A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Study Protocol
2.2. Diets
2.3. DNA Extraction, PCR Amplification and Sequencing
2.4. Data Processing and Analyses
2.5. Short Chain Fatty Acids Extraction and Analysis
3. Results
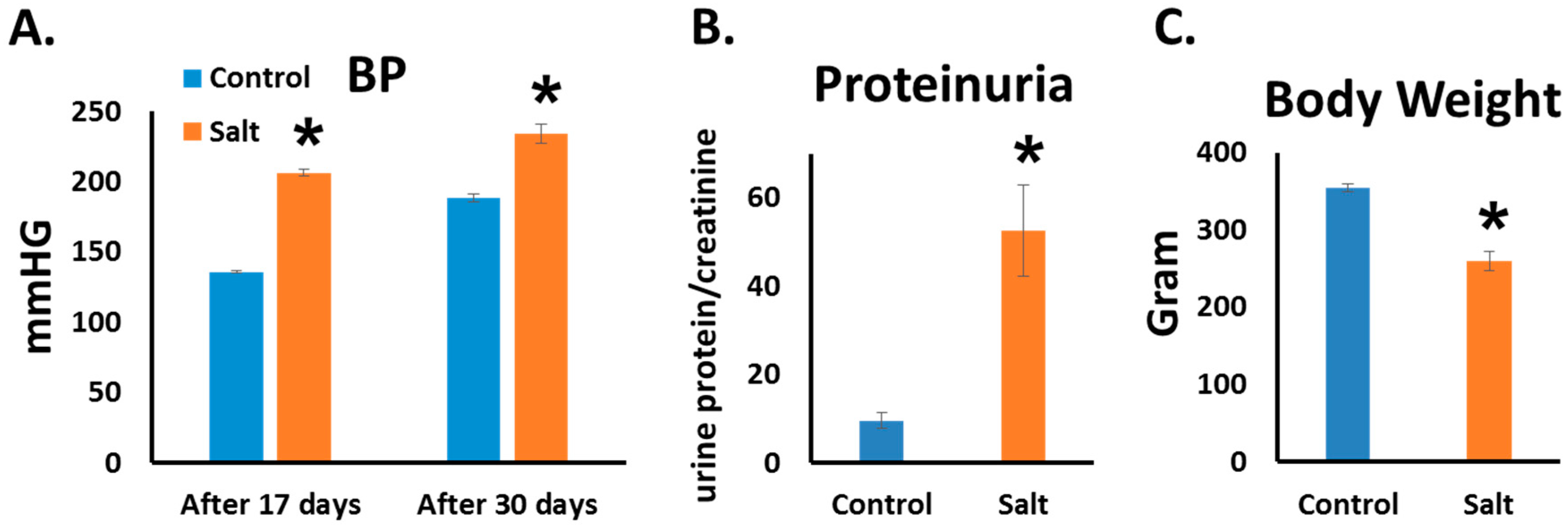
3.1. Blood Pressure and Proteinuria
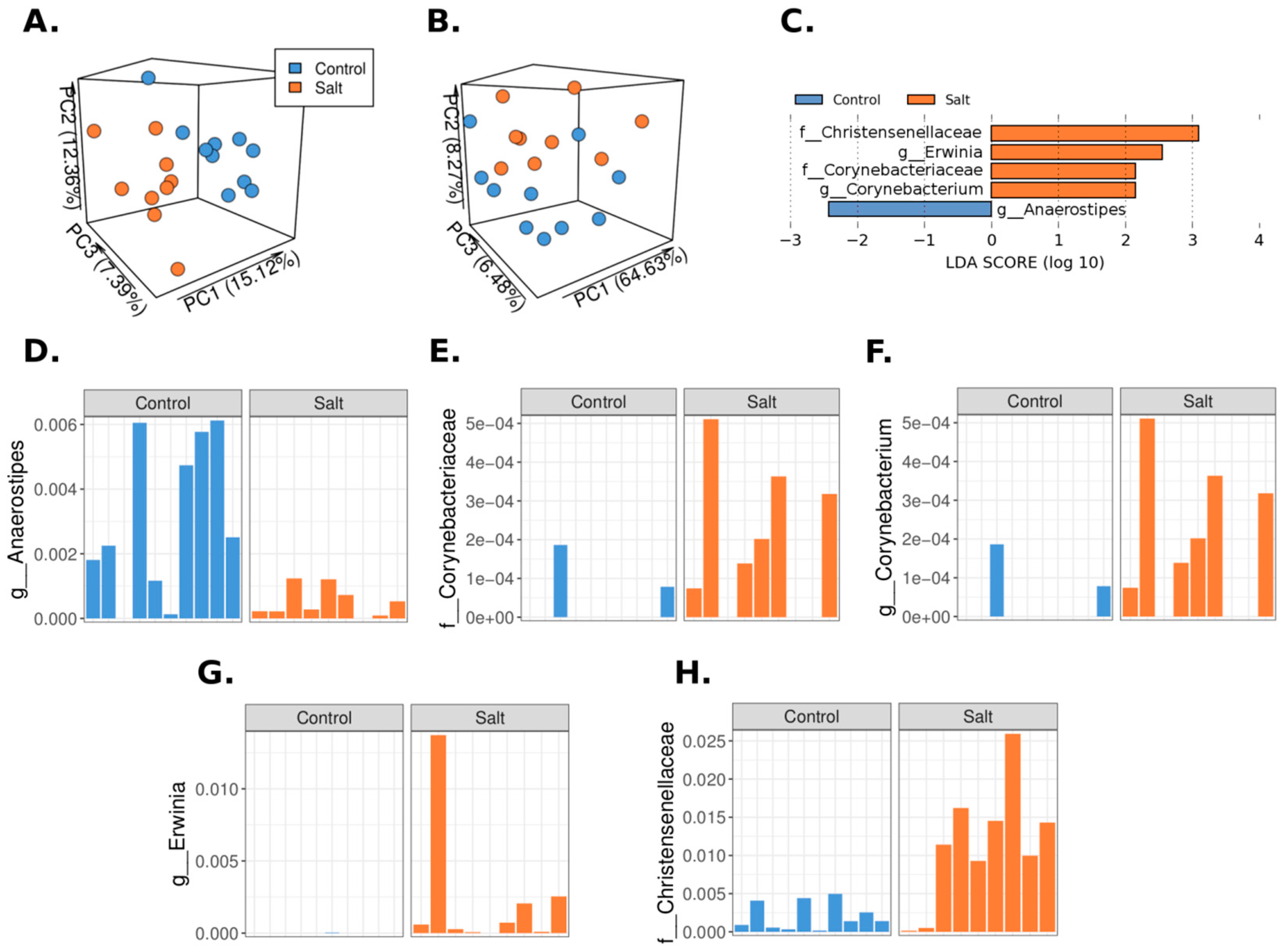
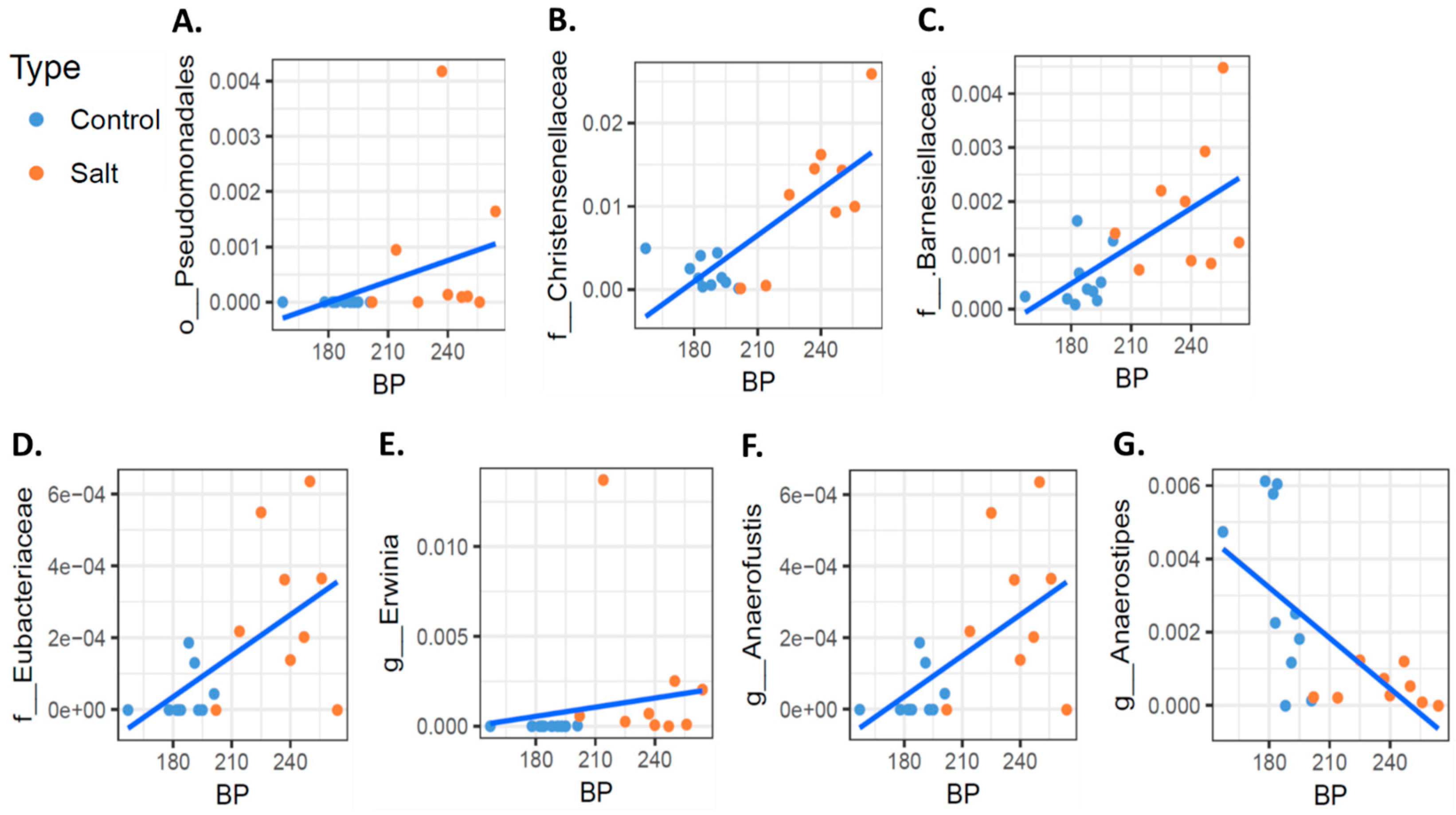
3.2. The Microbiome Profile
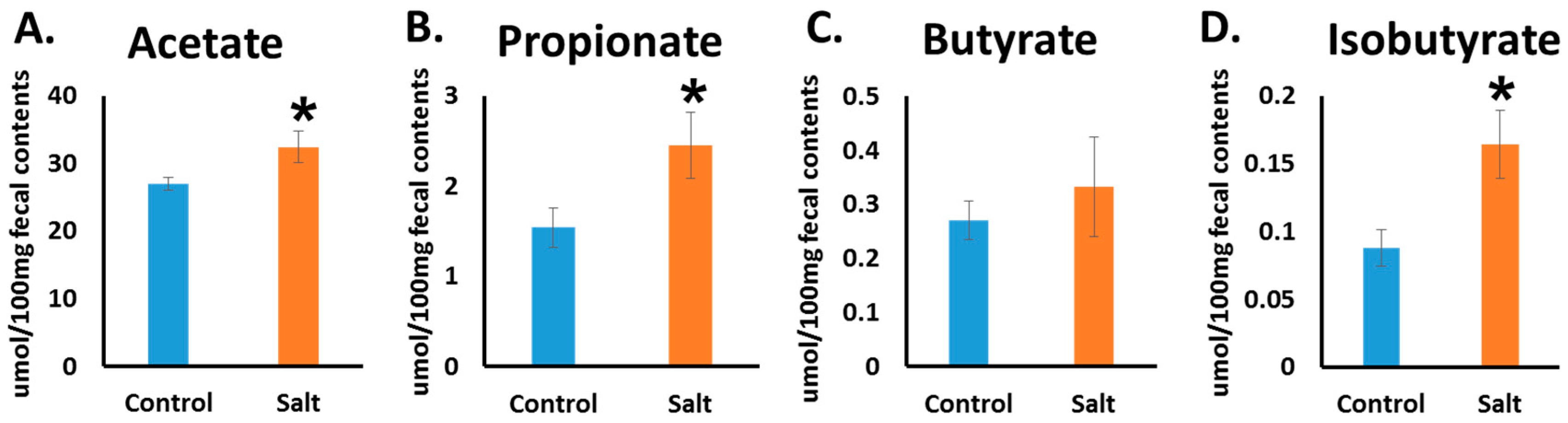
3.3. Short Chain Fatty Acid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cifkova, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Prim. 2018, 4, 18014. [Google Scholar] [CrossRef] [PubMed]
- Rucker, A.J.; Rudemiller, N.P.; Crowley, S.D. Salt, Hypertension, and Immunity. Annu. Rev. Physiol. 2018, 80, 283–307. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.; Mente, A.; Yusuf, S. Sodium intake and cardiovascular health. Circ. Res. 2015, 116, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; Biddinger, S.B.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.Z.; Mackay, C.R.; Kaye, D.M. Beyond gut feelings: How the gut microbiota regulates blood pressure. Nat. Rev. Cardiol. 2018, 15, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Luo, H.; Wang, J.; Tang, W.; Lu, J.; Wu, S.; Xiong, Z.; Yang, G.; Chen, Z.; Lan, T.; et al. Enteric dysbiosis-linked gut barrier disruption triggers early renal injury induced by chronic high salt feeding in mice. Exp. Mol. Med. 2017, 49, e370. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Huang, Z.; Yu, K.; Ding, R.; Ye, K.; Dai, C.; Xu, X.; Zhou, G.; Li, C. High-Salt Diet Has a Certain Impact on Protein Digestion and Gut Microbiota: A Sequencing and Proteome Combined Study. Front. Microbiol. 2017, 8, 1838. [Google Scholar] [CrossRef] [PubMed]
- Miranda, P.M.; De Palma, G.; Serkis, V.; Lu, J.; Louis-Auguste, M.P.; McCarville, J.L.; Verdu, E.F.; Collins, S.M.; Bercik, P. High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mahler, A.; Balogh, A.; Marko, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Iwai, J.; Dahl, L.K.; Knudsen, K.D. Genetic influence on the renin-angiotensin system: Low renin activities in hypertension-prone rats. Circ. Res. 1973, 32, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Nishiyama, A.; Abe, Y.; Navar, L.G. Enhancement of intrarenal angiotensinogen in Dahl salt-sensitive rats on high salt diet. Hypertension (Dallas TX 1979) 2003, 41, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Mell, B.; Jala, V.R.; Mathew, A.V.; Byun, J.; Waghulde, H.; Zhang, Y.; Haribabu, B.; Vijay-Kumar, M.; Pennathur, S.; Joe, B. Evidence for a link between gut microbiota and hypertension in the Dahl rat. Physiol. Genom. 2015, 47, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Macfarlane, G.T.; Macfarlane, S. Bacteria, colonic fermentation, and gastrointestinal health. J. AOAC Int. 2012, 95, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L.; Protzko, R.J.; Gevorgyan, H.; Peterlin, Z.; Sipos, A.; Han, J. Olfactory receptor responding to gut microbiota- derived signals plays a role in renin secretion and blood pressure regulation. Proc. Natl. Acad. Sci. USA 2013, 110, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Raizada, M.K.; Joe, B.; Bryan, N.S.; Chang, E.B.; Dewhirst, F.E.; Borisy, G.G.; Galis, Z.S.; Henderson, W.; Jose, P.A.; Ketchum, C.J.; et al. Report of the National Heart, Lung, and Blood Institute Working Group on the Role of Microbiota in Blood Pressure Regulation: Current Status and Future Directions. Hypertension (Dallas TX 1979) 2017. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Di Segni, A.; BenShoshan, M.; Asaf, R.; Squires, J.E.; Farage Barhom, S.; Glick Saar, E.; Cesarkas, K.; Smollan, G.; Weiss, B.; et al. Fecal microbial characterization of hospitalized patients with suspected infectious diarrhea shows significant dysbiosis. Sci. Rep. 2017, 7, 1088. [Google Scholar] [CrossRef] [PubMed]
- Di Segni, A.; Braun, T.; BenShoshan, M.; Farage Barhom, S.; Glick Saar, E.; Cesarkas, K.; Squires, J.E.; Keller, N.; Haberman, Y. Guided Protocol for Fecal Microbial Characterization by 16S rRNA-Amplicon Sequencing. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; Bousvaros, A.; et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012, 13, R79. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.-O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, A.; Volkov, A.; Voloshin, K.; Shemesh, C.; Barshack, I.; Grossman, E. Melatonin prevents kidney injury in a high salt diet-induced hypertension model by decreasing oxidative stress. J. Pineal Res. 2016, 60, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.-Y.; Xia, G.-H.; Lu, J.-Q.; Chen, M.-X.; Zhen, X.; Wang, S.; You, C.; Nie, J.; Zhou, H.-W.; Yin, J. Impaired renal function and dysbiosis of gut microbiota contribute to increased trimethylamine-N-oxide in chronic kidney disease patients. Sci. Rep. 2017, 7, 1445. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Prieto, I.; Abriouel, H.; Villarejo, A.B.; Ramirez-Sanchez, M.; Cobo, A.; Benomar, N.; Galvez, A.; Martinez-Canamero, M. Changes in Gut Microbiota Linked to a Reduction in Systolic Blood Pressure in Spontaneously Hypertensive Rats Fed an Extra Virgin Olive Oil-Enriched Diet. Plant Foods Hum. Nutr. 2018, 73, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Goel, R.; Kumar, A.; Qi, Y.; Lobaton, G.; Hosaka, K.; Mohammed, M.; Handberg, E.M.; Richards, E.M.; Pepine, C.J.; et al. Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin. Sci. (Lond). 2018, 132, 701–718. [Google Scholar] [CrossRef] [PubMed]
- Feng, D.; Yang, C.; Geurts, A.M.; Kurth, T.; Liang, M.; Lazar, J.; Mattson, D.L.; O’Connor, P.M.; Cowley, A.W.J. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012, 15, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Pluznick, J.L. Microbial Short-Chain Fatty Acids and Blood Pressure Regulation. Curr. Hypertens. Rep. 2017, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Hold, G.L.; Duncan, S.H.; Gruhl, B.; Collins, M.D.; Lawson, P.A.; Flint, H.J.; Blaut, M. Anaerostipes caccae gen. nov., sp. nov., a new saccharolytic, acetate-utilising, butyrate-producing bacterium from human faeces. Syst. Appl. Microbiol. 2002, 25, 46–51. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bier, A.; Braun, T.; Khasbab, R.; Di Segni, A.; Grossman, E.; Haberman, Y.; Leibowitz, A. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients 2018, 10, 1154. https://doi.org/10.3390/nu10091154
Bier A, Braun T, Khasbab R, Di Segni A, Grossman E, Haberman Y, Leibowitz A. A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients. 2018; 10(9):1154. https://doi.org/10.3390/nu10091154
Chicago/Turabian StyleBier, Ariel, Tzipi Braun, Rawan Khasbab, Ayelet Di Segni, Ehud Grossman, Yael Haberman, and Avshalom Leibowitz. 2018. "A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model" Nutrients 10, no. 9: 1154. https://doi.org/10.3390/nu10091154
APA StyleBier, A., Braun, T., Khasbab, R., Di Segni, A., Grossman, E., Haberman, Y., & Leibowitz, A. (2018). A High Salt Diet Modulates the Gut Microbiota and Short Chain Fatty Acids Production in a Salt-Sensitive Hypertension Rat Model. Nutrients, 10(9), 1154. https://doi.org/10.3390/nu10091154