Human Milk Lipidomics: Current Techniques and Methodologies


Abstract
1. Introduction
2. Sampling
2.1. Sampling with Respect to the Feed
2.2. Sampling over 24 h
2.3. Sampling through Stages of Lactation
2.4. Ideal Sampling Routine
3. Storage
3.1. Freezing
3.2. Preservatives
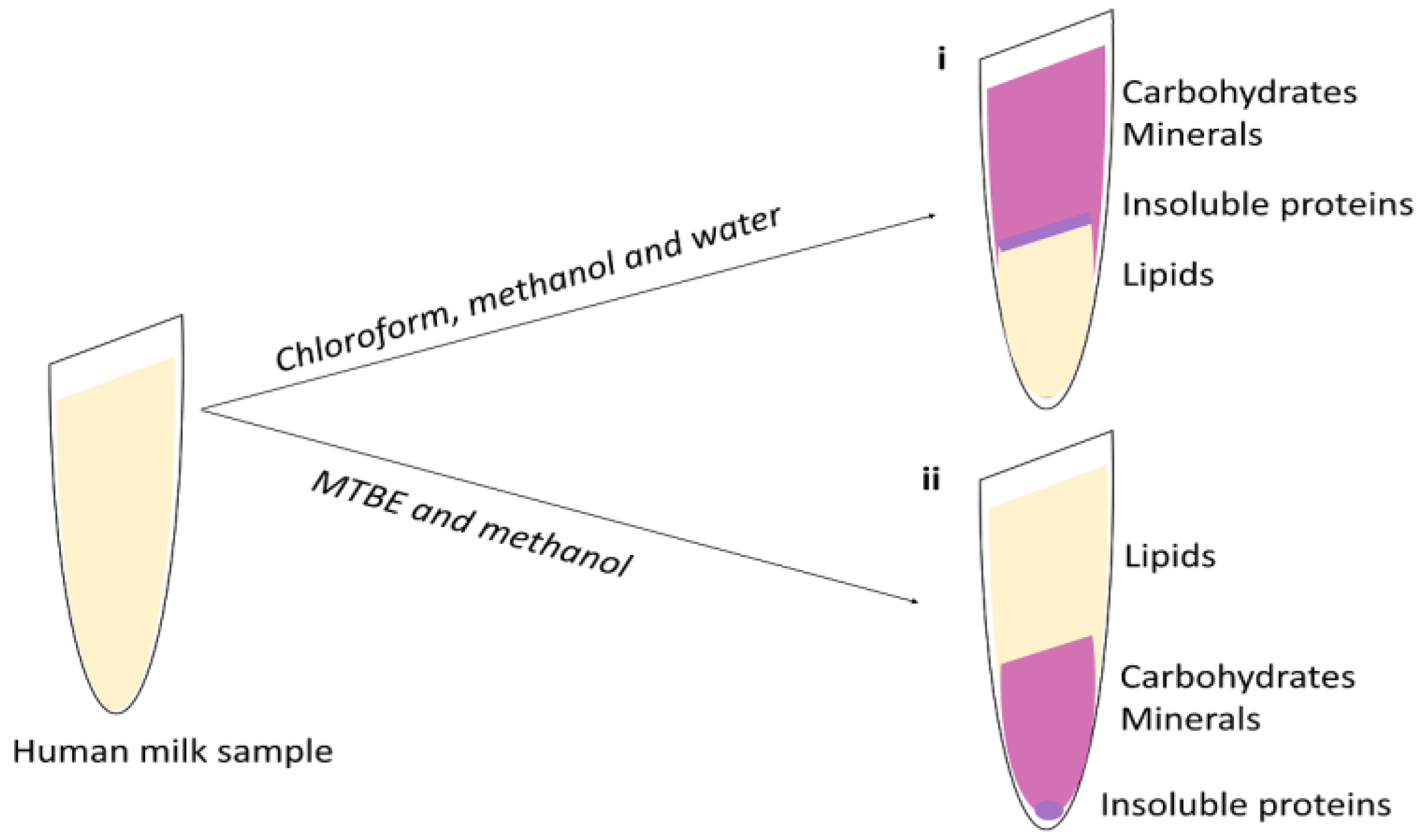
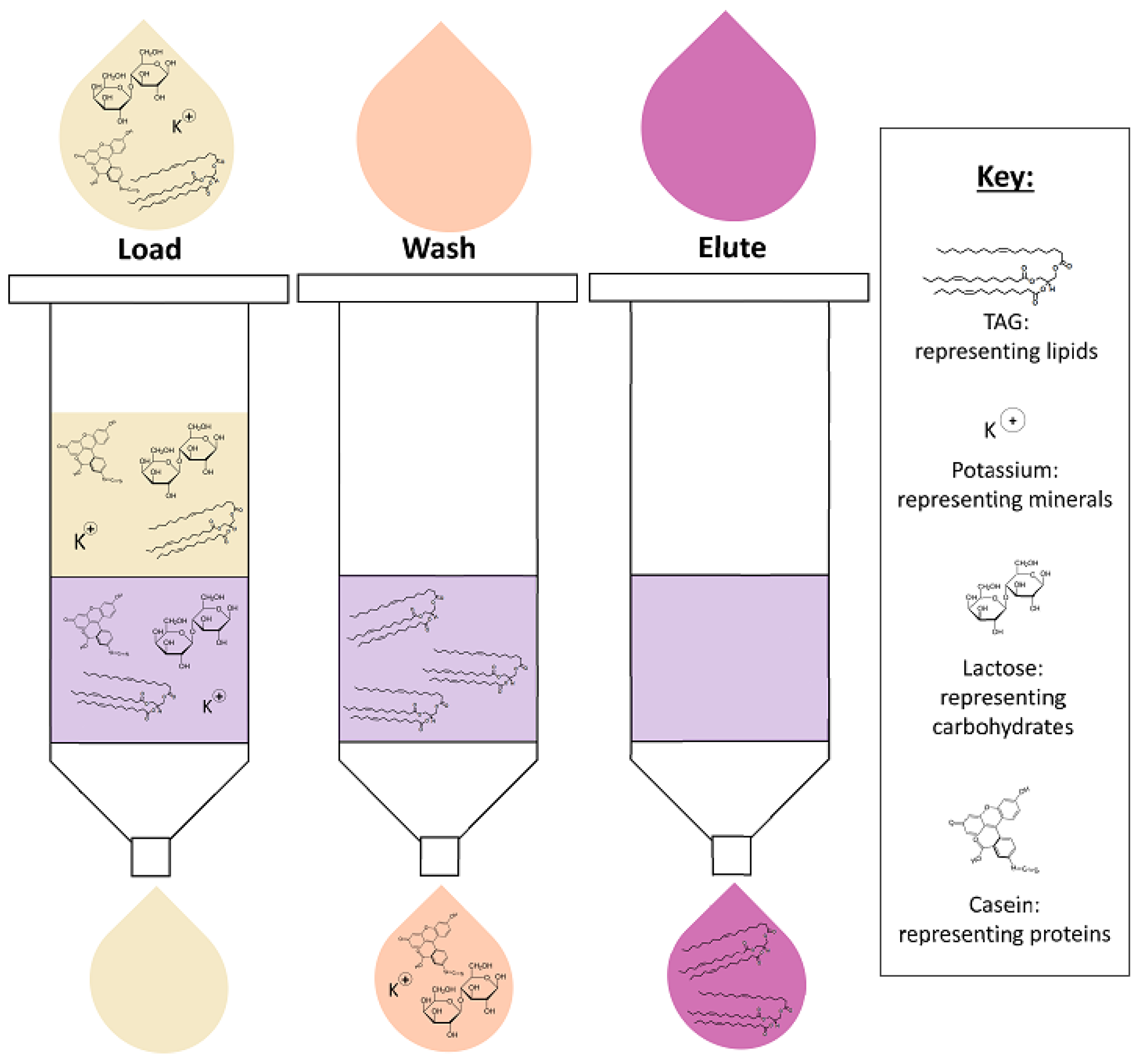
4. Lipid Extraction
4.1. Liquid-Liquid Extraction
4.2. Solid-Phase Extraction
5. Lipid Transesterification
6. Quality Control
6.1. In Sample
6.2. Out of Sample
7. Analytical Instrumentation for Lipidomic Analysis
7.1. Separation Methods
7.1.1. Gas Chromatography
7.1.2. Liquid Chromatography
7.1.3. Supercritical Fluid Chromatography
7.1.4. Thin-Layer Chromatography
7.2. Mass Spectrometry
7.3. Nuclear Magnetic Resonance Spectroscopy
8. Limitations and Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Eidelman, A.I. Breastfeeding and the use of human milk: An analysis of the American academy of pediatrics 2012 breastfeeding policy statement. Breastfeed. Med. 2012, 7, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Layde, P.M.; Webster, L.A.; Baughman, A.L.; Wingo, P.A.; Rubin, G.L.; Ory, H.W. The independent associations of parity, age at first full term pregnancy, and duration of breastfeeding with the risk of breast cancer. Cancer and steroid hormone study group. J. Clin. Epidemiol. 1989, 42, 963–973. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Breast cancer and breastfeeding: Collaborative reanalysis of individual data from 47 epidemiological studies in 30 countries, including 50302 women with breast cancer and 96973 women without the disease. Lancet 2002, 360, 187–195. [Google Scholar] [CrossRef]
- Victora, C.G.; Bahl, R.; Barros, A.J.; Franca, A.V.; Horton, S.; Krasevec, J.; Murch, S.; Sankar, M.J.; Walker, N.; Rollins, N.C. Breastfeeding in the 21st century: Epidemiology, mechanisms, and lifelong effect. Lancet 2016, 387, 475–490. [Google Scholar] [CrossRef]
- Jenness, R. The composition of human milk. Semin. Perinatol. 1979, 3, 225–339. [Google Scholar] [PubMed]
- Jensen, R.G. Lipids in human milk. Lipids 1999, 34, 1243–1271. [Google Scholar] [CrossRef] [PubMed]
- Koletzko, B.; Agostoni, C.; Bergmann, R.; Ritzenthaler, K.; Shamir, R. Physiological aspects of human milk lipids and implications for infant feeding: A workshop report. Acta Paediatr. 2011, 100, 1405–1415. [Google Scholar] [CrossRef] [PubMed]
- Innis, S.M. Dietary triacylglycerol structure and its role in infant nutrition. Adv. Nutr. 2011, 2, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, L.; Fewtrell, M.; Agostoni, C. Dietary arachidonic acid in perinatal nutrition: A commentary. Pediatr. Res. 2015, 77, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Alexandre-Gouabau, M.-C.; Moyon, T.; Cariou, V.; Antignac, J.-P.; Qannari, E.M.; Croyal, M.; Soumah, M.; Guitton, Y.; David-Sochard, A.; Billard, H.; et al. Breast milk lipidome is associated with early growth trajectory in preterm infants. Nutrients 2018, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.G. The lipids in human milk. Prog. Lipid Res. 1996, 35, 53–92. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Kwan, K.Y.; Tong, K.K.; Ratnayake, W.M.N.; Li, H.Q.; Leung, S.S.F. Breast milk fatty acid composition: A comparative study between Hong Kong and Chongqing Chinese. Lipids 1997, 32, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wu, K.; Yu, Z.; Ren, Y.; Zhao, Y.; Jiang, Y.; Xu, X.; Li, W.; Jin, Y.; Yuan, J.; et al. Changes in fatty acid composition of human milk over lactation stages and relationship with dietary intake in Chinese women. Food Funct. 2016, 7, 3154–3162. [Google Scholar] [CrossRef] [PubMed]
- Makrides, M.; Simmer, K.; Neumann, M.; Gibson, R. Changes in the polyunsaturated fatty acids of breast milk from mothers of full-term infants over 30 wk of lactation. Am. J. Clin. Nutr. 1995, 61, 1231–1233. [Google Scholar] [CrossRef] [PubMed]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: Lipid Maps structure database. Nucleic Acids Res. 2007, 35, D527–D532. [Google Scholar] [CrossRef] [PubMed]
- Dotson, K.D.; Jerrell, J.P.; Picciano, M.F.; Perkins, E.G. High-performance liquid chromatography of human milk triacylglycerols and gas chromatography of component fatty acids. Lipids 1992, 27, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Kallio, M.J.; Siimes, M.A.; Perheentupa, J.; Salmenperä, L.; Miettinen, T.A. Cholesterol and its precursors in human milk during prolonged exclusive breast-feeding. Am. J. Clin. Nutr. 1989, 50, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Andreas, N.J.; Hyde, M.J.; Gomez-Romero, M.; Lopez-Gonzalvez, M.A.; Villaseñor, A.; Wijeyesekera, A.; Barbas, C.; Modi, N.; Holmes, E.; Garcia-Perez, I. Multiplatform characterization of dynamic changes in breast milk during lactation. Electrophoresis 2015, 36, 2269–2285. [Google Scholar] [CrossRef] [PubMed]
- Insull, W. The fatty acids of human milk from mothers on diets taken ad libitum. Biochem. J. 1959, 72, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, F.; Cruz-Hernandez, C.; Flück, B.; Tavazzi, I.; Thakkar, S.K.; Destaillats, F.; Braun, M. Quantification of phospholipids classes in human milk. Lipids 2013, 48, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Gay, M.C.L.; Lai, C.T.; Trengove, R.D.; Hartmann, P.E.; Geddes, D.T. Comparison of gravimetric, creamatocrit and esterified fatty acid methods for determination of total fat content in human milk. Food Chem. 2017, 217, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Saarela, T.; Kokkonen, J.; Koivisto, M. Macronutrient and energy contents of human milk fractions during the first six months of lactation. Acta Paediatr. 2007, 94, 1176–1181. [Google Scholar] [CrossRef]
- Khan, S.; Hepworth, A.R.; Prime, D.K.; Lai, C.T.; Trengove, N.J.; Hartmann, P.E. Variation in fat, lactose, and protein composition in breast milk over 24 hours: Associations with infant feeding patterns. J. Hum. Lact. 2013, 29, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Makrides, M.; Neumann, M.A.; Gibson, R.A. Effect of maternal docosahexaenoic acid (DHA) supplementation on breast milk composition. Eur. J. Clin. Nutr. 1996, 50, 352–357. [Google Scholar] [PubMed]
- Ruan, C.; Liu, X.; Man, H.; Ma, X.; Lu, G.; Duan, G.; DeFrancesco, C.A.; Connor, W.E. Milk composition in women from five different regions of China: The great diversity of milk fatty acids. J. Nutr. 1995, 125, 2993–2998. [Google Scholar] [PubMed]
- Mitoulas, L.R.; Kent, J.C.; Cox, D.B.; Owens, R.A.; Sherriff, J.L.; Hartmann, P.E. Variation in fat, lactose and protein in human milk over 24 h and throughout the first year of lactation. Br. J. Nutr. 2002, 88, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Haug, M.; Dieterich, I.; Laubach, C.; Reinhardt, D.; Harzer, G. Capillary Gas Chromatography of Fatty Acid Methyl Esters from Human Milk Lipid Subclasses. J. Chromatogr. A 1983, 279, 549–553. [Google Scholar] [CrossRef]
- van Beusekom, C.M.; Nijeboer, H.J.; van der Veere, C.N.; Luteyn, A.J.; Offringa, P.J.; Muskiet, F.A.J.; Boersma, E.R. Indicators of long chain polyunsaturated fatty acid status of exclusively breastfed infants at delivery and after 20–22 days. Early Hum. Dev. 1993, 32, 207–218. [Google Scholar] [CrossRef]
- Liu, Z.; Cocks, B.G.; Rochfort, S. Comparison of molecular species distribution of DHA-containing triacylglycerols in milk and different infant formulas by liquid Chromatography-Mass spectrometry. J. Agric. Food Chem. 2016, 64, 2134–2144. [Google Scholar] [CrossRef] [PubMed]
- Rosenlund, H.; Fagerstedt, S.; Alm, J.; Mie, A. Breastmilk fatty acids in relation to sensitization-the ALADDIN birth cohort. Allergy 2016, 71, 1444–1452. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.S.; Rahman, I.A.; Lai, C.T.; Hepworth, A.; Trengove, N.; Hartmann, P.E.; Geddes, D.T. Changes in fatty acid composition of human milk in response to cold-like symptoms in the lactating mother and infant. Nutrients 2017, 9, 1034. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Hernandez, C.; Goeuriot, S.; Giuffrida, F.; Thakkar, S.K.; Destaillats, F. Direct quantification of fatty acids in human milk by Gas Chromatography. J. Chromatogr. A 2013, 1284, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Daly, S.E.; Di Rosso, A.; Owens, R.A.; Hartmann, P.E. Degree of breast emptying explains changes in the fat content, but not fatty acid composition, of human milk. Exp. Physiol. 1993, 78, 741–755. [Google Scholar] [CrossRef]
- Prime, D.K.; Kent, J.C.; Hepworth, A.R.; Trengove, N.J.; Hartmann, P.E. Dynamics of milk removal during simultaneous breast expression in women. Breastfeed. Med. 2012, 7, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.C.; Ramsay, D.T.; Doherty, D.; Larsson, M.; Hartmann, P.E. Response of breasts to different stimulation patterns of an electric breast pump. J. Hum. Lact. 2003, 19, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.G.; Lammi-Keefe, C.J.; Koletzko, B. Representative sampling of human milk and the extraction of fat for analysis of environmental lipophilic contaminants. Toxicol. Environ. Chem. 1997, 62, 229–247. [Google Scholar] [CrossRef]
- Lubetzky, R.; Littner, Y.; Mimouni, F.B.; Dollberg, S.; Mandel, D. Circadian variations in fat content of expressed breast milk from mothers of preterm infants. J. Am. Coll. Nutr. 2006, 25, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.C.; Mitoulas, L.R.; Cregan, M.D.; Ramsay, D.T.; Doherty, D.A.; Hartmann, P.E. Volume and frequency of breastfeedings and fat content of breast milk throughout the day. Pediatrics 2006, 117, E387–e395. [Google Scholar] [CrossRef] [PubMed]
- Jackson, D.A.; Imong, S.M.; Silprasert, A.; Ruckphaopunt, S.; Woolridge, M.W.; Baum, J.D.; Amatayakul, K. Circadian variation in fat concentration of breast-milk in a rural northern Thai population. Br. J. Nutr. 1988, 59, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hassiotou, F.; Hepworth, A.R.; Williams, T.M.; Twigger, A.J.; Perrella, S.; Lai, C.T.; Filgueira, L.; Geddes, D.T.; Hartmann, P.E. Breastmilk cell and fat contents respond similarly to removal of breastmilk by the infant. PLoS ONE 2013, 8, E78232. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-Y.; Pelletier, G.; Hollywood, R.; Ratnayake, W.M. Trans fatty acid isomers in Canadian human milk. Lipids 1995, 30, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Pons, S.M.; Bargallo, A.C.; Folgoso, C.C.; Lopez Sabater, M.C. Triacylglycerol composition in colostrum, transitional and mature human milk. Eur. J. Clin. Nutr. 2000, 54, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Scopesi, F.; Ciangherotti, S.; Lantieri, P.B.; Risso, D.; Bertini, I.; Campone, F.; Pedrotti, A.; Bonacci, W.; Serra, G. Maternal dietary pufas intake and human milk content relationships during the first month of lactation. Clin. Nutr. 2001, 20, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Minda, H.; Kovacs, A.; Funke, S.; Szasz, M.; Burus, I.; Molnar, S.; Marosvolgyi, T.; Decsi, T. Changes of fatty acid composition of human milk during the first month of lactation: A day-to-day approach in the first week. Ann. Nutr. Metab. 2004, 48, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Mosley, E.E.; Wright, A.L.; McGuire, M.K.; McGuire, M.A. Trans fatty acids in milk produced by women in the United States. Am. J. Clin. Nutr. 2005, 82, 1292–1297. [Google Scholar] [CrossRef] [PubMed]
- Dreiucker, J.; Vetter, W. Fatty acids patterns in camel, moose, cow and human milk as determined with GC/MS after silver ion solid phase extraction. Food Chem. 2011, 126, 762–771. [Google Scholar] [CrossRef]
- Kelishadi, R.; Hadi, B.; Iranpour, R.; Khosravi-Darani, K.; Mirmoghtadaee, P.; Farajian, S.; Poursafa, P. A study on lipid content and fatty acid of breast milk and its association with mother’s diet composition. J. Res. Med. Sci. 2012, 17, 824–827. [Google Scholar] [PubMed]
- Daud, A.Z.; Mohd-Esa, N.; Azlan, A.; Chan, Y.M. The ‘trans’ fatty acid content in human milk and its association with maternal diet among lactating mothers in Malaysia. Asia Pac. J. Clin. Nutr. 2013, 22, 431–442. [Google Scholar] [PubMed]
- Saphier, O.; Blumenfeld, J.; Silberstein, T.; Tzor, T.; Burg, A. Fatty acid composition of breastmilk of Israeli mothers. Indian Pediatr. 2013, 50, 1044–1046. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.-K.; Yeung, C.-Y.; Jim, W.-T.; Lin, S.-P.; Wang, T.J.; Huang, S.-F.; Liu, H.-L. Comparison of free fatty acid content of human milk from Taiwanese mothers and infant formula. Taiwan. J. Obstet. Gynecol. 2013, 52, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Villasenor, A.; Garcia-Perez, I.; Garcia, A.; Posma, J.M.; Fernandez-Lopez, M.; Nicholas, A.J.; Modi, N.; Holmes, E.; Barbas, C. Breast Milk metabolome characterization in a single-phase extraction, multiplatform analytical approach. Anal. Chem. 2014, 86, 8245–8252. [Google Scholar] [CrossRef] [PubMed]
- Sokol, E.; Ulven, T.; Færgeman, N.J.; Ejsing, C.S. Comprehensive and quantitative profiling of lipid species in human milk, cow milk and a phospholipid-enriched milk formula by GC and MS/MS(All). Eur. J. Lipid Sci. Technol. 2015, 117, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Ten-Doménech, I.; Beltrán-Iturat, E.; Herrero-Martínez, J.M.; Sancho-Llopis, J.V.; Simó-Alfonso, E.F. Triacylglycerol analysis in human milk and other mammalian species: Small-scale sample preparation, characterization, and statistical classification using HPLC-ELSD profiles. J. Agric. Food Chem. 2015, 63, 5761–5770. [Google Scholar]
- Liu, G.; Ding, Z.; Li, X.; Chen, X.; Wu, Y.; Xie, L. Relationship between polyunsaturated fatty acid levels in maternal diets and human milk in the first month post-partum. J. Hum. Nutr. Diet. 2016, 29, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Garwolińska, D.; Hewelt-Belka, W.; Namieśnik, J.; Kot-Wasik, A. Rapid characterization of the human breast milk lipidome using a Solid-Phase Microextraction and liquid Chromatography-Mass Spectrometry-based approach. J. Proteom. Res. 2017, 16, 3200–3208. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Domellöf, M.; Zivkovic, A.M.; Larsson, G.; Öhman, A.; Nording, M.L. NMR-based metabolite profiling of human milk: A pilot study of methods for investigating compositional changes during lactation. Biochem. Biophys. Res. Commun. 2016, 469, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Tu, A.; Ma, Q.; Bai, H.; Du, Z. A comparative study of triacylglycerol composition in Chinese human milk within different lactation stages and imported infant formula by SFC Coupled with Q-TOF-MS. Food Chem. 2017, 221, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Zechner, R. Rapid and simple isolation procedure for lipoprotein lipase from human milk. Biochim. Biophys. Acta (BBA) Lipids Lipid MeTable 1990, 1044, 20–25. [Google Scholar] [CrossRef]
- Freudenberg, E. Lipase of human milk; Studies on its enzymological and nutritional significance. Bibl. Paediatr. 1953, 54, 1–68. [Google Scholar]
- Berkow, S.E.; Freed, L.M.; Hamosh, M.; Bitman, J.; Wood, D.L.; Happ, B.; Hamosh, P. Lipases and lipids in human milk: Effect of freeze-thawing and storage. Pediatr. Res. 1984, 18, 1257–1262. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Chen, C.-H.; Lin, M.-C. The macronutrients in human milk change after storage in various containers. Pediatr. Neonatol. 2012, 53, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lara, N.R.; Escuder-Vieco, D.; Garcia-Algar, O.; De la Cruz, J.; Lora, D.; Pallas-Alonso, C. Effect of freezing time on macronutrients and energy content of breastmilk. Breastfeed. Med. 2012, 7, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Lev, H.M.; Ovental, A.; Mandel, D.; Mimouni, F.B.; Marom, R.; Lubetzky, R. Major losses of fat, carbohydrates and energy content of preterm human milk frozen at −80 °C. J. Perinatol. 2014, 34, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Fusch, G.; Rochow, N.; Choi, A.; Fusch, S.; Poeschl, S.; Ubah, A.O.; Lee, S.-Y.; Raja, P.; Fusch, C. Rapid measurement of macronutrients in breast milk: How reliable are infrared milk analyzers? Clin. Nutr. 2015, 34, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Bitman, J.; Wood, D.L.; Mehta, N.R.; Hamosh, P.; Hamosh, M. Lipolysis of triglycerides of human milk during storage at low temperatures: A note of caution. J. Pediatr. Gastroenterol. Nutr. 1983, 2, 521–524. [Google Scholar] [CrossRef] [PubMed]
- Tijerina-Saenz, A.; Innis, S.M.; Kitts, D.D. Antioxidant capacity of human milk and its association with vitamins A and E and fatty acid composition. Acta Paediatr. 2009, 98, 1793–1798. [Google Scholar] [CrossRef] [PubMed]
- Boris, J.; Jensen, B.; Dalby Salvig, J.; Secher, N.J.; Olsen, S.F. A randomized controlled trial of the effect of fish oil supplementation in late pregnancy and early lactation on the n-3 fatty acid content in human breast milk. Lipids 2004, 39, 1191–1196. [Google Scholar] [CrossRef] [PubMed]
- Magnusardottir, A.R.; Skuladottir, G.V. Effects of storage time and added antioxidant on fatty acid composition of red blood cells at −20 °C. Lipids 2006, 41, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Bio.l Chem. 1957, 226, 497–509. [Google Scholar]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Gutierrez, V.; Perez-Camino, M.C. Update on solid-phase extraction for the analysis of lipid classes and related compounds. J. Chromatogr. A 2000, 885, 321–341. [Google Scholar] [CrossRef]
- Chappell, J.E.; Clandinin, M.T.; McVey, M.A.; Chance, G.W. Free fatty acid content of human milk: Physiologic significance and artifactual determinants. Lipids 1985, 20, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Meurant, G. Handbook of Milk Composition, 1st ed.; Jensen, R.G., Ed.; Academic Press: San Diego, CA, USA, 1995. [Google Scholar]
- Orata, F. Derivatization reactions and reagents for Gas Chromatography analysis. Adv. Gas Chromatogr. 2012, 5, 83–108. [Google Scholar]
- Morrison, W.R.; Smith, L.M. Preparation of fatty acid methyl esters and dimethylacetals from lipids with boron fluoride—Methanol. J. Lipid Res. 1964, 5, 600–608. [Google Scholar] [PubMed]
- Christie, W.W. Preparation of ester derivatives of fatty acids for chromatographic analysis. Adv. Lipid Method. 1993, 2, 69–111. [Google Scholar]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and considerations for the use of system suitability and quality control samples in mass spectrometry assays applied in untargeted clinical metabolomic studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, X. Lipidomics: Techniques, applications, and outcomes related to biomedical sciences. Trends Biochem. Sci. 2016, 41, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Jurowski, K.; Kochan, K.; Walczak, J.; Baranska, M.; Piekoszewski, W.; Buszewski, B. Analytical techniques in lipidomics: State of the art. Crit. Rev. Anal. Chem. 2017, 47, 418–437. [Google Scholar] [CrossRef] [PubMed]
- Ackman, R.G. Misidentification of fatty acid methyl ester peaks in liquid canola shortening. J. Am. Oil Chem. Soc. 1990, 67, 1028. [Google Scholar] [CrossRef]
- Hawrysh, Z.J.; Shand, P.J.; Lin, C.; Tokarska, B.; Hardin, R.T. Efficacy of tertiary butylhydroquinone on the storage and heat stability of liquid canola shortening. J. Am. Oil Chem. Soc. 1990, 67, 585–590. [Google Scholar] [CrossRef]
- Waston, A.D. Thematic review series: Systems biology approaches to metabolic and cardiovascular disorders. Lipidomics: A global approach to lipid analysis in biological systems. J. Lipid Res. 2006, 47, 2101–2111. [Google Scholar]
- Sommer, U.; Herscovitz, H.; Welty, F.K.; Costello, C.E. LC-MS-based method for the qualitative and quantitative analysis of complex lipid mixtures. J. Lipid Res. 2006, 47, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Laakso, P.; Manninen, P. Identification of milk fat triacylglycerols by capillary supercritical fluid chromatography-atmospheric pressure chemical ionization mass spectrometry. Lipids 1997, 32, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.T. Supercritical fluid chromatography. Anal. Chem. 2010, 82, 4925–4935. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Lutz, N.W.; Confort-Gouny, S.; Cozzone, P.J.; Armand, M.; Bernard, M. Phospholipid fingerprints of milk from different mammalians determined by 31p NMR: Towards specific interest in human health. Food Chem. 2012, 135, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Sundekilde, U.K.; Larsen, L.B.; Bertram, H.C. NMR-based milk metabolomics. Metabolites 2013, 3, 204–222. [Google Scholar] [CrossRef] [PubMed]
- Guillén, M.D.; Ruiz, A. 1H nuclear magnetic resonance as a fast tool for determining the composition of acyl chains in acylglycerol mixtures. Eur. J. Lipid Sci. Technol. 2003, 105, 502–507. [Google Scholar]
- Wishart, D.S. Metabolomics: Applications to food science and nutrition research. Trends Food Sci. Technol. 2008, 19, 482–493. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Lipids Identified | Sampling | Storage | Sample Preparation | Quality Control | Instrumentation | Reference |
|---|---|---|---|---|---|---|
| Fatty acids ranging from 10:0 to 22:6, including some unknown at the time | 6 hospital participants, mid-feed samples (for 24 hours, pooled); 5 participants at home, random samples | 4 °C (prior to pooling); −15 °C | 1 or 2 mL human milk ➔LLE 95% ethanol-ethyl ether ➔Hydrolysis 5% methanolic-KOH ➔Derivatisation 5% methanolic-HCl | In: - Out: - | GC–FID Reoplex 400/Apiezon M column (Carrier gas: nitrogen) | Insull et al. (1959) [19] |
| Fatty acids ranging from 12:0 to 22:6 | 15 participant random samples (pooled) | - | 4 mL human milk ➔TLC pre-separation ➔LLE chloroform:methanol (9:1) ➔Derivatisation BF3 | In: - Out: - | GC–FID, 50 m CP-Sil-88 column (Carrier gas: nitrogen) | Haug et al. (1983) [27] |
| Fatty acids ranging from 6:0 to 26:0 | 7 participants, sampled on day 20–22 (mid-feed) | On ice ≤2 hours; 20 °C | - | In: C17:0 Out: - | GC–FID | van Beusekom et al. (1993) [28] |
| Polyunsaturated fatty acids ranging from 18:2 to 22:6; total saturated FAs; total monounsaturated FAs | 23 participants 7-day samples from a single feed at weeks 6, 16, 30 (each time-point pooled) | −20 °C prior to delivery to laboratory | - mL human milk ➔Extracted - ➔Derivatisation 1% methanolic-H2SO4 | In: - Out: - | GC 50 m BPX-70 column | Makrides et al. (1995) [14] |
| Fatty acids ranging from 10:0 to 22:6 including cis and trans isomers and some unknown at the time | 198 samples, 3–4 weeks, mid-feed for a day (pooled) | - | 5 g human milk ➔LLE chloroform:methanol (2:1) ➔0.02% BHT preservative ➔Derivatisation methanolic-BF3 | In: Triheptadecanoin (in extraction solvent) Out: - | GC–FID 100 m SP-2560 column (Carrier gas: hydrogen) | Chen et al. (1995) [41] |
| Fatty acids ranging from 10:0 to 22:6 | Samples from 84 participants at day 3 and weeks 2, 4, and 6 | −20 °C | 2 g human milk ➔LLE chloroform:methanol (2:1) ➔Derivatisation methanolic-BF3 | In: Triheptadecanoin (in extraction solvent) Out: - | GC–FID 100 m SP-2560 column (Carrier gas: hydrogen) | Chen et al. (1997) [12] |
| 31 Triglycerides | Pre- and post-feed samples from 11 participants between days 1–3, days 7–10, days 25–60 (47 samples) | −80 °C | 1.5 mL human milk ➔LLE dicholoromethane-methanol (2:1) | In: C33:0 (after extraction) Out: - | LC–LSD, 250 mm Spherisorb ODS-2 column (Solvents: acetonitrile, dichloromethane, acetone) | Pons et al. (2000) [42] |
| Fatty acids ranging from 14:0 to 22:6 | 34 participants, samples on days 1, 4, 7, 14, 21, 28, at any time of day | −20 °C | ≤2 mL human milk ➔LLE chloroform:methanol (2:1) ➔BHT preservative ➔Derivatisation methanolic-BF3 | In: - Out: - | GC | Scopesi et al. (2001) [43] |
| Fatty acids ranging from 14:0 to 22:6 | 18 participants, days 1, 2, 3, 4, 5, 6, 7, 14, 28 between 0800–1000 | 4–8 °C (for <4 hours), deep freeze, 1 freeze-thaw cycle | 100 µL human milk ➔LLE chloroform:methanol ➔Derivatisation – | In: Pentadecanoic acid Out: - | GC–FID 40 m Cyanopropyl DB-23 column | Minda et al. (2004) [44] |
| 1. Fatty acids ranging from 4:0 to 22:6 2. 18:1 t isomers | 81 samples, from complete breast expression, between 0600 and 0800 in the first month | Room temperature (4 hours); Lipid layer frozen at −20 °C | 2 g human milk lipid layer ➔LLE chloroform:methanol (2:1) ➔Derivatisation sodium methoxide | In: - Out: - | 1. GC–FID 100 m CP-Sil-88 column 2. GC–MS DB225 MS column | Mosley et al. (2005) [45] |
| Groups of FAMES and approximately 36 × specific FAMEs | 1 random sample | −20 °C | 1 mg human milk fat ➔LLE cyclohexane/ethylacetate, ➔Hydrolysis methanolic-KOH ➔Derivatisation BF3 ➔SPE fractionation Ag+-SPE | In: 14:0 and 17:0 Out: - | GC–EI–MS, 60 m SP2331 cyanosiloxane column (Carrier gas: helium) | Dreiucker et al. (2011) [46] |
| DHA and AA and other fatty acids | 52 participants | - | 1 mL human milk ➔Hydrolysis methanolic-KOH ➔Derivatisation H2SO4 ➔LLE hexane | In: C19:0 Out: - | GC–FID 50 m fused-silica CPSIL88 column (Carrier gas: helium) | Kelishadi et al. (2012) [47] |
| Fatty acids from 12:0 to 18:2 | 101 participant random samples over 3 days | −80 °C | 20 µL human milk fat ➔Transesterification methanolic-BF3 | In: Tridecanoic acid (in extraction solvent) Out: - | GC 100 m HP88 column (Carrier gas: helium) | Akmar et al. (2013) [48] |
| Total saturated and unsaturated fatty acids, 18:2 n6, 18:3 n3, 20:4 n6, 22:6 n3 | 29 mid-feed samples (8–12 weeks post-partum) between 1200 and 1500 | −80 °C | 100 µL human milk ➔Hydrolysis methanolic-NaOCH3 ➔Derivatisation methanolic-BF3 | In: - Out: - | GC–FID 40 m RTX-2330 (Carrier gas: helium) | Saphier et al. (2013) [49] |
| Free fatty acids between C10 and C24 | 23 term and 15 preterm participants/38 post-feed samples during days 0–7 day, 8–21, >21 | Frozen | 500 µL human milk ➔LLE chloroform methanol ➔transesterification methanolic-HCl | In: C17:0 Out: - | GC–MS 30 m Ultra Alloy-5 column (Carrier gas: helium) | Chuang et al. (2013) [50] |
| Fatty acids between 4:0 and 22:6 | 50 participants 4 weeks post-partum, provided one full breast expression | −80 °C | 250 µL human milk ➔Transesterification methanolic-HCl | In: 11:0 FAME, 13:0 TAG Out: - | GC–FID 100m CP-Sil 88 column (Carrier gas: hydrogen) | Cruz-Hernandez et al. (2013) [32] |
| Phospholipid classes | 50 participants, pre-, mid-, post-feed samples at 4 weeks | −80 °C | 250 mg human milk ➔LLE chloroforom:methanol (2:1) ➔Filtration PTFE filter | In: Phosphatidylglyceol Out: - | NP HPLC (ELSD) 2 x 250 mm Nucleosil 50-5 columns (Solvents: acetonitrile/methanol) NMR | Giuffrida et al. (2013) [20] |
| Polar and lipidic metabolites Tentative 287 lipids (positive mode), 126 lipids (negative mode) | 52 samples between days 1 and 76, pooled. 10 participant samples at week 1, 9 participant samples at week 4 | −80 °C (long term) −20 °C (short term) | 50 µL human milk ➔LLE MTBE ➔Transesterification methanolic-HCl, BSTFA | In: C18:0 after extraction Out: Pooled HM | GC–Q–MS 30 m 122-5332G DVB5-MS column (Carrier gas: helium) LC–QTOF–MS (ESI) 15 cm EC-C8 column (Solvents: methanol water) | Villasenor et al. (2014) [51] |
| 1. Fatty acids between 10:0 and 20:4 2. Triglycerides between 32:0 and 54:5 | 2 samples 4 random weeks post-partum | - | 200 µL human milk ➔LLE (1) chloroform:methanol (2:1) ➔Transesterification with acid ➔LLE (2) chloroform: methanol: isopropanol (1:2:4) | In: 17:1–17:1–17:1 TAG, 17:0–14:1 PE, 17:0–14:1 PS, 17:0–14:1 PI, 18:1;2/17:0 SM (after extraction, for MS/MS) Out: - | 1. GC–FID 60 m TRFRAME column (Carrier gas: helium) 2. MS/MS Triple TOF (positive and negative mode) | Sokol et al. (2015) [52] |
| Over 40 triglycerides | 15 between-feed samples over days 1–5, 6–15 and >16 | 150 uL human milk ➔dichloromethane:methanol (2:1) ➔BHT preservative | In: - Out: - | HPLC–APCI–MS 150 mm Kinetex C18 column (Solvents: acetonitrile/n-pentanol) | Ten-Domenech et al. (2015) [53] | |
| Fatty acids ranging from 10:0 to 22:6 | 477 participants gave pre-feed samples on days 1, 14, 42 between 1000 and 1100 | −20 °C; −80 °C | 200 µL human milk ➔LLE chloroform: methanol (1:1) ➔BHT preservative ➔Hydrolysis methanolic-KOH ➔Derivatisation methanolic-BF3 ➔SPE Sep-pak silica column | In: - Out: - | GC–FID 60 m DB-23 Fused silica column (Carrier gas: nitrogen) | Jiang et al. (2016) [13] |
| 8 long-chain polyunsaturated fatty acids | 514 participants, between 0900 and 1100 for first 22–25 days | −80 °C | 0.2 mL human milk fat ➔Transesterification methanolic-CH3COCl | In: C17:0 Daturic acid Out: - | GC–FID 100 mm SP2560 column (Carrier gas: nitrogen) | Liu et al. (2016) [54] |
| 1. Identified putative DHA-TAGs 2. Verified 56 DHA-TAGs ranging from C45H74O6 to C67H116O6 | 1 sample | - | 0.2 mL human milk ➔LLE chloroform:methanol (2:1) | In: - Out: - | 1. LC–ESI–triple quadrupole MS 250 mm synergi polar RP column 2. LC–ESI–LTQ–ORBI MS 2x 150 mm Poroshell 120 EC-C18 (Solvents: acetonitrile/water) | Liu et al. (2016) [29] |
| Polyunsaturated fatty acids | 225 participants, provided pre- and/or post-feed milk at their own discretion, at 2 months | 4˚C (≤24 hours); −80˚C | 200 uL human milk ➔Transesterification - | In: - Out: - | GC–FID | Rosenlund et al. (2016) [30] |
| Groups of fatty acids, Glycerophospholipids, Prenol lipids, Glycerolipids, Sphingolipids, Sterol lipids | 1 participant provided samples, at 1 year | −80˚C | 1 mL human milk ➔SPME C18, isopropanol elution | In: - Out: - | LC–ESI–QTOF–MS 50 mm SB-C18 column (Solvents: methanol, water, hexane, isopropanol) | Garwolinska et al. (2017) [55] |
| sn-glycero-3-phosphocholine (and other lipid derivatives) | 37 mothers provided 15 (morning and evening) samples on days 9, 12, 24, 31, 60, 85, 86, 87 | −20˚C (2-8 days); −80˚C | - mL human milk ➔LLE methanol:water | In: - Out: - | NMR | Wu et al. (2016) [56] |
| 64 Triglycerides ranging from C33H62O6 to C65H120O6 | 27 participants provided a day 7 and day 42 sample | −20˚C | 0.1 mL human milk ➔LLE hexane ➔Filtration 0.22 µm nylon filter | In: - Out: 4 commercial QC 18:2/18:2/18:2; 18:1/18:1/18:1; 16:0/16:0/16:0; 18:1/16:0/18:1 for calibration curves | SFC ESI–QTOF 100 mm BEH-2-Ethylpyridine column (Solvents: supercritical CO2, methanol, acetonitrile) | Tu et al. (2017) [57] |
| Fatty acids ranging from 8:0 to 20:3 | 26 participants, left and a right sample at the same time on 3 consecutive days | −20˚C (≤1 week); −80˚C | - mL human milk ➔LLE chloroform:methanol (2:1) ➔Transesterification methanolic-H2SO4 | In: - Out: - | GC–FID 50 mm BPX-70 column (Carrier gas: helium) | Gardner et al. (2017) [31] |
| 1. Fatty acids ranging from 8:0 to 22:6 2. 2 × Ceramides; 7 × GlucosylCeramide; 22 × Phosphatidylcholine; 25 × Phosphatidylethanolamine; 5 × Phosphatidylglycerol; 2 × Phosphatidylinositol; 2 × Phosphatidylserine; Retinol; 9 × Diglycerides; 49×Triglycerideas; 11 × Sphingomyeline; 10 × Eicosanoids; 2×Cardiolipines; 10 × LysoPhosphatidylcholine/Phosphatidylethanolamine | 118 participants gave samples over 24 h (each participant pooled). | −80˚C | - mL human milk ➔LLE chloroform:methanol (1:1) ➔Transesterification - | In: - Out: pooled QC (10 participants pooled samples) | 1. GC–FID 30 m fused silica column 2. LC-ESI-HRMS in positive and negative mode 100 mm CSH C18 column (Solvents: acetonitrile, water, isopropanol) | Alexandre-Gouabau et al. (2018) [10] |
| Sampling | During Feed | Time of Day | Lactation Stage | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Study | i) Participant n ii) Sample n | Pre-Feed (g/L) | Post-Feed (g/L) | Morning (g/L) | Noon (g/L) | Afternoon (g/L) | Evening (g/L) | 1 (g/L) | 2 (g/L) | 3 (g/L) | 4 (g/L) | 5 (g/L) | 6 (g/L) | 9 (g/L) | 12 (g/L) |
| Mitoulas et al., 2002 [26] | i) 17 initially ii) 76 | - | - | - | - | - | - | 39.9 (SE 1.4) | 35.2 (SE 1.4) | - | 35.4 (SE 1.4) | - | 37.3 (SE 1.4) | 40.7 (SE 1.4) | 40.9 (SE 3.3) |
| Saarela et al., 2005 [22] | i) 20 ii) 483 | 21.0 (SD 8.4) | 57.1 (SD 4.5) | - | - | - | - | 19.7 (SD 8.2) | 23.5 (SD 8.8) | 21.0 (SD 8.4) | 16.2 (SD 9.4) | 11.4 (SD 6.2) | 18.8 (SD 4.2) | - | - |
| Jackson et al., 1988 [39] | i) 25 ii) - | 0.35–21.85 (SD 1.92) | - | 17.9–50.6 31.4 (SD 6.6) | - | - | 20.7–45.7 31.4 (SD 6.6) | - | - | - | - | - | - | - | - |
| Khan et al., 2013 [23] | i) 15 ii) - | 32 (SD 12) | 56 (SD 17) | 18.4–69.2 29.3 (SD 10.9) | 22.1–80.6 35 (SD 12.9) | 21.2–72 31.6 (SD 10.4) | 15.9–63.3 28.1 (SD 12.2) | - | - | - | - | - | - | - | - |
| Separation/Detection Method | Advantages | Disadvantages |
|---|---|---|
| Gas chromatography | 1. Fatty acid methyl ester analysis is well characterized 2. Flame ionisation detector is robust and easy to maintain | 1. Sample derivatisation is required 2. Destructive 3. Isomers separation requires longer column and run time 4. Flame ionisation detector lacks mass selectivity |
| Liquid chromatography | 1. No sample derivatisation required 2. Large selection of column chemistry available | 1. Solvent system must be compatible with detector type |
| Supercritical fluid chromatography | 1. No derivatisation required 2. Compatible with almost any detector type 3. Relatively inexpensive 4. Low waste output 5. Faster separation than in GC/LC 6. Higher resolution than in GC/LC | 1. Polar lipid separation requires organic modifier |
| Thin-layer chromatography | 1. Inexpensive | 1. Qualitative lipid class separation only 2. Low separating resolution compared to GC and LC. |
| Mass spectrometry | 1. High sensitivity and specificity 2. Qualitative and quantitative (with standards) | 1. Expensive 2. Destructive |
| NMR spectroscopy | 1. Non-destructive 2. Highly reproducible | 1. Expensive 2. Signal overlapping in complex samples 3. Lower sensitivity than MS 4. Requires larger samples volume |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, A.D.; Gay, M.C.L.; Trengove, R.D.; Geddes, D.T. Human Milk Lipidomics: Current Techniques and Methodologies. Nutrients 2018, 10, 1169. https://doi.org/10.3390/nu10091169
George AD, Gay MCL, Trengove RD, Geddes DT. Human Milk Lipidomics: Current Techniques and Methodologies. Nutrients. 2018; 10(9):1169. https://doi.org/10.3390/nu10091169
Chicago/Turabian StyleGeorge, Alexandra D., Melvin C. L. Gay, Robert D. Trengove, and Donna T. Geddes. 2018. "Human Milk Lipidomics: Current Techniques and Methodologies" Nutrients 10, no. 9: 1169. https://doi.org/10.3390/nu10091169
APA StyleGeorge, A. D., Gay, M. C. L., Trengove, R. D., & Geddes, D. T. (2018). Human Milk Lipidomics: Current Techniques and Methodologies. Nutrients, 10(9), 1169. https://doi.org/10.3390/nu10091169