Role of Dietary Lipids in Modulating Inflammation through the Gut Microbiota

{kind=link}
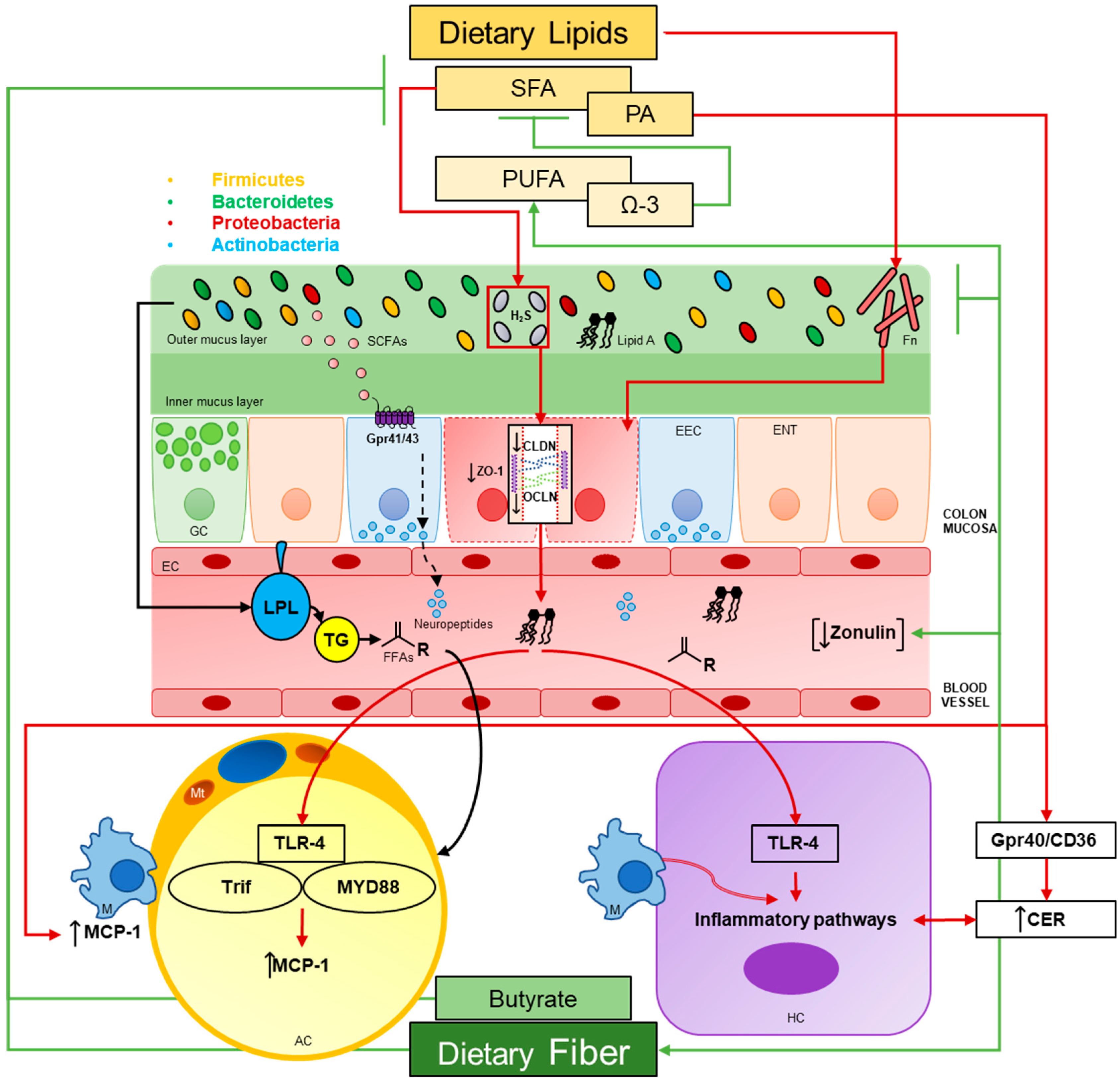
Abstract
1. Introduction
2. Role of Microorganisms in Energy Regulation
2.1. SCFAs
2.2. Studies Investigating SCFA in Humans
2.3. Increased Energy Harvesting
2.4. Dietary Protein and Energy Regulation
2.5. Studies Investigating Increased Dietary Protein Intake in Humans
3. LPS, Inflammation and Dietary Lipids
3.1. Gut Permeability
3.1.1. HFDs
3.1.2. Microbial Secretory Metabolites
3.1.3. Gut Permeability in Humans
3.1.4. Fusobacterium and Colorectal Cancer
3.2. White Adipose Tissue
3.3. Liver
3.3.1. Total Fat Content
3.3.2. Ceramide De Novo Synthesis and PA
3.3.3. Butyrate Supplementation
3.3.4. Bile Acids
3.3.5. NAFLD in Humans
3.4. LPS as Immunosuppressive
4. Endocannabinoid System and the Microbiota-Emerging Area
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Goulet, O. Potential role of the intestinal microbiota in programming health and disease. Nutr. Rev. 2015, 73 (Suppl. 1), 32–40. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef] [PubMed]
- Gummesson, A.; Carlsson, L.M.; Storlien, L.H.; Backhed, F.; Lundin, P.; Lofgren, L.; Stenlof, K.; Lam, Y.Y.; Fagerberg, B.; Carlsson, B. Intestinal permeability is associated with visceral adiposity in healthy women. Obesity 2011, 19, 2280–2282. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.D.; Grice, D.M.; Wade, B.; Kerr, C.A.; Bauer, D.C.; Li, D.; Hannan, G.N. Gut permeability, its interaction with gut microflora and effects on metabolic health are mediated by the lymphatics system, liver and bile acid. Future Microbiol. 2015, 10, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Pereira, S.S.; Alvarez-Leite, J.I. Low-grade inflammation, obesity, and diabetes. Curr. Obes. Rep. 2014, 3, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yin, A.; Li, H.; Wang, R.; Wu, G.; Shen, J.; Zhang, M.; Wang, L.; Hou, Y.; Ouyang, H.; et al. Dietary modulation of gut microbiota contributes to alleviation of both genetic and simple obesity in children. EBioMedicine 2015, 2, 968–984. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef]
- Strober, W. Impact of the gut microbiome on mucosal inflammation. Trends Immunol. 2013, 34, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojarvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, F.; Ding, X.; Wu, G.; Lam, Y.Y.; Wang, X.; Fu, H.; Xue, X.; Lu, C.; Ma, J.; et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 2018, 359, 1151–1156. [Google Scholar] [CrossRef] [PubMed]
- Krajmalnik-Brown, R.; Ilhan, Z.E.; Kang, D.W.; DiBaise, J.K. Effects of gut microbes on nutrient absorption and energy regulation. Nutr. Clin. Pract. 2012, 27, 201–214. [Google Scholar] [CrossRef]
- Molloy, M.J.; Bouladoux, N.; Belkaid, Y. Intestinal microbiota: Shaping local and systemic immune responses. Semin. Immunol. 2012, 24, 58–66. [Google Scholar] [CrossRef]
- Salyers, A.A.; West, S.E.; Vercellotti, J.R.; Wilkins, T.D. Fermentation of mucins and plant polysaccharides by anaerobic bacteria from the human colon. Appl. Environ. Microbiol. 1977, 34, 529–533. [Google Scholar]
- Sonnenburg, E.D.; Sonnenburg, J.L. Starving our microbial self: The deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014, 20, 779–786. [Google Scholar] [CrossRef]
- Cockburn, D.W.; Koropatkin, N.M. Polysaccharide degradation by the intestinal microbiota and its influence on human health and disease. J. Mol. Biol. 2016, 428, 3230–3252. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [PubMed]
- Layden, B.T.; Angueira, A.R.; Brodsky, M.; Durai, V.; Lowe, W.L., Jr. Short chain fatty acids and their receptors: New metabolic targets. Transl. Res. 2013, 161, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Sudo, N.; Chida, Y.; Aiba, Y.; Sonoda, J.; Oyama, N.; Yu, X.N.; Kubo, C.; Koga, Y. Postnatal microbial colonization programs the hypothalamic-pituitary-adrenal system for stress response in mice. J. Physiol. 2004, 558, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.M.; de Souza, R.; Kendall, C.W.; Emam, A.; Jenkins, D.J. Colonic health: Fermentation and short chain fatty acids. J. Clin. Gastroenterol. 2006, 40, 235–243. [Google Scholar] [CrossRef]
- Soldavini, J.; Kaunitz, J.D. Pathobiology and potential therapeutic value of intestinal short-chain fatty acids in gut inflammation and obesity. Dig. Dis. Sci. 2013, 58, 2756–2766. [Google Scholar] [CrossRef]
- Macfarlane, S.; Macfarlane, G.T. Regulation of short-chain fatty acid production. Proc. Nutr. Soc. 2003, 62, 67–72. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef]
- Dalby, M.J.; Ross, A.W.; Walker, A.W.; Morgan, P.J. Dietary uncoupling of gut microbiota and energy harvesting from obesity and glucose tolerance in mice. Cell Rep. 2017, 21, 1521–1533. [Google Scholar] [CrossRef]
- Riviere, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon bacteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Redondo-Blanco, S.; Guitiérrez-del-Río, I.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: A review. J. Funct. Foods 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Cao, Y.; Shen, J.; Ran, Z.H. Association between faecalibacterium prausnitzii reduction and inflammatory bowel disease: A meta-analysis and systematic review of the literature. Gastroenterol. Res. Pract. 2014, 2014, 872725. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Khan, M.T.; Paglia, G.; Rodionov, D.A.; Harmsen, H.J.; Thiele, I. Functional metabolic map of faecalibacterium prausnitzii, a beneficial human gut microbe. J. Bacteriol. 2014, 196, 3289–3302. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.C.; Wisniewski, P.J.; Noji, M.; McGuinness, L.R.; Haggblom, M.M.; Lightfoot, S.A.; Joseph, L.B.; Kerkhof, L.J. The effect of diet and exercise on intestinal integrity and microbial diversity in mice. PLoS ONE 2016, 11, e0150502. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Macfarlane, G.T.; Cummings, J.H. Sulphate reducing bacteria and hydrogen metabolism in the human large intestine. Gut 1993, 34, 437–439. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from europe and rural africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Roshanravan, N.; Mahdavi, R.; Alizadeh, E.; Jafarabadi, M.A.; Hedayati, M.; Ghavami, A.; Alipour, S.; Alamdari, N.M.; Barati, M.; Ostadrahimi, A. Effect of butyrate and inulin supplementation on glycemic status, lipid profile and glucagon-like peptide 1 level in patients with type 2 diabetes: A randomized double-blind, placebo-controlled trial. Horm. Metab. Res. 2017, 49, 886–891. [Google Scholar] [CrossRef]
- Roshanravan, N.; Mahdavi, R.; Alizadeh, E.; Ghavami, A.; Rahbar Saadat, Y.; Mesri Alamdari, N.; Alipour, S.; Dastouri, M.R.; Ostadrahimi, A. The effects of sodium butyrate and inulin supplementation on angiotensin signaling pathway via promotion of akkermansia muciniphila abundance in type 2 diabetes; a randomized, double-blind, placebo-controlled trial. J. Cardiovasc. Thorac. Res. 2017, 9, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a ppargamma-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef]
- Towle, H.C. Glucose and camp: Adversaries in the regulation of hepatic gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 13476–13478. [Google Scholar] [CrossRef]
- Zhou, J.; Martin, R.J.; Tulley, R.T.; Raggio, A.M.; McCutcheon, K.L.; Shen, L.; Danna, S.C.; Tripathy, S.; Hegsted, M.; Keenan, M.J. Dietary resistant starch upregulates total glp-1 and pyy in a sustained day-long manner through fermentation in rodents. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1160–E1166. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Kyrou, I.; Tan, B.K.; Dimitriadis, G.K.; Ramanjaneya, M.; Tripathi, G.; Patel, V.; James, S.; Kawan, M.; Chen, J.; et al. Short-chain fatty acid acetate stimulates adipogenesis and mitochondrial biogenesis via gpr43 in brown adipocytes. Endocrinology 2016, 157, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding g protein-coupled receptor, gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Milligan, G.; Stoddart, L.A.; Smith, N.J. Agonism and allosterism: The pharmacology of the free fatty acid receptors ffa2 and ffa3. Br. J. Pharmacol. 2009, 158, 146–153. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef]
- Gaudichon, C.; Bos, C.; Morens, C.; Petzke, K.J.; Mariotti, F.; Everwand, J.; Benamouzig, R.; Dare, S.; Tome, D.; Metges, C.C. Ileal losses of nitrogen and amino acids in humans and their importance to the assessment of amino acid requirements. Gastroenterology 2002, 123, 50–59. [Google Scholar] [CrossRef]
- Gibson, J.A.; Sladen, G.E.; Dawson, A.M. Protein absorption and ammonia production: The effects of dietary protein and removal of the colon. Br. J. Nutr. 1976, 35, 61–65. [Google Scholar] [CrossRef]
- Roager, H.M.; Hansen, L.B.; Bahl, M.I.; Frandsen, H.L.; Carvalho, V.; Gobel, R.J.; Dalgaard, M.D.; Plichta, D.R.; Sparholt, M.H.; Vestergaard, H.; et al. Colonic transit time is related to bacterial metabolism and mucosal turnover in the gut. Nat. Microbiol. 2016, 1, 16093. [Google Scholar] [CrossRef]
- Macfarlane, G.T.; Cummings, J.H.; Macfarlane, S.; Gibson, G.R. Influence of retention time on degradation of pancreatic enzymes by human colonic bacteria grown in a 3-stage continuous culture system. J. Appl. Bacteriol. 1989, 67, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Geypens, B.; Claus, D.; Evenepoel, P.; Hiele, M.; Maes, B.; Peeters, M.; Rutgeerts, P.; Ghoos, Y. Influence of dietary protein supplements on the formation of bacterial metabolites in the colon. Gut 1997, 41, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Nyangale, E.P.; Mottram, D.S.; Gibson, G.R. Gut microbial activity, implications for health and disease: The potential role of metabolite analysis. J. Proteome Res. 2012, 11, 5573–5585. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Blouin, J.M.; Santacruz, A.; Lan, A.; Andriamihaja, M.; Wilkanowicz, S.; Benetti, P.H.; Tome, D.; Sanz, Y.; Blachier, F.; et al. High-protein diet modifies colonic microbiota and luminal environment but not colonocyte metabolism in the rat model: The increased luminal bulk connection. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G459–G470. [Google Scholar] [CrossRef] [PubMed]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Windey, K.; De Preter, V.; Louat, T.; Schuit, F.; Herman, J.; Vansant, G.; Verbeke, K. Modulation of protein fermentation does not affect fecal water toxicity: A randomized cross-over study in healthy subjects. PLoS ONE 2012, 7, e52387. [Google Scholar] [CrossRef]
- Vidal-Lletjos, S.; Beaumont, M.; Tome, D.; Benamouzig, R.; Blachier, F.; Lan, A. Dietary protein and amino acid supplementation in inflammatory bowel disease course: What impact on the colonic mucosa? Nutrients 2017, 9, 310. [Google Scholar] [CrossRef]
- Alexander, C.; Rietschel, E.T. Bacterial lipopolysaccharides and innate immunity. J. Endotoxin Res. 2001, 7, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Raetz, C.R.; Whitfield, C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective lps signaling in c3h/hej and c57bl/10sccr mice: Mutations in tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (tlr4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for tlr4 as the lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar]
- Beutler, B.; Cerami, A. Tumor necrosis, cachexia, shock, and inflammation: A common mediator. Annu. Rev. Biochem. 1988, 57, 505–518. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1 and interleukin-1 antagonism. Blood 1991, 77, 1627–1652. [Google Scholar] [PubMed]
- Medzhitov, R.; Janeway, C., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.D.; Ramos, R.A.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. Cd14, a receptor for complexes of lipopolysaccharide (lps) and lps binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Moore, K.J.; Andersson, L.P.; Ingalls, R.R.; Monks, B.G.; Li, R.; Arnaout, M.A.; Golenbock, D.T.; Freeman, M.W. Divergent response to lps and bacteria in cd14-deficient murine macrophages. J. Immunol. 2000, 165, 4272–4280. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Shan, Y.; Fan, Y.; Fan, C.; Chen, S.; Sun, J.; Zhu, L.; Qin, L.; Yu, M.; Lin, Z. Nf-kappab inhibition attenuates lps-induced tlr4 activation in monocyte cells. Mol. Med. Rep. 2016, 14, 4505–4510. [Google Scholar] [CrossRef]
- Hooper, L.V.; Macpherson, A.J. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat. Rev. Immunol. 2010, 10, 159–169. [Google Scholar] [CrossRef]
- Lam, Y.Y.; Ha, C.W.; Campbell, C.R.; Mitchell, A.J.; Dinudom, A.; Oscarsson, J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PLoS ONE 2012, 7, e34233. [Google Scholar] [CrossRef]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Berge, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C.; et al. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.; Hoffmann, J.M.; Oscarsson, J.; Dinudom, A.; Mather, T.J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Effects of dietary fat profile on gut permeability and microbiota and their relationships with metabolic changes in mice. Obesity 2015, 23, 1429–1439. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in il10-/- mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.K.; Boudry, G.; Lemay, D.G.; Raybould, H.E. Changes in intestinal barrier function and gut microbiota in high-fat diet-fed rats are dynamic and region dependent. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G840–G851. [Google Scholar] [CrossRef] [PubMed]
- Ellis, T.N.; Kuehn, M.J. Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 2010, 74, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Bang, J.Y.; Park, G.W.; Choi, D.S.; Kang, J.S.; Kim, H.J.; Park, K.S.; Lee, J.O.; Kim, Y.K.; Kwon, K.H.; et al. Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 2007, 7, 3143–3153. [Google Scholar] [CrossRef]
- Horstman, A.L.; Kuehn, M.J. Bacterial surface association of heat-labile enterotoxin through lipopolysaccharide after secretion via the general secretory pathway. J. Biol. Chem. 2002, 277, 32538–32545. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.W.; Kim, M.R.; Lee, E.Y.; Kim, J.H.; Kim, Y.S.; Jeon, S.G.; Yang, J.M.; Lee, B.J.; Pyun, B.Y.; Gho, Y.S.; et al. Extracellular vesicles derived from staphylococcus aureus induce atopic dermatitis-like skin inflammation. Allergy 2011, 66, 351–359. [Google Scholar] [CrossRef]
- Kim, Y.S.; Choi, E.J.; Lee, W.H.; Choi, S.J.; Roh, T.Y.; Park, J.; Jee, Y.K.; Zhu, Z.; Koh, Y.Y.; Gho, Y.S.; et al. Extracellular vesicles, especially derived from gram-negative bacteria, in indoor dust induce neutrophilic pulmonary inflammation associated with both th1 and th17 cell responses. Clin. Exp. Allergy 2013, 43, 443–454. [Google Scholar] [CrossRef]
- Kuehn, M.J.; Kesty, N.C. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005, 19, 2645–2655. [Google Scholar] [CrossRef]
- Chelakkot, C.; Choi, Y.; Kim, D.K.; Park, H.T.; Ghim, J.; Kwon, Y.; Jeon, J.; Kim, M.S.; Jee, Y.K.; Gho, Y.S.; et al. Akkermansia muciniphila-derived extracellular vesicles influence gut permeability through the regulation of tight junctions. Exp. Mol. Med. 2018, 50, e450. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Lee, W.H.; Choi, E.J.; Choi, J.P.; Heo, Y.J.; Gho, Y.S.; Jee, Y.K.; Oh, Y.M.; Kim, Y.K. Extracellular vesicles derived from gram-negative bacteria, such as Escherichia coli, induce emphysema mainly via il-17a-mediated neutrophilic inflammation. J. Immunol. 2015, 194, 3361–3368. [Google Scholar] [CrossRef] [PubMed]
- Mangell, P.; Nejdfors, P.; Wang, M.; Ahrne, S.; Westrom, B.; Thorlacius, H.; Jeppsson, B. Lactobacillus plantarum 299v inhibits Escherichia coli-induced intestinal permeability. Dig. Dis. Sci. 2002, 47, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Choi, K.H.; Kim, Y.S.; Hong, B.S.; Kim, O.Y.; Kim, J.H.; Yoon, C.M.; Koh, G.Y.; Kim, Y.K.; Gho, Y.S. Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS ONE 2010, 5, e11334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jouret, F.; Rinehart, J.; Sfakianos, J.; Mellman, I.; Lifton, R.P.; Young, L.H.; Caplan, M.J. Amp-activated protein kinase (ampk) activation and glycogen synthase kinase-3beta (gsk-3beta) inhibition induce Ca2+-independent deposition of tight junction components at the plasma membrane. J. Biol. Chem. 2011, 286, 16879–16890. [Google Scholar] [CrossRef]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by amp-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. Amp-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Fujio, S.; Navarrete, P.; Ugalde, J.A.; Magne, F.; Carrasco-Pozo, C.; Tralma, K.; Quezada, M.; Hurtado, C.; Covarrubias, N.; et al. Impact of dietary lipids on colonic function and microbiota: An experimental approach involving orlistat-induced fat malabsorption in human volunteers. Clin. Transl. Gastroenterol. 2016, 7, e161. [Google Scholar] [CrossRef]
- Carriere, F.; Renou, C.; Ransac, S.; Lopez, V.; De Caro, J.; Ferrato, F.; De Caro, A.; Fleury, A.; Sanwald-Ducray, P.; Lengsfeld, H.; et al. Inhibition of gastrointestinal lipolysis by orlistat during digestion of test meals in healthy volunteers. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G16–G28. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (frap) as a measure of “antioxidant power”: The frap assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Mokkala, K.; Roytio, H.; Munukka, E.; Pietila, S.; Ekblad, U.; Ronnemaa, T.; Eerola, E.; Laiho, A.; Laitinen, K. Gut microbiota richness and composition and dietary intake of overweight pregnant women are related to serum zonulin concentration, a marker for intestinal permeability. J. Nutr. 2016, 146, 1694–1700. [Google Scholar] [CrossRef] [PubMed]
- Genser, L.; Aguanno, D.; Soula, H.A.; Dong, L.; Trystram, L.; Assmann, K.; Salem, J.E.; Vaillant, J.C.; Oppert, J.M.; Laugerette, F.; et al. Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. J. Pathol. 2018, 246, 217–230. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Amasheh, S.; Richter, J.F.; Milatz, S.; Gunzel, D.; Westphal, J.K.; Huber, O.; Schulzke, J.D.; Fromm, M. Tricellulin forms a barrier to macromolecules in tricellular tight junctions without affecting ion permeability. Mol. Biol. Cell 2009, 20, 3713–3724. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Miquel, S.; Benevides, L.; Bridonneau, C.; Robert, V.; Hudault, S.; Chain, F.; Berteau, O.; Azevedo, V.; Chatel, J.M.; et al. Functional characterization of novel faecalibacterium prausnitzii strains isolated from healthy volunteers: A step forward in the use of f. Prausnitzii as a next-generation probiotic. Front. Microbiol. 2017, 8, 1226. [Google Scholar] [CrossRef] [PubMed]
- Neut, C.; Bulois, P.; Desreumaux, P.; Membre, J.M.; Lederman, E.; Gambiez, L.; Cortot, A.; Quandalle, P.; van Kruiningen, H.; Colombel, J.F. Changes in the bacterial flora of the neoterminal ileum after ileocolonic resection for crohn’s disease. Am. J. Gastroenterol. 2002, 97, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Ohkusa, T.; Sato, N.; Ogihara, T.; Morita, K.; Ogawa, M.; Okayasu, I. Fusobacterium varium localized in the colonic mucosa of patients with ulcerative colitis stimulates species-specific antibody. J. Gastroenterol. Hepatol. 2002, 17, 849–853. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.; Kaplan, G.G.; Beck, P.L.; Rioux, K.; Panaccione, R.; Devinney, R.; Lynch, T.; Allen-Vercoe, E. Invasive potential of gut mucosa-derived fusobacterium nucleatum positively correlates with ibd status of the host. Inflamm. Bowel Dis. 2011, 17, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L.; Pardoll, D.M. Perspective: Alpha-bugs, their microbial partners, and the link to colon cancer. J. Infect. Dis. 2011, 203, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Hashemi Goradel, N.; Heidarzadeh, S.; Jahangiri, S.; Farhood, B.; Mortezaee, K.; Khanlarkhani, N.; Negahdari, B. Fusobacterium nucleatum and colorectal cancer: A mechanistic overview. J. Cell. Physiol. 2019, 234, 2337–2344. [Google Scholar] [CrossRef]
- Yang, G.Y.; Shamsuddin, A.M. Gal-galnac: A biomarker of colon carcinogenesis. Histol. Histopathol. 1996, 11, 801–806. [Google Scholar] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Yang, Y.; Xia, Y.; Okugawa, Y.; Yang, J.; Liang, Y.; Chen, H.; Zhang, P.; Wang, F.; Han, H.; et al. Novel evidence for an oncogenic role of microrna-21 in colitis-associated colorectal cancer. Gut 2016, 65, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.S.; Nishihara, R.; Cao, Y.; Song, M.; Mima, K.; Qian, Z.R.; Nowak, J.A.; Kosumi, K.; Hamada, T.; Masugi, Y.; et al. Association of dietary patterns with risk of colorectal cancer subtypes classified by fusobacterium nucleatum in tumor tissue. JAMA Oncol. 2017, 3, 921–927. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in african americans and rural africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef]
- Lago, F.; Dieguez, C.; Gomez-Reino, J.; Gualillo, O. Adipokines as emerging mediators of immune response and inflammation. Nat. Clin. Pract. Rheumatol. 2007, 3, 716–724. [Google Scholar] [CrossRef]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M. Adipose tissue dysfunction in obesity. Exp. Clin. Endocrinol. Diabetes 2009, 117, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Khadir, A.; Kavalakatt, S.; Abubaker, J.; Cherian, P.; Madhu, D.; Al-Khairi, I.; Abu-Farha, M.; Warsame, S.; Elkum, N.; Dehbi, M.; et al. Physical exercise alleviates er stress in obese humans through reduction in the expression and release of grp78 chaperone. Metabolism 2016, 65, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. Tlr4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.W.; Olefsky, J.M. Jnk and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3t3-l1 adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Wang, Y.; Sul, H.S. Lipolysis in adipocytes. Int. J. Biochem. Cell Biol. 2010, 42, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.R.; Lobo, S.; Bernlohr, D.A. Fatty acid flux in adipocytes: The in’s and out’s of fat cell lipid trafficking. Mol. Cell. Endocrinol. 2010, 318, 24–33. [Google Scholar] [CrossRef]
- Unger, R.H.; Clark, G.O.; Scherer, P.E.; Orci, L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 2010, 1801, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.E.; Gabler, N.K.; Walker-Daniels, J.; Spurlock, M.E. The c-jun n-terminal kinase mediates the induction of oxidative stress and insulin resistance by palmitate and toll-like receptor 2 and 4 ligands in 3t3-l1 adipocytes. Horm. Metab. Res. 2009, 41, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Kopp, A.; Buechler, C.; Neumeier, M.; Weigert, J.; Aslanidis, C.; Scholmerich, J.; Schaffler, A. Innate immunity and adipocyte function: Ligand-specific activation of multiple toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity 2009, 17, 648–656. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Martinez, K.; Chuang, C.C.; LaPoint, K.; McIntosh, M. Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: Mechanisms of action and implications. J. Nutr. 2009, 139, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. Mcp-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Cullberg, K.B.; Larsen, J.O.; Pedersen, S.B.; Richelsen, B. Effects of lps and dietary free fatty acids on mcp-1 in 3t3-l1 adipocytes and macrophages in vitro. Nutr. Diabetes 2014, 4, e113. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Fak, F.; Backhed, F. Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J. Intern. Med. 2010, 268, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Caesar, R.; Tremaroli, V.; Kovatcheva-Datchary, P.; Cani, P.D.; Backhed, F. Crosstalk between gut microbiota and dietary lipids aggravates wat inflammation through tlr signaling. Cell Metab. 2015, 22, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Meli, R.; Mattace Raso, G.; Calignano, A. Role of innate immune response in Non-Alcoholic Fatty Liver Disease: Metabolic complications and therapeutic tools. Front. Immunol. 2014, 5, 177. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; Network, N.C.R. Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in nafld: Distinct clinicopathologic meanings. Hepatology 2011, 53, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of nonalcoholic steatohepatitis: Interactions between liver parenchymal and nonparenchymal cells. BioMed Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef] [PubMed]
- Ferolla, S.M.; Silva, L.C.; Ferrari Mde, L.; da Cunha, A.S.; Martins Fdos, S.; Couto, C.A.; Ferrari, T.C. Dietary approach in the treatment of nonalcoholic fatty liver disease. World J. Hepatol. 2015, 7, 2522–2534. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Nutritional modulation of non-alcoholic fatty liver disease and insulin resistance. Nutrients 2015, 7, 9127–9138. [Google Scholar] [CrossRef] [PubMed]
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular mechanisms and the role of saturated fatty acids in the progression of non-alcoholic fatty liver disease. Prog. Lipid Res. 2013, 52, 165–174. [Google Scholar] [CrossRef]
- Elias, S.; Wisam, S.; Luai, A.; Massad, B.; Nimer, A. Lipotoxicity in obesity: Benefit of olive oil. Adv. Exp. Med. Biol. 2017, 960, 607–617. [Google Scholar]
- Meidan, E.; Kolesnikov, Y.; Tirosh, O. High fat diets composed of palm stearin and olive oil equally exacerbate liver inflammatory damage and metabolic stress in mice. Mol. Nutr. Food Res. 2018, 62, e1700915. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Z.; Ru, J.H.; Lopes-Virella, M.F.; Lyons, T.J.; Huang, Y. Saturated fatty acid combined with lipopolysaccharide stimulates a strong inflammatory response in hepatocytes in vivo and in vitro. Am. J. Physiol. Endocrinol. Metab. 2018. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.C. Gastrointestinal microflora in mammalian nutrition. Annu. Rev. Nutr. 1986, 6, 155–178. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, A. Contributions of colonic short-chain fatty acid receptors in energy homeostasis. Front. Endocrinol. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Mattace Raso, G.; Simeoli, R.; Russo, R.; Iacono, A.; Santoro, A.; Paciello, O.; Ferrante, M.C.; Canani, R.B.; Calignano, A.; Meli, R. Effects of sodium butyrate and its synthetic amide derivative on liver inflammation and glucose tolerance in an animal model of steatosis induced by high fat diet. PLoS ONE 2013, 8, e68626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Pan, Q.; Xin, F.Z.; Zhang, R.N.; He, C.X.; Chen, G.Y.; Liu, C.; Chen, Y.W.; Fan, J.G. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 2017, 23, 60–75. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Lv, L.; Wu, W.; Li, Y.; Shi, D.; Fang, D.; Guo, F.; Jiang, H.; Yan, R.; Ye, W.; et al. Butyrate protects mice against methionine-choline-deficient diet-induced non-alcoholic steatohepatitis by improving gut barrier function, attenuating inflammation and reducing endotoxin levels. Front. Microbiol. 2018, 9, 1967. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191. [Google Scholar] [CrossRef]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor fxr/bar. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A g protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Patnaude, L.A.; Pelletier, J.; Souza, D.J.; Lukas, S.M.; King, F.J.; Hill, J.D.; Stefanopoulos, D.E.; Ryan, K.; Desai, S.; et al. A GPBAR1 (TGR5) small molecule agonist shows specific inhibitory effects on myeloid cell activation in vitro and reduces experimental autoimmune encephalitis (EAE) in vivo. PLoS ONE 2014, 9, e100883. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile acids control inflammation and metabolic disorder through inhibition of nlrp3 inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Raymond, K. Nuclear receptors, bile-acid detoxification, and cholestasis. Lancet 2006, 367, 454–456. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Joyce, S.A.; Shanahan, F.; Hill, C.; Gahan, C.G. Bacterial bile salt hydrolase in host metabolism: Potential for influencing gastrointestinal microbe-host crosstalk. Gut Microbes 2014, 5, 669–674. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile acids and dysbiosis in non-alcoholic fatty liver disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Cusi, K. Nonalcoholic fatty liver disease in type 2 diabetes mellitus. Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 141–149. [Google Scholar] [CrossRef]
- Lee, J.J.; Lambert, J.E.; Hovhannisyan, Y.; Ramos-Roman, M.A.; Trombold, J.R.; Wagner, D.A.; Parks, E.J. Palmitoleic acid is elevated in fatty liver disease and reflects hepatic lipogenesis. Am. J. Clin. Nutr. 2015, 101, 34–43. [Google Scholar] [CrossRef]
- Jacobs, S.; Jager, S.; Jansen, E.; Peter, A.; Stefan, N.; Boeing, H.; Schulze, M.B.; Kroger, J. Associations of erythrocyte fatty acids in the de novo lipogenesis pathway with proxies of liver fat accumulation in the epic-potsdam study. PLoS ONE 2015, 10, e0127368. [Google Scholar] [CrossRef] [PubMed]
- Cansancao, K.; Silva Monteiro, L.; Carvalho Leite, N.; Davalos, A.; Tavares do Carmo, M.D.G.; Arantes Ferreira Peres, W. Advanced liver fibrosis is independently associated with palmitic acid and insulin levels in patients with non-alcoholic fatty liver disease. Nutrients 2018, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Lemas, D.J.; Johnson, L.K.; Weir, T.; Lenz, L.L.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, A.; Yanez, A.; Price, J.G.; Chow, A.; Merad, M.; Goodridge, H.S.; Mazmanian, S.K. Gut microbiota promote hematopoiesis to control bacterial infection. Cell Host Microbe 2014, 15, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hamalainen, A.M.; et al. Variation in microbiome lps immunogenicity contributes to autoimmunity in humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef]
- Hajjar, A.M.; Ernst, R.K.; Tsai, J.H.; Wilson, C.B.; Miller, S.I. Human toll-like receptor 4 recognizes host-specific lps modifications. Nat. Immunol. 2002, 3, 354–359. [Google Scholar] [CrossRef]
- Needham, B.D.; Carroll, S.M.; Giles, D.K.; Georgiou, G.; Whiteley, M.; Trent, M.S. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc. Natl. Acad. Sci. USA 2013, 110, 1464–1469. [Google Scholar] [CrossRef]
- Coats, S.R.; Do, C.T.; Karimi-Naser, L.M.; Braham, P.H.; Darveau, R.P. Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human tlr4 via interaction with the human md-2 lipopolysaccharide binding site. Cell. Microbiol. 2007, 9, 1191–1202. [Google Scholar] [CrossRef]
- Herath, T.D.; Wang, Y.; Seneviratne, C.J.; Lu, Q.; Darveau, R.P.; Wang, C.Y.; Jin, L. Porphyromonas gingivalis lipopolysaccharide lipid a heterogeneity differentially modulates the expression of IL-6 and IL-8 in human gingival fibroblasts. J. Clin. Periodontol. 2011, 38, 694–701. [Google Scholar] [CrossRef]
- D’Hennezel, E.; Abubucker, S.; Murphy, L.O.; Cullen, T.W. Total lipopolysaccharide from the human gut microbiome silences tolllike receptor signaling. Am. Soc. Microbiol. 2018, 2, e00046-00017. [Google Scholar] [CrossRef]
- Truong, D.T.; Franzosa, E.A.; Tickle, T.L.; Scholz, M.; Weingart, G.; Pasolli, E.; Tett, A.; Huttenhower, C.; Segata, N. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 2015, 12, 902–903. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Mehrpouya-Bahrami, P.; Chitrala, K.N.; Ganewatta, M.S.; Tang, C.; Murphy, E.A.; Enos, R.T.; Velazquez, K.T.; McCellan, J.; Nagarkatti, M.; Nagarkatti, P. Blockade of CB1 cannabinoid receptor alters gut microbiota and attenuates inflammation and diet-induced obesity. Sci. Rep. 2017, 7, 15645. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, L.A.; Lolait, S.J.; Brownstein, M.J.; Young, A.C.; Bonner, T.I. Structure of a cannabinoid receptor and functional expression of the cloned cdna. Nature 1990, 346, 561–564. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Muccioli, G.G. The endocannabinoid system in inflammatory bowel diseases: From pathophysiology to therapeutic opportunity. Trends Mol. Med. 2012, 18, 615–625. [Google Scholar] [CrossRef]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Sharkey, K.A.; Wiley, J.W. The role of the endocannabinoid system in the brain-gut axis. Gastroenterology 2016, 151, 252–266. [Google Scholar] [CrossRef]
- Trautmann, S.M.; Sharkey, K.A. The endocannabinoid system and its role in regulating the intrinsic neural circuitry of the gastrointestinal tract. Int. Rev. Neurobiol. 2015, 125, 85–126. [Google Scholar]
- Muccioli, G.G.; Naslain, D.; Backhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 392. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Crosstalk between the gut microbiota and the endocannabinoid system: Impact on the gut barrier function and the adipose tissue. Clin. Microbiol. Infect. 2012, 18 (Suppl. 4), 50–53. [Google Scholar] [CrossRef]
- Horvath, B.; Mukhopadhyay, P.; Hasko, G.; Pacher, P. The endocannabinoid system and plant-derived cannabinoids in diabetes and diabetic complications. Am. J. Pathol. 2012, 180, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Rousseaux, C.; Thuru, X.; Gelot, A.; Barnich, N.; Neut, C.; Dubuquoy, L.; Dubuquoy, C.; Merour, E.; Geboes, K.; Chamaillard, M.; et al. Lactobacillus acidophilus modulates intestinal pain and induces opioid and cannabinoid receptors. Nat. Med. 2007, 13, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Ringel-Kulka, T.; Goldsmith, J.R.; Carroll, I.M.; Barros, S.P.; Palsson, O.; Jobin, C.; Ringel, Y. Lactobacillus acidophilus ncfm affects colonic mucosal opioid receptor expression in patients with functional abdominal pain—A randomised clinical study. Aliment. Pharmacol. Ther. 2014, 40, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving glp-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Geurts, L.; Lazarevic, V.; Derrien, M.; Everard, A.; Van Roye, M.; Knauf, C.; Valet, P.; Girard, M.; Muccioli, G.G.; Francois, P.; et al. Altered gut microbiota and endocannabinoid system tone in obese and diabetic leptin-resistant mice: Impact on apelin regulation in adipose tissue. Front. Microbiol. 2011, 2, 149. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wisniewski, P.J.; Dowden, R.A.; Campbell, S.C. Role of Dietary Lipids in Modulating Inflammation through the Gut Microbiota. Nutrients 2019, 11, 117. https://doi.org/10.3390/nu11010117
Wisniewski PJ, Dowden RA, Campbell SC. Role of Dietary Lipids in Modulating Inflammation through the Gut Microbiota. Nutrients. 2019; 11(1):117. https://doi.org/10.3390/nu11010117
Chicago/Turabian StyleWisniewski, Paul J., Robert A. Dowden, and Sara C. Campbell. 2019. "Role of Dietary Lipids in Modulating Inflammation through the Gut Microbiota" Nutrients 11, no. 1: 117. https://doi.org/10.3390/nu11010117
APA StyleWisniewski, P. J., Dowden, R. A., & Campbell, S. C. (2019). Role of Dietary Lipids in Modulating Inflammation through the Gut Microbiota. Nutrients, 11(1), 117. https://doi.org/10.3390/nu11010117