Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene Limit BE(2)-C Neuroblastoma Cell Growth and Neurosphere Formation In Vitro



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Neurosphere Formation Assay
2.3. Reagents
2.4. EGCG and IIF Treatments
2.5. MTT Assay
2.6. Combination Index (CI)
2.7. RNA Isolation
2.8. Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
2.9. Quantitative Polymerase Chain Reaction (qPCR)
2.10. Western Blot
2.11. Zymography
2.12 Statistical Analysis
3. Results
3.1. RAR and RXR Expression Changed after EGCG and IIF Treatments
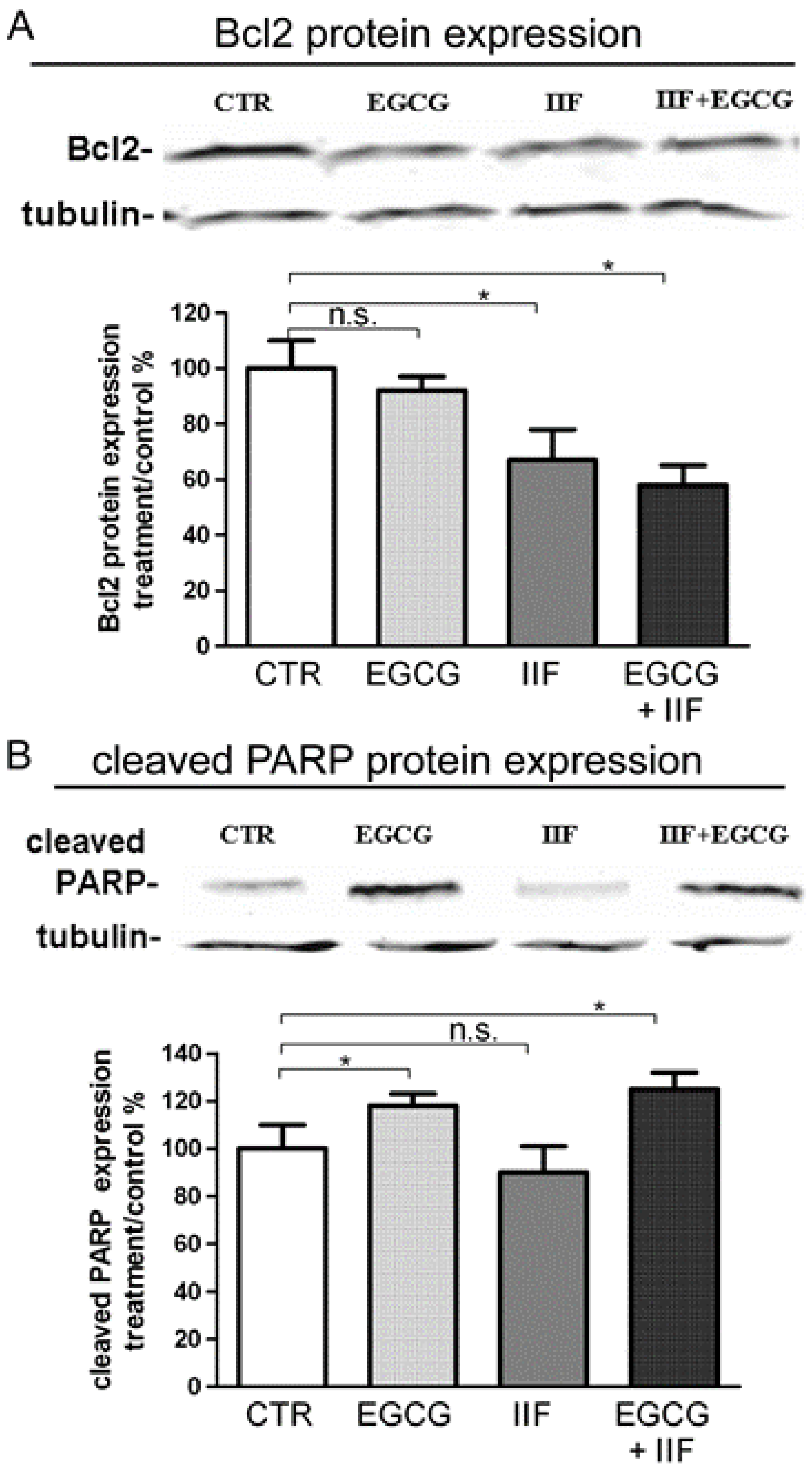
3.2. Synergistic Effect of EGCG and IIF in Combination Enhanced Cytotoxicity and Activated Apoptosis in BE(2)-C Cells
3.3. EGCG and IIF Downregulated N-MYC Expression
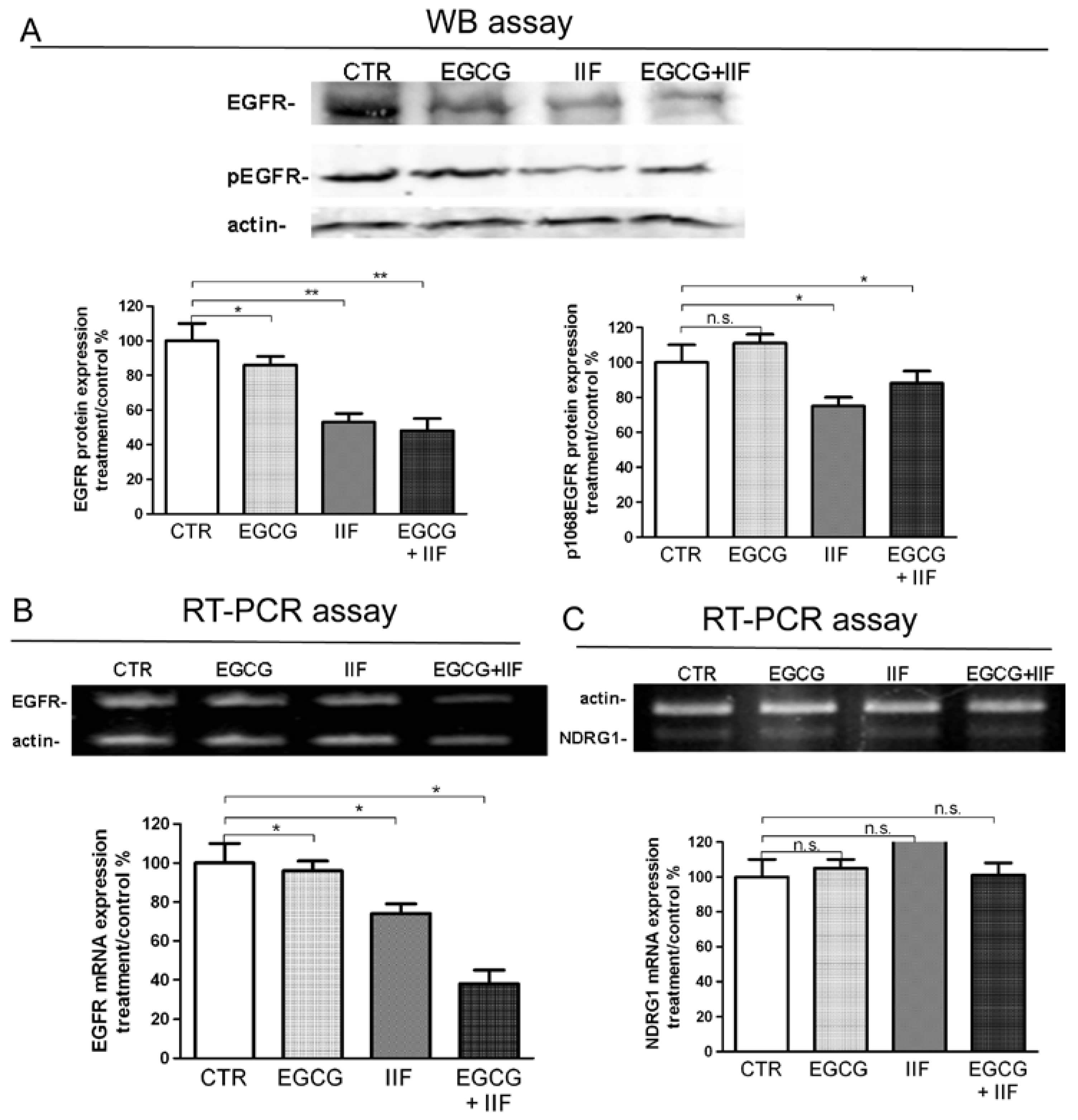
3.4. Molecular Targets of EGCG and IIF and N-MYC Downregulation
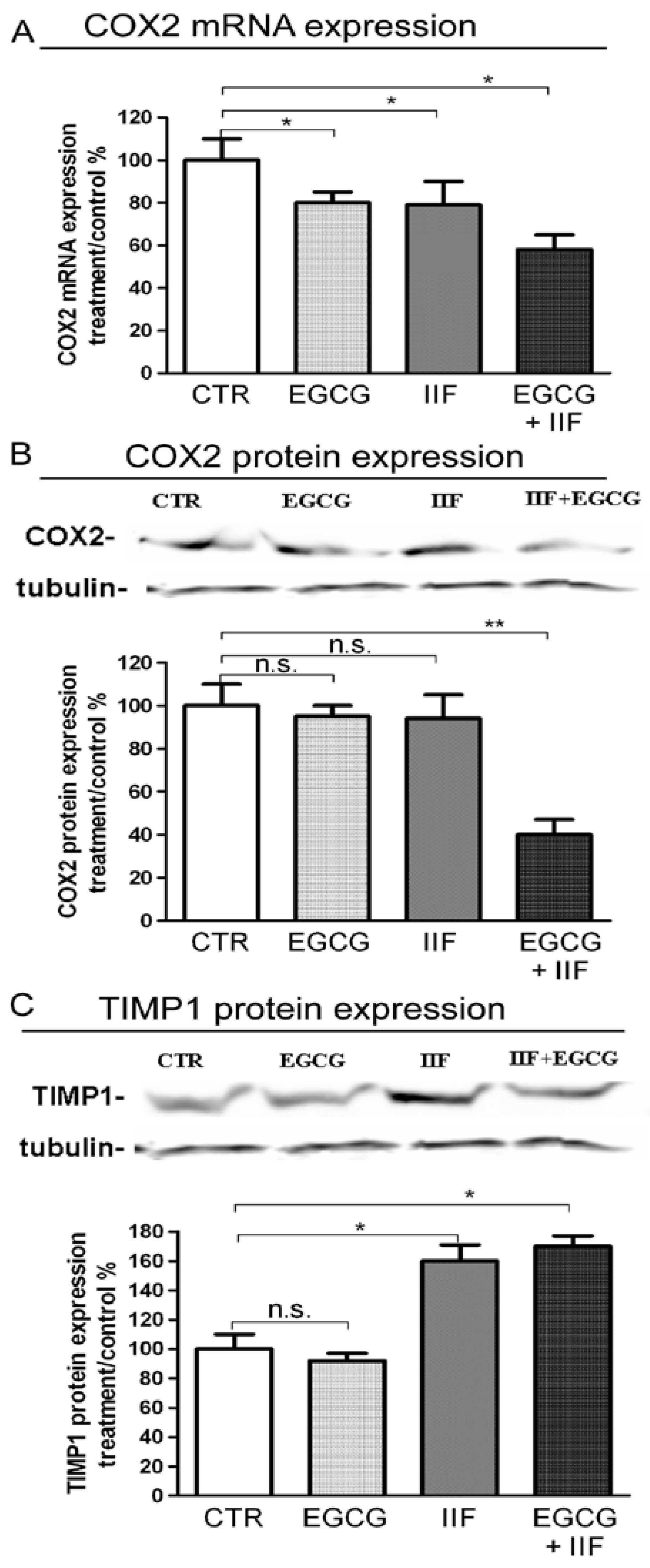
3.5. EGCG and IIF Limited Invasion and Metastatic Capability
3.6. EGCG and IIF Impaired Sphere Formation in BE(2)-C Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548–e557. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, M.A.; Desai, A.V.; Glade Bender, J.L.; Cohn, S.L. Emerging and investigational therapies for neuroblastoma. Expert Opin. Orphan Drugs 2017, 5, 355–368. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.P.; Matthay, K.K.; Villablanca, J.G.; Maurer, B.J. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003, 197, 185–192. [Google Scholar] [CrossRef]
- Peinemann, F.; van Dalen, E.C.; Enk, H.; Berthold, F. Retinoic acid postconsolidation therapy for high-risk neuroblastoma patients treated with autologous haematopoietic stem cell transplantation. Cochrane Database Syst. Rev. 2017, 8, CD010685. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Biagi, C.; Zama, D.; Vendemini, F.; Martoni, A.; Morello, W.; Gasperini, P.; Pession, A. Retinoids in pediatric onco-hematology: The model of acute promyelocytic leukemia and neuroblastoma. Adv. Ther. 2012, 29, 747–762. [Google Scholar] [CrossRef] [PubMed]
- Duffy, D.J.; Krstic, A.; Halasz, M.; Schwarzl, T.; Konietzny, A.; Iljin, K.; Higgins, D.G.; Kolch, W. Retinoic acid and TGF-β signalling cooperate to overcome MYCN-induced retinoid resistance. Genome Med. 2017, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lindner, S.; Bohrer, A.; Maurer, J.; De Preter, K.; Lefever, S.; Heukamp, L.; Schulte, S.; Molenaar, J.; Versteeg, R.; et al. MYCN and ALKF1174L are sufficient to drive neuroblastoma development from neural crest progenitor cells. Oncogene 2013, 32, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Weiss, W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013, 3, a014415. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Djelloul, S.; Raymond, E. New paradigms in anticancer therapy: Targeting multiple signaling pathways with kinase inhibitors. Semin. Oncol. 2006, 33, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Govoni, M.; Ciavarella, C.; Orlandi, M.; Papi, A. A RXR ligand 6-OH-11-O-hydroxyphenanthrene with antitumour properties enhances (-)-epigallocatechin-3-gallate activity in three human breast carcinoma cell lines. BioMed Res. Int. 2014, 2014, 853086. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Govoni, M.; Ciavarella, C.; Spisni, E.; Orlandi, M.; Farabegoli, F. Epigallocatechin-3-gallate Increases RXRγ-mediated Pro-apoptotic and Anti-invasive Effects in Gastrointestinal Cancer Cell Lines. Curr. Cancer Drug Targets 2016, 16, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Farabegoli, F.; Govoni, M.; Spisni, E.; Papi, A. EGFR inhibition by (-)-epigallocatechin-3-gallate and IIF treatments reduces breast cancer cell invasion. Biosci. Rep. 2017, 37, BSR20170168. [Google Scholar] [CrossRef] [PubMed]
- Bartolini, G.; Orlandi, M.; Ammar, K.; Magrini, E.; Ferreri, A.M.; Rocchi, P. Effect of a new derivative of retinoic acid on proliferation and differentiation in human neuroblastoma cells. Anticancer Res. 2003, 23, 1495–1499. [Google Scholar] [PubMed]
- Das, A.; Banik, N.L.; Ray, S.K. Retinoids induce differentiation and downregulate telomerase activity and N-MYC to increase sensitivity to flavonoids for apoptosis in human malignant neuroblastoma SH-SY5Y cells. Int. J. Oncol. 2009, 34, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, M.; Khandkar, M.; Banik, N.L.; Ray, S.K. Alterations in expression of specific microRNAs by combination of 4-HPR and EGCG inhibited growth of human malignant neuroblastoma cells. Brain Res. 2012, 1454, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.F.; Beierle, E.A. Cancer Stem Cells and Their Interaction with the Tumor Microenvironment in Neuroblastoma. Cancers 2015, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Ferreri, A.M.; Rocchi, P.; Guerra, F.; Orlandi, M. Epigenetic modifiers as anticancer drugs: Effectiveness of valproic acid in neural crest-derived tumor cells. Anticancer Res. 2010, 30, 535–540. [Google Scholar] [PubMed]
- Goldsmith, K.C.; Lestini, B.J.; Gross, M.; Lp, L.; Bhumbla, A.; Zhang, X.; Zhao, H.; Liu, X.; Hogarty, M.D. Mitochondrial Bcl-2 family dynamics define therapy response and resistance in neuroblastoma. Cell Death Differ. 2010, 17, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar] [PubMed]
- McCurrach, M.E.; Connor, T.M.; Knudson, C.M.; Korsmeyer, S.J.; Lowe, S.W. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 2345–2349. [Google Scholar] [CrossRef] [PubMed]
- Perini, G.; Diolaiti, D.; Porro, A.; Della Valle, G. In vivo transcriptional regulation of N-MYC target genes is controlled by E-box methylation. Proc. Natl. Acad. Sci. USA 2005, 102, 12117–12122. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Mendonca, J.; Ying, J.; Kim, H.S.; Verdone, J.E.; Zarif, J.C.; Carducci, M.; Hammers, H.; Pienta, K.J.; Kachhap, S. The prostate metastasis suppressor gene NDRG1 differentially regulates cell motility and invasion. Mol. Oncol. 2017, 11, 655–669. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, Z.; Menezes, S.V.; Sahni, S.; Kalinowski, D.S.; Bae, D.H.; Lane, D.J.; Richardson, D.R. The Metastasis Suppressor, N-MYC Downstream-regulated Gene-1 (NDRG1), Down-regulates the ErbB Family of Receptors to Inhibit Downstream Oncogenic Signaling Pathways. J. Biol. Chem. 2016, 291, 1029–1052. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumimoto, T.; Williams, P.; Yoneda, T. The SK-N-AS human neuroblastoma cell line develops osteolytic bone metastases with increased angiogenesis and COX-2 expression. J. Bone Oncol. 2014, 3, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Panda, C.K. Molecular aspects of cancer chemopreventive and therapeutic efficacies of tea and tea polyphenols. Nutrition 2017, 43–44, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Handgretinger, R.; Schlegel, P. Emerging role of immunotherapy for childhood cancers. Chin. Clin. Oncol. 2018, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Orlandi, M. Role of nuclear receptors in breast cancer stem cells. World J. Stem Cells 2016, 8, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Ziegler, D.S.; Trahair, T.N.; Marshall, G.M.; Haber, M.; Norris, M.D. Too many targets, not enough patients: Rethinking neuroblastoma clinical trials. Nat. Rev. Cancer 2018, 18, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, N.; Hartomo, T.B.; Pham, T.V.; Lee, M.J.; Yamamoto, T.; Morikawa, S.; Hasegawa, D.; Takeda, H.; Kawasaki, K.; Kosaka, Y.; et al. Epigallocatechin gallate inhibits sphere formation of neuroblastoma BE(2)-C cells. Environ. Health Prev. Med. 2012, 17, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Craig, B.T.; Rellinger, E.J.; Alvarez, A.L.; Dusek, H.L.; Qiao, J.; Chung, D.H. Induced differentiation inhibits sphere formation in neuroblastoma. Biochem. Biophys. Res. Commun. 2016, 477, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Noujaim, D.; van Golen, C.M.; van Golen, K.L.; Grauman, A.; Feldman, E.L. N-MYC and Bcl-2 coexpression induces MMP-2 secretion and activation in human neuroblastoma cells. Oncogene 2002, 21, 4549–4557. [Google Scholar] [CrossRef] [PubMed]
- Uryu, K.; Nishimura, R.; Kataoka, K.; Sato, Y.; Nakazawa, A.; Suzuki, H.; Yoshida, K.; Seki, M.; Hiwatari, M.; Isobe, T.; et al. Identification of the genetic and clinical characteristics of neuroblastomas using genome-wide analysis. Oncotarget 2017, 8, 107513–107529. [Google Scholar] [CrossRef] [PubMed]
- Light, J.E.; Koyama, H.; Minturn, J.E.; Ho, R.; Simpson, A.M.; Iyer, R.; Mangino, J.L.; Kolla, V.; London, W.B.; Brodeur, G.M. Clinical significance of NTRK family gene expression in neuroblastomas. Pediatr. Blood Cancer 2012, 59, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, S.; Kadomatsu, K. Origin and initiation mechanisms of neuroblastoma. Cell Tissue Res. 2018, 372, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Furman, W.L.; McGregor, L.M.; McCarville, M.B.; Onciu, M.; Davidoff, A.M.; Kovach, S.; Hawkins, D.; McPherson, V.; Houghton, P.J.; Billups, C.A.; et al. A single-arm pilot phase II study of gefitinib and irinotecan in children with newly diagnosed high-risk neuroblastoma. Investig. New Drugs 2012, 30, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Tatenhorst, L.; Terwel, D.; Hermes, M.; Kummer, M.P.; Orlandi, M.; Heneka, M.T. PPARgamma and RXRgamma ligands act synergistically as potent antineoplastic agents in vitro and in vivo glioma models. J. Neurochem. 2009, 109, 1779–1790. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farabegoli, F.; Govoni, M.; Spisni, E.; Papi, A. Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene Limit BE(2)-C Neuroblastoma Cell Growth and Neurosphere Formation In Vitro. Nutrients 2018, 10, 1141. https://doi.org/10.3390/nu10091141
Farabegoli F, Govoni M, Spisni E, Papi A. Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene Limit BE(2)-C Neuroblastoma Cell Growth and Neurosphere Formation In Vitro. Nutrients. 2018; 10(9):1141. https://doi.org/10.3390/nu10091141
Chicago/Turabian StyleFarabegoli, Fulvia, Marzia Govoni, Enzo Spisni, and Alessio Papi. 2018. "Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene Limit BE(2)-C Neuroblastoma Cell Growth and Neurosphere Formation In Vitro" Nutrients 10, no. 9: 1141. https://doi.org/10.3390/nu10091141
APA StyleFarabegoli, F., Govoni, M., Spisni, E., & Papi, A. (2018). Epigallocatechin-3-gallate and 6-OH-11-O-Hydroxyphenanthrene Limit BE(2)-C Neuroblastoma Cell Growth and Neurosphere Formation In Vitro. Nutrients, 10(9), 1141. https://doi.org/10.3390/nu10091141