The Role of n-3 Long Chain Polyunsaturated Fatty Acids in Cardiovascular Disease Prevention, and Interactions with Statins




Abstract
1. Introduction
2. Statins: Mode of Action
3. Epidemiology of Statin Use
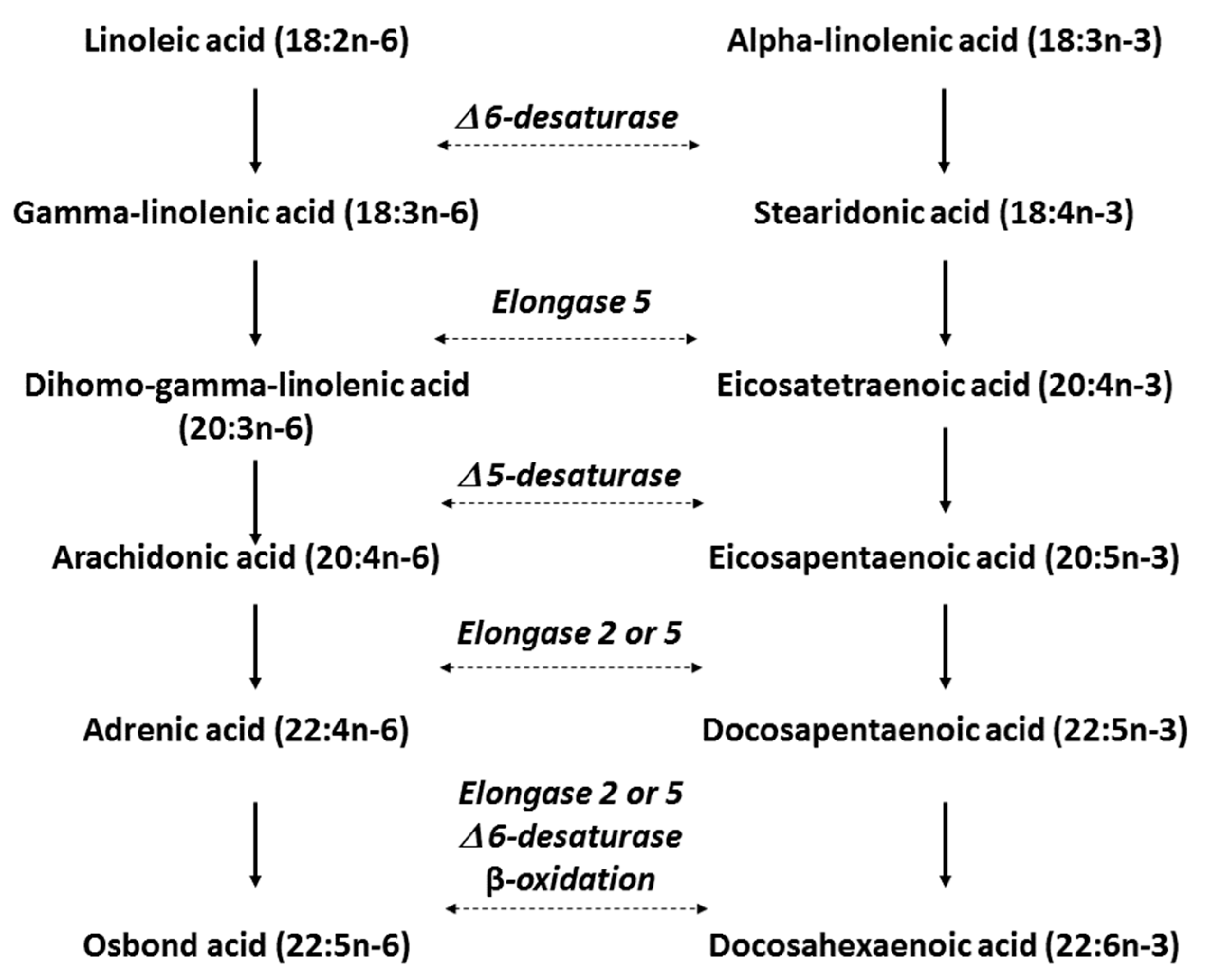
4. n-3 LC PUFAs: Mode of Action in CVD Prevention
5. Use of n-3 LC PUFAs as Dietary Supplements in the General Population
6. Interactions between LC PUFAs and Statins, and Effects on Dyslipidemia, CVD and Mortality
6.1. Effects of Dietary Fatty Acids and Statin Co-Administration on Dyslipidemia
6.2. Effects of Statins on n-3 LC PUFA Concentrations
6.3. Interactions between Statins and n-3 LC PUFAs on Mitochondrial Function
6.4. Inhibition of CYP Enzymes by Statins and Effects on Eicosanoid Production
6.5. Effects of Statins and n-3 LC PUFAs on Clinical and Mechanistic Endpoints
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD Mortality Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef]
- Bowry, A.D.; Lewey, J.; Dugani, S.B.; Choudhry, N.K. The Burden of Cardiovascular Disease in Low- and Middle-Income Countries: Epidemiology and Management. Can. J. Cardiol. 2015, 31, 1151–1159. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine. Promoting Cardiovascular Health in the Developing World: A Critical Challenge to Achieve Global Health; National Academies Press (US): Washington, DC, USA, 2010. [Google Scholar]
- Vos, T.; Barber, R.M.; Bell, B.; Bertozzi-Villa, A.; Biryukov, S.; Bolliger, I.; Charlson, F.; Davis, A.; Degenhardt, L.; Dicker, D.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef]
- Ford, E.S.; Ajani, U.A.; Croft, J.B.; Critchley, J.A.; Labarthe, D.R.; Kottke, T.E.; Giles, W.H.; Capewell, S. Explaining the decrease in U.S. deaths from coronary disease, 1980–2000. N. Engl. J. Med. 2007, 356, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
- Koskinas, K.C.; Windecker, S.; Raber, L. Regression of coronary atherosclerosis: Current evidence and future perspectives. Trends Cardiovasc. Med. 2016, 26, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.; Huffman, M.D.; Macedo, A.F.; Moore, T.H.; Burke, M.; Davey Smith, G.; Ward, K.; Ebrahim, S. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2013, 1, CD004816. [Google Scholar] [CrossRef] [PubMed]
- Superko, H.R.; King, S., 3rd. Lipid management to reduce cardiovascular risk: A new strategy is required. Circulation 2008, 117, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Breslow, J.L. n-3 fatty acids and cardiovascular disease. Am. J. Clin. Nutr. 2006, 83, 1477S–1482S. [Google Scholar] [CrossRef] [PubMed]
- Bang, H.O.; Dyerberg, J.; Sinclair, H.M. The composition of the Eskimo food in north western Greenland. Am. J. Clin. Nutr. 1980, 33, 2657–2661. [Google Scholar] [CrossRef] [PubMed]
- Daviglus, M.L.; Stamler, J.; Orencia, A.J.; Dyer, A.R.; Liu, K.; Greenland, P.; Walsh, M.K.; Morris, D.; Shekelle, R.B. Fish consumption and the 30-year risk of fatal myocardial infarction. N. Engl. J. Med. 1997, 336, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Kromhout, D.; Bosschieter, E.B.; de Lezenne Coulander, C. The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N. Engl. J. Med. 1985, 312, 1205–1209. [Google Scholar] [CrossRef] [PubMed]
- Burr, M.L.; Fehily, A.M.; Gilbert, J.F.; Rogers, S.; Holliday, R.M.; Sweetnam, P.M.; Elwood, P.C.; Deadman, N.M. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: Diet and reinfarction trial (DART). Lancet 1989, 2, 757–761. [Google Scholar] [CrossRef]
- Singh, R.B.; Niaz, M.A.; Sharma, J.P.; Kumar, R.; Rastogi, V.; Moshiri, M. Randomized, double-blind, placebo-controlled trial of fish oil and mustard oil in patients with suspected acute myocardial infarction: the Indian experiment of infarct survival—4. Cardiovasc. Drugs Ther. 1997, 11, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Lancet 1999, 354, 447–455. [Google Scholar] [CrossRef]
- Kromhout, D.; Yasuda, S.; Geleijnse, J.M.; Shimokawa, H. Fish oil and omega-3 fatty acids in cardiovascular disease: do they really work? Eur. Heart J. 2012, 33, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.C.; Palacios, O.M.; Bell, M.; Toth, P.P. Use of supplemental long-chain omega-3 fatty acids and risk for cardiac death: An updated meta-analysis and review of research gaps. J. Clin. Lipidol. 2017, 11, 1152.e2–1160.e2. [Google Scholar] [CrossRef] [PubMed]
- Sethi, A.; Bajaj, A.; Khosla, S.; Arora, R.R. Statin Use Mitigate the Benefit of Omega-3 Fatty Acids Supplementation-A Meta-Regression of Randomized Trials. Am. J. Ther. 2016, 23, e737–e748. [Google Scholar] [CrossRef] [PubMed]
- Istvan, E. Statin inhibition of HMG-CoA reductase: A 3-dimensional view. Atheroscler. Suppl. 2003, 4, 3–8. [Google Scholar] [CrossRef]
- Zhou, Q.; Liao, J.K. Pleiotropic effects of statins—Basic research and clinical perspectives. Circ. J. 2010, 74, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M. Primary prevention of cardiovascular disease with statins: Assessing the evidence base behind clinical guidance. Clin. Pharm. 2016, 8, 57. [Google Scholar]
- Neuvonen, P.J. Drug interactions with HMG-CoA reductase inhibitors (statins): the importance of CYP enzymes, transporters and pharmacogenetics. Curr. Opin. Investig. Drugs 2010, 11, 323–332. [Google Scholar] [PubMed]
- Maji, D.; Shaikh, S.; Solanki, D.; Gaurav, K. Safety of statins. Indian J. Endocrinol. Metab. 2013, 17, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Shitara, Y.; Sugiyama, Y. Pharmacokinetic and pharmacodynamic alterations of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: drug-drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol. Ther. 2006, 112, 71–105. [Google Scholar] [CrossRef] [PubMed]
- Vaquero, M.P.; Sanchez Muniz, F.J.; Jimenez Redondo, S.; Prats Olivan, P.; Higueras, F.J.; Bastida, S. Major diet-drug interactions affecting the kinetic characteristics and hypolipidaemic properties of statins. Nutr. Hosp. 2010, 25, 193–206. [Google Scholar] [PubMed]
- Corsini, A.; Ceska, R. Drug-drug interactions with statins: Will pitavastatin overcome the statins’ Achilles’ heel? Curr. Med. Res. Opin. 2011, 27, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Fulcher, J.; O’Connell, R.; Voysey, M.; Emberson, J.; Blackwell, L.; Mihaylova, B.; Simes, J.; Collins, R.; Kirby, A.; Colhoun, H.; et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Cziraky, M.J.; Watson, K.E.; Talbert, R.L. Targeting low HDL-cholesterol to decrease residual cardiovascular risk in the managed care setting. J. Manag. Care Pharm. 2008, 14, S3–S28; quiz S30–S21. [Google Scholar] [CrossRef] [PubMed]
- Nishikido, T.; Oyama, J.; Keida, T.; Ohira, H.; Node, K. High-dose statin therapy with rosuvastatin reduces small dense LDL and MDA-LDL: The Standard versus high-dose therApy with Rosuvastatin for lipiD lowering (SARD) trial. J. Cardiol. 2016, 67, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.; Reynolds, K.; Smith, D.; Muntner, P. Trends in statin use and low-density lipoprotein cholesterol levels among US adults: impact of the 2001 National Cholesterol Education Program guidelines. Ann. Pharmacother. 2008, 42, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Falkingbridge, S. The Cardiovascular Market Outlook to 2017: Competitive Landscape, Global Market Analysis, Key Trends, and Pipeline Analysis; Informa: London, UK, 2012; pp. 58–61. [Google Scholar]
- Robinson, J.G.; Booth, B. Statin use and lipid levels in older adults: National Health and Nutrition Examination Survey, 2001 to 2006. J. Clin. Lipidol. 2010, 4, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Walley, T.; Folino-Gallo, P.; Stephens, P.; Van Ganse, E. Trends in prescribing and utilization of statins and other lipid lowering drugs across Europe 1997-2003. Br. J. Clin. Pharmacol. 2005, 60, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Food and Agriculture Organization of the United Nations. Fats and Fatty Acids in Human Nutrition: Report of an Expert Consultation; Food and Agriculture Organization of the United Nations: Rome, Italy, 2010; Available online: http://www.fao.org/3/a-i1953e.pdf (accessed on 5 April 2016).
- Stark, K.D.; Van Elswyk, M.E.; Higgins, M.R.; Weatherford, C.A.; Salem, N., Jr. Global survey of the omega-3 fatty acids, docosahexaenoic acid and eicosapentaenoic acid in the blood stream of healthy adults. Prog. Lipid Res. 2016, 63, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Arterburn, L.M.; Hall, E.B.; Oken, H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am. J. Clin. Nutr. 2006, 83, 1467S–1476S. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.D.; Aristizabal Henao, J.J.; Metherel, A.H.; Pilote, L. Translating plasma and whole blood fatty acid compositional data into the sum of eicosapentaenoic and docosahexaenoic acid in erythrocytes. Prostaglandins Leukot. Essent. Fatty Acids 2016, 104, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Brenna, J.T.; Salem, N., Jr.; Sinclair, A.J.; Cunnane, S.C.; for the International Society for the Study of Fatty Acids and Lipids; ISSFAL. alpha-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fatty Acids 2009, 80, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Brossard, N.; Croset, M.; Pachiaudi, C.; Riou, J.P.; Tayot, J.L.; Lagarde, M. Retroconversion and metabolism of (13C)22:6n-3 in humans and rats after intake of a single dose of (13C)22:6n-3-triacylglycerols. Am. J. Clin. Nutr. 1996, 64, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Conquer, J.A.; Holub, B.J. Supplementation with an algae source of docosahexaenoic acid increases (n-3) fatty acid status and alters selected risk factors for heart disease in vegetarian subjects. J. Nutr. 1996, 126, 3032–3039. [Google Scholar] [CrossRef] [PubMed]
- Conquer, J.A.; Holub, B.J. Dietary docosahexaenoic acid as a source of eicosapentaenoic acid in vegetarians and omnivores. Lipids 1997, 32, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Calo, L.; Martino, A.; Tota, C. The anti-arrhythmic effects of n-3 PUFAs. Int. J. Cardiol. 2013, 170, S21–S27. [Google Scholar] [CrossRef] [PubMed]
- Leaf, A.; Kang, J.X.; Xiao, Y.F.; Billman, G.E. Clinical prevention of sudden cardiac death by n-3 polyunsaturated fatty acids and mechanism of prevention of arrhythmias by n-3 fish oils. Circulation 2003, 107, 2646–2652. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, H. Update on marine omega-3 fatty acids: management of dyslipidemia and current omega-3 treatment options. Atherosclerosis 2013, 230, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, T.A.; Glickstein, S.B.; Rowe, J.D.; Soni, P.N. Effects of eicosapentaenoic acid and docosahexaenoic acid on low-density lipoprotein cholesterol and other lipids: A review. J. Clin. Lipidol. 2012, 6, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H. Omega-3 fatty acids: New insights into the pharmacology and biology of docosahexaenoic acid, docosapentaenoic acid, and eicosapentaenoic acid. Curr. Opin. Lipidol. 2013, 24, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.S.; Keske, M.A.; Hoffman, J.P.; Nelson, E.B. Clinical overview of algal-docosahexaenoic acid: effects on triglyceride levels and other cardiovascular risk factors. Am. J. Ther. 2009, 16, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.D.; Miller, P.E.; Van Elswyk, M.E.; Kuratko, C.N.; Bylsma, L.C. A Meta-Analysis of Randomized Controlled Trials and Prospective Cohort Studies of Eicosapentaenoic and Docosahexaenoic Long-Chain Omega-3 Fatty Acids and Coronary Heart Disease Risk. Mayo Clin. Proc. 2017, 92, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Yakoob, M.Y.; Shi, P.; Longstreth, W.T., Jr.; Lemaitre, R.N.; Siscovick, D.; Rexrode, K.M.; Willett, W.C.; Mozaffarian, D. Omega-3 Fatty Acids and Incident Ischemic Stroke and Its Atherothrombotic and Cardioembolic Subtypes in 3 US Cohorts. Stroke 2017, 48, 2678–2685. [Google Scholar] [CrossRef] [PubMed]
- Woodman, R.J.; Mori, T.A.; Burke, V.; Puddey, I.B.; Barden, A.; Watts, G.F.; Beilin, L.J. Effects of purified eicosapentaenoic acid and docosahexaenoic acid on platelet, fibrinolytic and vascular function in hypertensive type 2 diabetic patients. Atherosclerosis 2003, 166, 85–93. [Google Scholar] [CrossRef]
- Casula, M.; Soranna, D.; Catapano, A.L.; Corrao, G. Long-term effect of high dose omega-3 fatty acid supplementation for secondary prevention of cardiovascular outcomes: A meta-analysis of randomized, placebo controlled trials (corrected). Atheroscler. Suppl. 2013, 14, 243–251. [Google Scholar] [CrossRef]
- Rizos, E.C.; Ntzani, E.E.; Bika, E.; Kostapanos, M.S.; Elisaf, M.S. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: A systematic review and meta-analysis. JAMA 2012, 308, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Huang, W.; Bai, S.; Wu, Y.; Yu, J.; Zhu, X.; Qi, Z.; Shao, W.; Xie, P. BMI Affects the Relationship between Long Chain n-3 Polyunsaturated Fatty Acid Intake and Stroke Risk: A Meta-Analysis. Sci. Rep. 2015, 5, 14161. [Google Scholar] [CrossRef] [PubMed]
- Mollace, V.; Gliozzi, M.; Carresi, C.; Musolino, V.; Oppedisano, F. Re-assessing the mechanism of action of n-3 PUFAs. Int. J. Cardiol. 2013, 170, S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Innes, J.K.; Calder, P.C. The Differential Effects of Eicosapentaenoic Acid and Docosahexaenoic Acid on Cardiometabolic Risk Factors: A Systematic Review. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Frost and Sullivan. The State of the Global Omega-3 Ingredients Market in 2015. A Market at a Crossroad; Frost and Sullivan: New York, NY, USA, 2015; pp. 4–33. [Google Scholar]
- DSM Nutritional Products; TNS Canada Market Research. 2014 Omega-3 Usage & Attitude Study; DSM Nutritional Products: Heerlen, The Netherland, 2015; pp. 5–35. [Google Scholar]
- Burnett, A.J.; Livingstone, K.M.; Woods, J.L.; McNaughton, S.A. Dietary Supplement Use among Australian Adults: Findings from the 2011–2012 National Nutrition and Physical Activity Survey. Nutrients 2017, 9, 1248. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Gahche, J.J.; Miller, P.E.; Thomas, P.R.; Dwyer, J.T. Why US adults use dietary supplements. JAMA Intern. Med. 2013, 173, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N. Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot. Essent. Fatty Acids 2001, 65, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.A.; Woodman, R.J. The independent effects of eicosapentaenoic acid and docosahexaenoic acid on cardiovascular risk factors in humans. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Zhang, J.; Kris-Etherton, P.M. Cardiovascular disease risk of dietary stearic acid compared with trans, other saturated, and unsaturated fatty acids: a systematic review. Am. J. Clin. Nutr. 2010, 91, 46–63. [Google Scholar] [CrossRef] [PubMed]
- Proksch, E.; Feingold, K.R.; Elias, P.M. Epidermal HMG CoA reductase activity in essential fatty acid deficiency: barrier requirements rather than eicosanoid generation regulate cholesterol synthesis. J. Investig. Dermatol. 1992, 99, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Notarnicola, M.; Messa, C.; Refolo, M.G.; Tutino, V.; Miccolis, A.; Caruso, M.G. Polyunsaturated fatty acids reduce fatty acid synthase and hydroxy-methyl-glutaryl CoA-reductase gene expression and promote apoptosis in HepG2 cell line. Lipids Health Dis. 2011, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.T.; Chung, M.J.; Kim, S.H.; Choi, H.J.; Ham, S.S. Masou salmon (Oncorhynchus masou) ethanol extract decreases 3-hydroxy-3-methylglutaryl coenzyme A reductase expression in diet-induced obese mice. Nutr. Res. 2009, 29, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Shirai, N.; Suzuki, H. Effect of docosahexaenoic acid on brain 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity in male ICR mice. J. Nutr. Biochem. 2007, 18, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Ihara-Watanabe, M.; Umekawa, H.; Takahashi, T.; Furuichi, Y. Effects of dietary alpha- or gamma-linolenic acid on levels and fatty acid compositions of serum and hepatic lipids, and activity and mRNA abundance of 3-hydroxy-3-methylglutaryl CoA reductase in rats. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1999, 122, 213–220. [Google Scholar] [CrossRef]
- Wang, H.; Blumberg, J.B.; Chen, C.Y.; Choi, S.W.; Corcoran, M.P.; Harris, S.S.; Jacques, P.F.; Kristo, A.S.; Lai, C.Q.; Lamon-Fava, S.; et al. Dietary modulators of statin efficacy in cardiovascular disease and cognition. Mol. Aspects Med. 2014, 38, 1–53. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.I.; Hibbeln, J.R.; Mackey, R.H.; Muldoon, M.F. Statin treatment alters serum n-3 and n-6 fatty acids in hypercholesterolemic patients. Prostaglandins Leukot. Essent. Fatty Acids 2004, 71, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Nozue, T.; Michishita, I. Statin treatment alters serum n-3 to n-6 polyunsaturated fatty acids ratio in patients with dyslipidemia. Lipids Health Dis. 2015, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Jula, A.; Marniemi, J.; Ronnemaa, T.; Virtanen, A.; Huupponen, R. Effects of diet and simvastatin on fatty acid composition in hypercholesterolemic men: a randomized controlled trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Hashimoto, M.; Katakura, M.; Tanabe, Y.; Tsuchikura, S.; Hossain, S.; Shido, O. Effect of dietary n-3 fatty acids supplementation on fatty acid metabolism in atorvastatin-administered SHR.Cg-Leprcp/NDmcr rats, a metabolic syndrome model. Biomed. Pharmacother. 2017, 85, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Risé, P.; Pazzucconi, F.; Sirtori, C.R.; Galli, C. Statins enhance arachidonic acid synthesis in hypercholesterolemic patients. Nutr. Metab. Cardiovasc. Dis. 2001, 11, 88–94. [Google Scholar] [PubMed]
- Risé, P.; Ghezzi, S.; Galli, C. Relative potencies of statins in reducing cholesterol synthesis and enhancing linoleic acid metabolism. Eur. J. Pharmacol. 2003, 467, 73–75. [Google Scholar] [CrossRef]
- De Lorgeril, M.; Salen, P.; Guiraud, A.; Zeghichi, S.; Boucher, F.; de Leiris, J. Lipid-lowering drugs and essential omega-6 and omega-3 fatty acids in patients with coronary heart disease. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Ozguden-Akkoc, C.G.; Wang, Y.; Jump, D.B.; Olson, L.K. Role of fatty acid elongases in determination of de novo synthesized monounsaturated fatty acid species. J. Lipid Res. 2010, 51, 1871–1877. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Ando, J.; Shimada, K.; Nishizaki, Y.; Tani, S.; Ogawa, T.; Yamamoto, M.; Nagao, K.; Hirayama, A.; Yoshimura, M.; et al. The ratio of serum n-3 to n-6 polyunsaturated fatty acids is associated with diabetes mellitus in patients with prior myocardial infarction: A multicenter cross-sectional study. BMC Cardiovasc. Disord. 2017, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Risé, P.; Colombo, C.; Galli, C. Effects of simvastatin on the metabolism of polyunsaturated fatty acids and on glycerolipid, cholesterol, and de novo lipid synthesis in THP-1 cells. J. Lipid Res. 1997, 38, 1299–1307. [Google Scholar] [PubMed]
- Kurisu, S.; Ishibashi, K.; Kato, Y.; Mitsuba, N.; Dohi, Y.; Nishioka, K.; Kihara, Y. Effects of lipid-lowering therapy with strong statin on serum polyunsaturated fatty acid levels in patients with coronary artery disease. Heart Vessels 2013, 28, 34–38. [Google Scholar] [CrossRef] [PubMed]
- De Lorgeril, M.; Salen, P.; Defaye, P.; Rabaeus, M. Recent findings on the health effects of omega-3 fatty acids and statins, and their interactions: Do statins inhibit omega-3? BMC Med. 2013, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Serban, C.; Ursoniu, S.; Rysz, J.; Muntner, P.; Toth, P.P.; Jones, S.R.; Rizzo, M.; Glasser, S.P.; Watts, G.F.; et al. Statin therapy and plasma coenzyme Q10 concentrations—A systematic review and meta-analysis of placebo-controlled trials. Pharmacol. Res. 2015, 99, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Schunck, W.H.; Konkel, A.; Fischer, R.; Weylandt, K.H. Therapeutic potential of omega-3 fatty acid-derived epoxyeicosanoids in cardiovascular and inflammatory diseases. Pharmacol. Ther. 2018, 183, 177–204. [Google Scholar] [CrossRef] [PubMed]
- Fleming, I. The pharmacology of the cytochrome P450 epoxygenase/soluble epoxide hydrolase axis in the vasculature and cardiovascular disease. Pharmacol. Rev. 2014, 66, 1106–1140. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.H.; van Leeuwen, R.E.; van Thiel, G.C.; van Pelt, J.F.; Yap, S.H. Equally potent inhibitors of cholesterol synthesis in human hepatocytes have distinguishable effects on different cytochrome P450 enzymes. Biopharm. Drug Dispos. 2000, 21, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, K.L.; Endo, T.; Darwesh, A.M.; Samokhvalov, V.; Seubert, J.M. Cytochrome P450-derived eicosanoids and heart function. Pharmacol. Ther. 2017, 179, 47–83. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Kim, H.Y. Cytochrome P450 epoxygenase pathway of polyunsaturated fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Kiage, J.N.; Sampson, U.K.; Lipworth, L.; Fazio, S.; Mensah, G.A.; Yu, Q.; Munro, H.; Akwo, E.A.; Dai, Q.; Blot, W.J.; et al. Intake of polyunsaturated fat in relation to mortality among statin users and non-users in the Southern Community Cohort Study. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 1016–1024. [Google Scholar] [CrossRef] [PubMed]
- Eussen, S.R.; Geleijnse, J.M.; Giltay, E.J.; Rompelberg, C.J.; Klungel, O.H.; Kromhout, D. Effects of n-3 fatty acids on major cardiovascular events in statin users and non-users with a history of myocardial infarction. Eur. Heart J. 2012, 33, 1582–1588. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.J.; Temporelli, P.L.; Campia, U.; Vaduganathan, M.; Degli Esposti, L.; Buda, S.; Veronesi, C.; Butler, J.; Nodari, S. Effects of Polyunsaturated Fatty Acid Treatment on Postdischarge Outcomes After Acute Myocardial Infarction. Am. J. Cardiol. 2016, 117, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Macchia, A.; Romero, M.; D'Ettorre, A.; Tognoni, G.; Mariani, J. Exploratory analysis on the use of statins with or without n-3 PUFA and major events in patients discharged for acute myocardial infarction: an observational retrospective study. PLoS ONE 2013, 8, e62772. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Yokoyama, M.; Saito, Y.; Origasa, H.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; Kita, T.; et al. Incremental effects of eicosapentaenoic acid on cardiovascular events in statin-treated patients with coronary artery disease. Circ. J. 2009, 73, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of EPA on coronary artery disease in hypercholesterolemic patients with multiple risk factors: Sub-analysis of primary prevention cases from the Japan EPA Lipid Intervention Study (JELIS). Atherosclerosis 2008, 200, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Ando, K.; Daidoji, H.; Otaki, Y.; Sugawara, S.; Matsui, M.; Ikeno, E.; Hirono, O.; Miyawaki, H.; Yashiro, Y.; et al. A randomized controlled trial of eicosapentaenoic acid in patients with coronary heart disease on statins. J. Cardiol. 2017, 70, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Nosaka, K.; Miyoshi, T.; Iwamoto, M.; Kajiya, M.; Okawa, K.; Tsukuda, S.; Yokohama, F.; Sogo, M.; Nishibe, T.; Matsuo, N.; et al. Early initiation of eicosapentaenoic acid and statin treatment is associated with better clinical outcomes than statin alone in patients with acute coronary syndromes: 1-year outcomes of a randomized controlled study. Int. J. Cardiol. 2017, 228, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Alfaddagh, A.; Elajami, T.K.; Ashfaque, H.; Saleh, M.; Bistrian, B.R.; Welty, F.K. Effect of Eicosapentaenoic and Docosahexaenoic Acids Added to Statin Therapy on Coronary Artery Plaque in Patients With Coronary Artery Disease: A Randomized Clinical Trial. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Pang, J.; Barrett, P.H.; Sullivan, D.R.; Mori, T.A.; Burnett, J.R.; van Bockxmeer, F.M.; Watts, G.F. Effect of omega-3 fatty acid supplementation on arterial elasticity in patients with familial hypercholesterolaemia on statin therapy. Nutr. Metab. Cardiovasc. Dis. 2016, 26, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, C.M.; Braeckman, R.A.; Bays, H.E.; Kastelein, J.J.; Otvos, J.D.; Stirtan, W.G.; Doyle, R.T., Jr.; Soni, P.N.; Juliano, R.A. Effects of icosapent ethyl on lipoprotein particle concentration and size in statin-treated patients with persistent high triglycerides (the ANCHOR Study). J. Clin. Lipidol. 2015, 9, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Scolaro, B.; Nogueira, M.S.; Paiva, A.; Bertolami, A.; Barroso, L.P.; Vaisar, T.; Heffron, S.P.; Fisher, E.A.; Castro, I.A. Statin dose reduction with complementary diet therapy: A pilot study of personalized medicine. Mol. Metab. 2018, 11, 137–144. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Statin Name | Origin [23] | Structure [23] | Lipophilicity [23] | Generation [23] | CYP Metabolism [22,24,25] |
|---|---|---|---|---|---|
| Fluvastatin | Synthetic | Fluorophenyl group | Lipophilic | I | CYP2C9 |
| Atorvastatin | Synthetic | Fluorophenyl group | Lipophilic | II | CYP3A4 |
| Rosuvastatin | Synthetic | Fluorophenyl group | Lipophobic | III | CYP2C9 |
| Pitavastatin | Synthetic | Fluorophenyl group | Lipophilic | II | Marginal [26] |
| Lovastatin | Fungal | Butyryl group | Lipophilic | I | CYP3A4 |
| Pravastatin | Fungal | Butyryl group | Lipophobic | I | CYP2C9 |
| Simvastatin | Fungal | Butyryl group | Lipophilic | II | CYP3A4 |
| Study Name (n) | Study Type/Treatments | Main Results | Reference |
|---|---|---|---|
| Southern cohort community study (n = 69,559) | Prospective cohort study | Modest inverse associations between n-3 PUFA and n-6 PUFA intake with mortality among non-statin users but not among statin users. | [87] |
| (n = 14,704) | Retrospective cohort study | As compared with statins alone, combined treatment with statins and n-3 LC PUFAs was associated with an adjusted higher survival rate, survival free of atrial fibrillation and survival free of new heart failure development, but not with re-infarction. | [90] |
| JELIS (n = 18,645) | RCT Treatment: 1800 mg EPA + statin Control: statin alone | The incidence of MCE was significantly lower in the EPA group. Compared to patients with normal serum TG and HDL-C levels, those with abnormal had significantly higher CAD hazard ratio. In this higher risk group, EPA treatment suppressed the risk of CAD by 53%. | [91,92] |
| Alpha Omega (n = 4153) | Post hoc analysis of RCT Treatments: 400 mg EPA + DHA 2 g ALA Both Control: Placebo margarine | In statin users, n-3 fatty acids did not reduce cardiovascular events. In statin non-users, only 9% of those who received EPA-DHA plus ALA experienced an event compared with 18% in the placebo group. | [88] |
| CHERRY (n = 193) | RCT Treatment: 1,800 mg EPA + 4 mg pitavastatin Control: Pitavastatin (Pitavastatin) 4 mg | The prevalence rate of plaque regression was significantly higher in Pitavastatin/EPA group than in Pitavastatin group (50% vs. 24%, p < 0.001). | [93] |
| Kagawa hospital study (n = 241) | Prospective, open-label, randomized trial Treatment: 1,800 mg EPA + 2 mg Pitavastatin Control: 2 mg Pitavastatin | Significant reduction in composite endpoint of cardiovascular death, MI, stroke, or coronary revascularization at 1 year: 9.2% in the EPA group and 20.2% in the control group (absolute risk reduction, 11.0%; HR, 0.42; 95% CI, 0.21–087; p = 0.02), in acute coronary syndrome patients. | [94] |
| (n = 11,269) | Retrospective cohort study | n-3 LC PUFA supplement users had a reduced risk of all-cause mortality (HR 0.76 [0.59 to 0.97]). Statin use did not affect all-cause mortality reduction, however a reduction in recurrent myocardial infarction was only seen in statin users. | [89] |
| (n = 77,776) | Meta-regression | Lower control group statin use and higher DHA/EPA ratio was associated with higher reduction in total mortality. | [18] |
| HEARTS (n = 285) | RCT in patients with stable statin therapy Treatment: 1860 mg EPA + 1500 mg DHA Control: Placebo | EPA + DHA in addition to low-dose statin treatment prevented progression of atherosclerotic plaques, compared to low-dose statin treatment alone. | [95] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bird, J.K.; Calder, P.C.; Eggersdorfer, M. The Role of n-3 Long Chain Polyunsaturated Fatty Acids in Cardiovascular Disease Prevention, and Interactions with Statins. Nutrients 2018, 10, 775. https://doi.org/10.3390/nu10060775
Bird JK, Calder PC, Eggersdorfer M. The Role of n-3 Long Chain Polyunsaturated Fatty Acids in Cardiovascular Disease Prevention, and Interactions with Statins. Nutrients. 2018; 10(6):775. https://doi.org/10.3390/nu10060775
Chicago/Turabian StyleBird, Julia K., Philip C. Calder, and Manfred Eggersdorfer. 2018. "The Role of n-3 Long Chain Polyunsaturated Fatty Acids in Cardiovascular Disease Prevention, and Interactions with Statins" Nutrients 10, no. 6: 775. https://doi.org/10.3390/nu10060775
APA StyleBird, J. K., Calder, P. C., & Eggersdorfer, M. (2018). The Role of n-3 Long Chain Polyunsaturated Fatty Acids in Cardiovascular Disease Prevention, and Interactions with Statins. Nutrients, 10(6), 775. https://doi.org/10.3390/nu10060775