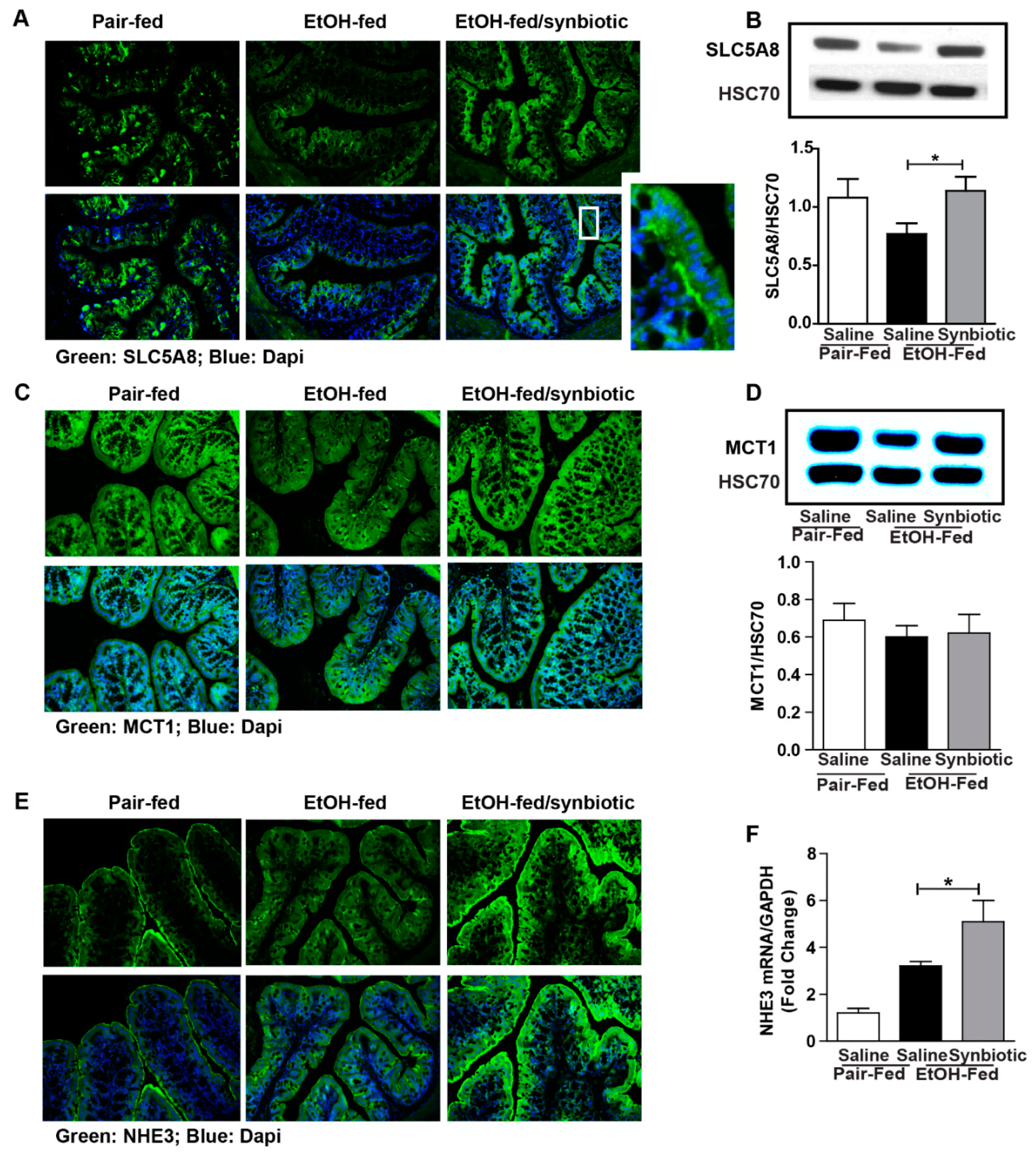
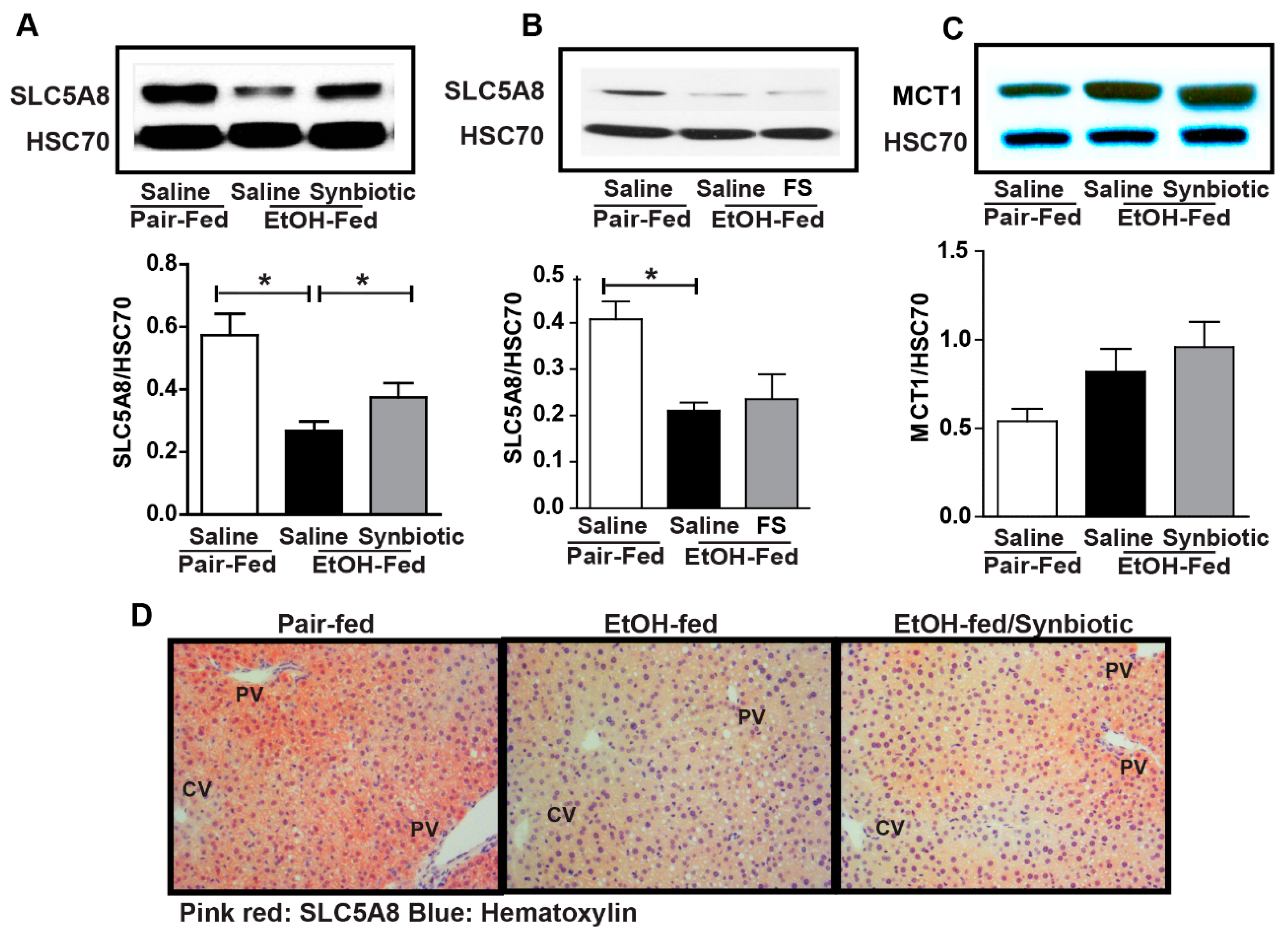
3.2. Synbiotic Supplementation Mitigated Losses in SCFA Transport Mechanisms Altered by Ethanol Exposure
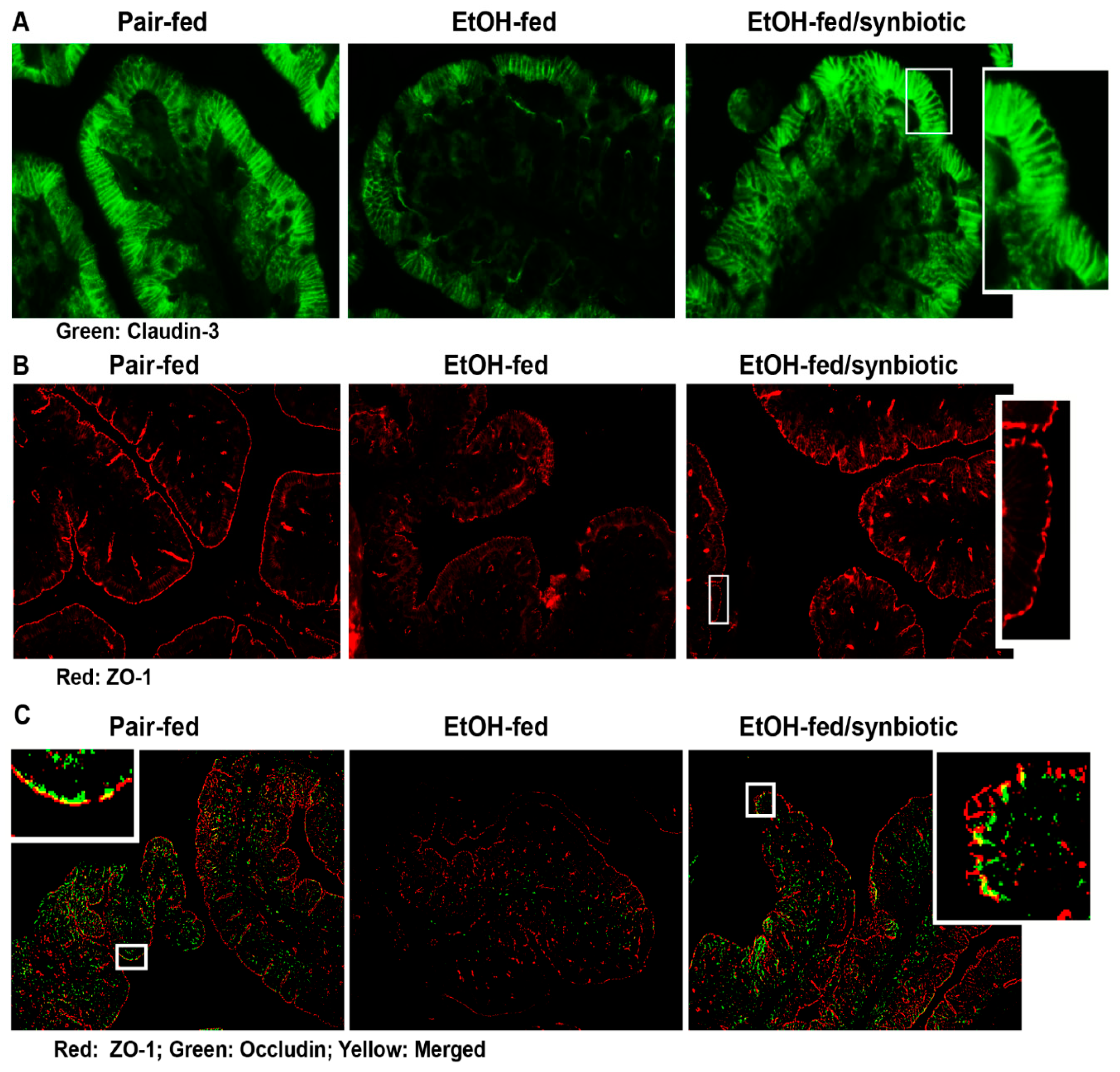
3.3. Ethanol-Induced Alterations of Tight Junction Proteins in Proximal Colon Was Mitigated with Synbiotic Supplementation
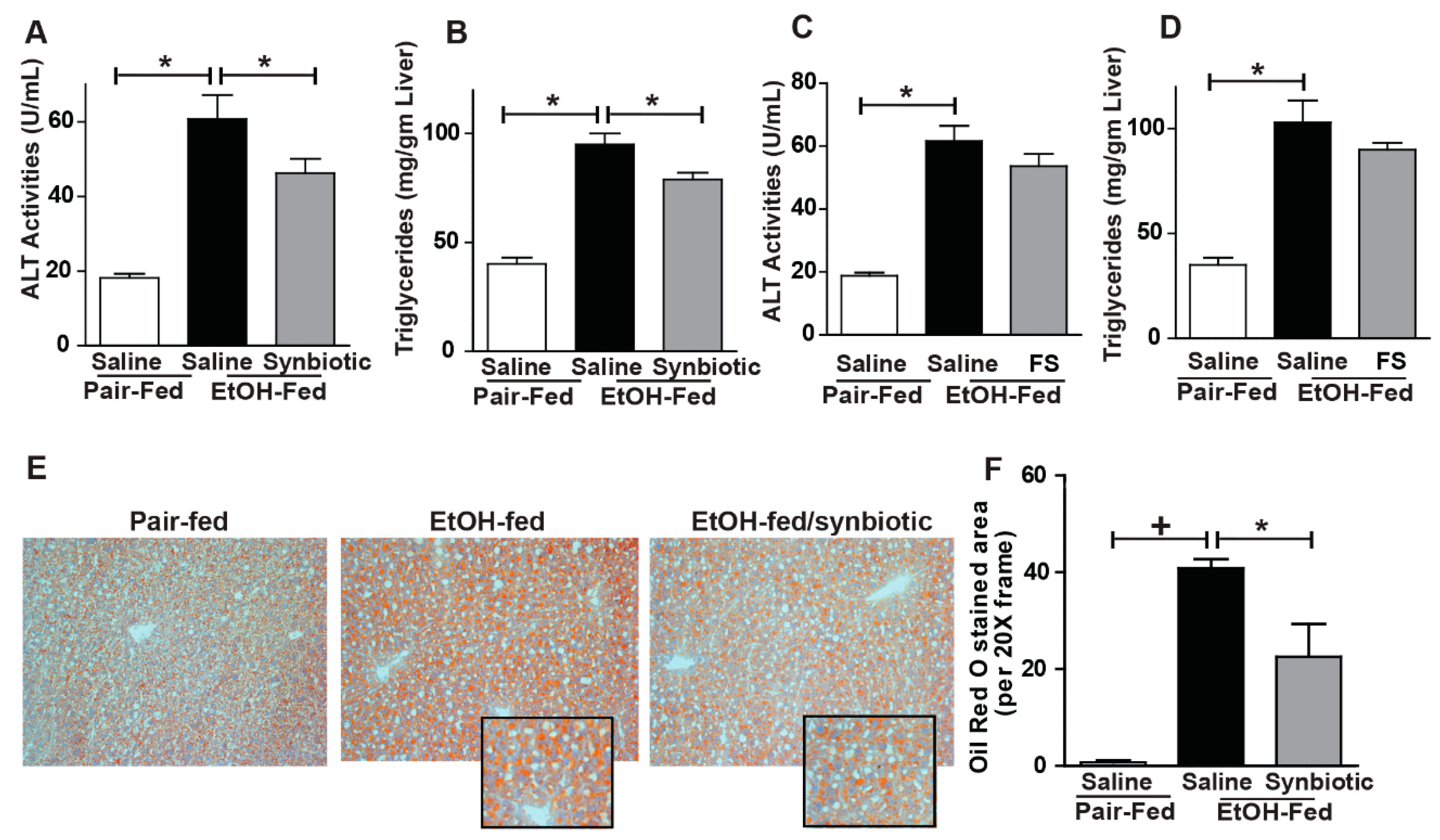
3.4. Synbiotic Supplementation Attenuated against Chronic-Binge Ethanol-Induced Steatosis and Liver Injury
3.5. Synbiotic Supplementation Reduces Chronic-Binge Ethanol-Induced TNFα Expression and 4-HNE-Adduct Accumulation in Mouse Liver
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boyle, M.; Masson, S.; Anstee, Q.M. The bidirectional impacts of alcohol consumption and the metabolic syndrome: Cofactors for progressive fatty liver disease. J. Hepatol. 2018, 68, 251–267. [Google Scholar] [CrossRef] [PubMed]
- Oshita, M.; Takei, Y.; Kawano, S.; Yoshihara, H.; Hijioka, T.; Fukui, H.; Goto, M.; Masuda, E.; Nishimura, Y.; Fusamoto, H.; Kamada, T. Roles of endothelin-1 and nitric oxide in the mechanism for ethanol-induced vasoconstriction in rat liver. J. Clin. Investig. 1993, 91, 1337–1342. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Kharbanda, K.K. Multi-organ alcohol-related damage: Mechanisms and treatment. Biomolecules 2016, 6, 20. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How Is Alcohol Metabolized by the Body? Annu. Rev. Nutr. 2006, 29, 245–254. [Google Scholar] [CrossRef]
- Li, F.; Duan, K.; Wang, C.; Mcclain, C.; Feng, W. Probiotics and Alcoholic Liver Disease: Treatment and Potential Mechanisms. Gastroenterol. Res. Pract. 2015, 2016. [Google Scholar] [CrossRef] [PubMed]
- Engen, P.A.; Green, S.J.; Voigt, R.M.; Forsyth, C.B.; Keshavarzian, A. The Gastrointestinal Microbiome. Alcohol. Res. 2015, 37, 223–236. [Google Scholar] [CrossRef]
- Xie, G.; Zhong, W.; Zheng, X.; Li, Q.; Qiu, Y.; Li, H.; Chen, H.; Zhou, Z.; Jia, W. Chronic ethanol consumption alters mammalian gastrointestinal content metabolites. J. Proteome Res. 2013, 12, 3297–3306. [Google Scholar] [CrossRef]
- Yang, F.; Wang, L.K.; Li, X.; Wang, L.W.; Han, X.Q.; Gong, Z.J. Sodium butyrate protects against toxin-induced acute liver failure in rats. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 309–315. [Google Scholar] [CrossRef]
- Sun, J.; Wu, Q.; Sun, H.; Qiao, Y. Inhibition of histone deacetylase by butyrate protects rat liver from ischemic reperfusion injury. Int. J. Mol. Sci. 2014, 15, 21069–21079. [Google Scholar] [CrossRef]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32. [Google Scholar] [CrossRef]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document: The international scientific association for probiotics and prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef]
- Kirpich, I.A.; Solovieva, N.V.; Leikhter, S.N.; Shidakova, N.A.; Lebedeva, O.V.; Sidorov, P.I.; Bazhukova, T.A.; Soloviev, A.G.; Barve, S.S.; McClain, C.J.; et al. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: A pilot study. Alcohol 2008, 42, 675–682. [Google Scholar] [CrossRef]
- Barone, R.; Rappa, F.; Macaluso, F.; Bavisotto, C.C.; Sangiorgi, C.; Di Paola, G.; Tomasello, G.; Di Felice, V.; Marcianò, V.; Farina, F.; et al. Alcoholic liver disease: A mouse model reveals protection by Lactobacillus fermentum. Clin. Transl. Gastroenterol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van De Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lin, C.; Zhang, Y.; Deng, Y.; Liu, C.; Yang, Q. Probiotic mixture of Lactobacillus and Bifidobacterium alleviates systemic adiposity and inflammation in non-alcoholic fatty liver disease rats through Gpr109a and the commensal metabolite butyrate. Inflammopharmacology 2018, 26, 1051–1055. [Google Scholar] [CrossRef]
- Carlson, J.L.; Erickson, J.M.; Lloyd, B.B.; Slavin, J.L. Health Effects and Sources of Prebiotic Dietary Fiber. Curr. Dev. Nutr. 2018, 1–8. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Cadnum, J.; Glueck, B.; Obrenovich, M.; Donskey, C.; Cresci, G.A.M. Faecalibacterium prausnitzii and a Prebiotic Protect Intestinal Health in a Mouse Model of Antibiotic and Clostridium difficile Exposure. J. Parenter. Enter. Nutr. 2018, 42, 1156–1167. [Google Scholar] [CrossRef]
- Mohammad, M.A.; Haymond, M.W. Regulation of lipid synthesis genes and milk fat production in human mammary epithelial cells during secretory activation. AJP Endocrinol. Metab. 2013, 305, E700–E716. [Google Scholar] [CrossRef]
- Liu, H.; Wang, J.; He, T.; Becker, S.; Zhang, G.; Li, D.; Ma, X. Butyrate: A double-edged sword for health? Adv. Nutr. 2018, 9, 21–29. [Google Scholar] [CrossRef]
- Gonçalves, P.; Martel, F. Regulation of colonic epithelial butyrate transport: Focus on colorectal cancer. Porto Biomed. J. 2016, 1, 83–91. [Google Scholar] [CrossRef]
- Velazquez, O.C.; Lederer, H.M.; Rombeau, J.L. Butyrate and the colonocyte: production, absorption, metabolism, and therapeutic implications. In Dietary Fiber in Health and Disease; Kritchevsky, D., Bonfield, C., Eds.; Plenum Press: New York, NY, USA, 1997. [Google Scholar]
- Mitic, L.L.; Anderson, J.M. Molecular architecture. Nat. New Biol. 1971. [Google Scholar] [CrossRef]
- Elamin, E.E.; Masclee, A.A.; Dekker, J.; Jonkers, D.M. Ethanol metabolism and its effects on the intestinal epithelial barrier. Nutr. Rev. 2013, 71, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Wang, K. Molecular mechanisms of hepatic apoptosis. Cell Death Dis. 2014, 5, e996. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Xu, G.S.; Li, Y.L.; Yang, J.H.; Yan, N.; Liu, L.; Yuan, S.; Luo, Z.P.; Sang, C.F.; Gu, S.; et al. Visible imaging measurement of position and displacement of the last closed flux surface in EAST tokamak. Fusion Eng. Des. 2017, 119, 42–50. [Google Scholar] [CrossRef]
- Khalesi, S.; Johnson, D.W.; Campbell, K.; Williams, S.; Fenning, A.; Saluja, S.; Irwin, C. Effect of probiotics and synbiotics consumption on serum concentrations of liver function test enzymes: A systematic review and meta-analysis. Eur. J. Nutr. 2018, 57, 2037–2053. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.H.; Macfarlane, G.T.; Englyst, H.N. Prebiotic digestion and fermentation. Am. J. Clin. Nutr. 2001, 73, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Rios-Covian, D.; Gueimonde, M.; Duncan, S.H.; Flint, H.J.; De Los Reyes-Gavilan, C.G. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. FEMS Microbiol. Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.M.; Dumon, H.J.W.; Lecannu, G.; Champ, M.M.J. Potato and high-amylose maize starches are not equivalent producers of butyrate for the colonic mucosa. Br. J. Nutr. 2000, 84, 689–696. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Miquel, S.; Leclerc, M.; Martin, R.; Chain, F.; Lenoir, M.; Raguideau, S.; Hudault, S. Identification of Metabolic Signatures Linked to Anti-Inflammatory Effects of Faecalibacterium prausnitzii. MBio 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Munukka, E.; Rintala, A.; Toivonen, R.; Nylund, M.; Yang, B.; Takanen, A.; Hänninen, A.; Vuopio, J.; Huovinen, P.; Jalkanen, S.; Pekkala, S. Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 2017, 11, 1667–1679. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2017, 19, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Belenguer, A.; Duncan, S.H.; Calder, A.G.; Holtrop, G.; Louis, P.; Lobley, G.E.; Flint, H.J. Two Routes of Metabolic Cross-Feeding between. Society 2006, 72, 3593–3599. [Google Scholar] [CrossRef]
- Den Besten, G.; Lange, K.; Havinga, R.; van Dijk, T.H.; Gerding, A.; van Eunen, K.; Muller, M.; Groen, A.K.; Hooiveld, G.J.; Bakker, B.M.; Reijngoud, D.-J. Gut-derived short-chain fatty acids are vividly assimilated into host carbohydrates and lipids. AJP Gastrointest. Liver Physiol. 2013, 305, G900–G910. [Google Scholar] [CrossRef]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915. [Google Scholar] [CrossRef]
- El Aidy, S.; Van Den Abbeele, P.; Van De Wiele, T.; Louis, P.; Kleerebezem, M. Intestinal colonization: How key microbial players become established in this dynamic process: Microbial metabolic activities and the interplay between the host and microbes Prospects & Overviews S. E. Aidy et al. BioEssays 2013, 35, 913–923. [Google Scholar] [CrossRef]
- Cresci, G.A.; Thangaraju, M.; Mellinger, J.D.; Liu, K.; Ganapathy, V. Colonic gene expression in conventional and germ-free mice with a focus on the butyrate receptor GPR109A and the butyrate transporter SLC5A8. J. Gastrointest. Surg. 2010, 14. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-chain fatty acid transporters: role in colonic homeostasis. Compr Physiol 2018, 8, 299–314. [Google Scholar] [CrossRef]
- Koho, N.; Maijala, V.; Norberg, H.; Nieminen, M.; Pösö, A.R. Expression of MCT1, MCT2 and MCT4 in the rumen, small intestine and liver of reindeer (Rangifer tarandus tarandus L.). Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2005, 141, 29–34. [Google Scholar] [CrossRef]
- Goncalves, P.; Arajo, J.R.; Martel, F. Characterization of butyrate uptake by nontransformed intestinal epithelial cell lines. J. Membr. Biol. 2011, 240, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, P.; Gregório, I.; Catarino, T.A.; Martel, F. The effect of oxidative stress upon the intestinal epithelial uptake of butyrate. Eur. J. Pharmacol. 2013, 699, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Kappo, K.A.; Petzke, K.J.; Kipp, A.P.; Blaut, M.; Klaus, S. Importance of propionate for the repression of hepatic lipogenesis and improvement of insulin sensitivity in high-fat diet-induced obesity. Mol. Nutr. Food Res. 2016, 60, 2611–2621. [Google Scholar] [CrossRef] [PubMed]
- Mollica, M.P.; Raso, G.M.; Cavaliere, G.; Trinchese, G.; De Filippo, C.; Aceto, S.; Prisco, M.; Pirozzi, C.; Di Guida, F.; Lama, A.; et al. Butyrate regulates liver mitochondrial function, efficiency, and dynamics in insulin-resistant obese mice. Diabetes 2017, 66, 1405–1418. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roychowdhury, S.; Glueck, B.; Han, Y.; Mohammad, M.A.; Cresci, G.A.M. A Designer Synbiotic Attenuates Chronic-Binge Ethanol-Induced Gut-Liver Injury in Mice. Nutrients 2019, 11, 97. https://doi.org/10.3390/nu11010097
Roychowdhury S, Glueck B, Han Y, Mohammad MA, Cresci GAM. A Designer Synbiotic Attenuates Chronic-Binge Ethanol-Induced Gut-Liver Injury in Mice. Nutrients. 2019; 11(1):97. https://doi.org/10.3390/nu11010097
Chicago/Turabian StyleRoychowdhury, Sanjoy, Bryan Glueck, Yingchun Han, Mahmoud Ali Mohammad, and Gail A. M. Cresci. 2019. "A Designer Synbiotic Attenuates Chronic-Binge Ethanol-Induced Gut-Liver Injury in Mice" Nutrients 11, no. 1: 97. https://doi.org/10.3390/nu11010097
APA StyleRoychowdhury, S., Glueck, B., Han, Y., Mohammad, M. A., & Cresci, G. A. M. (2019). A Designer Synbiotic Attenuates Chronic-Binge Ethanol-Induced Gut-Liver Injury in Mice. Nutrients, 11(1), 97. https://doi.org/10.3390/nu11010097