Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Obesity
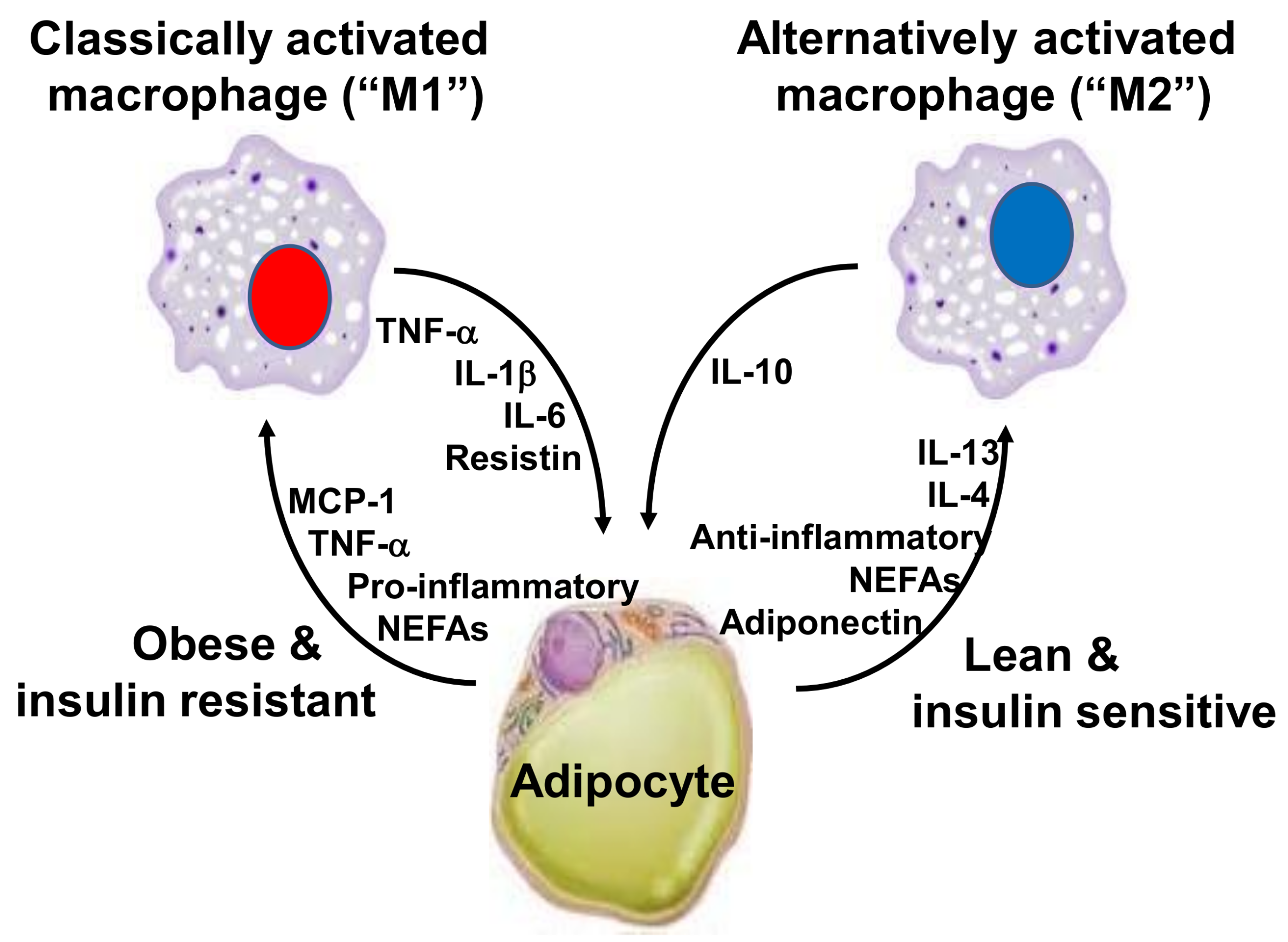
2. Inflammation, Adipose Tissue and Obesity
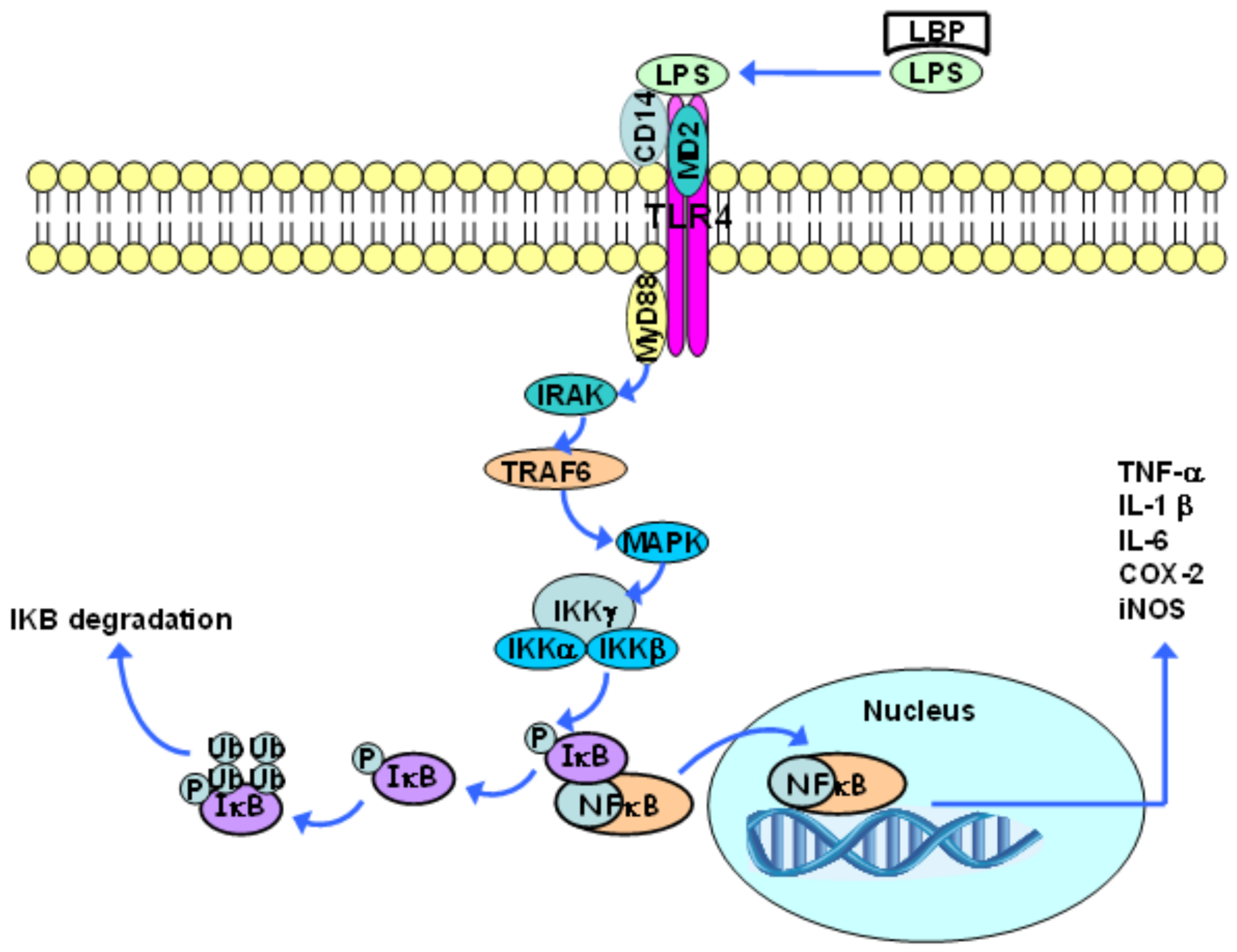
3. Toll-Like Receptor 4 and Inflammatory Response
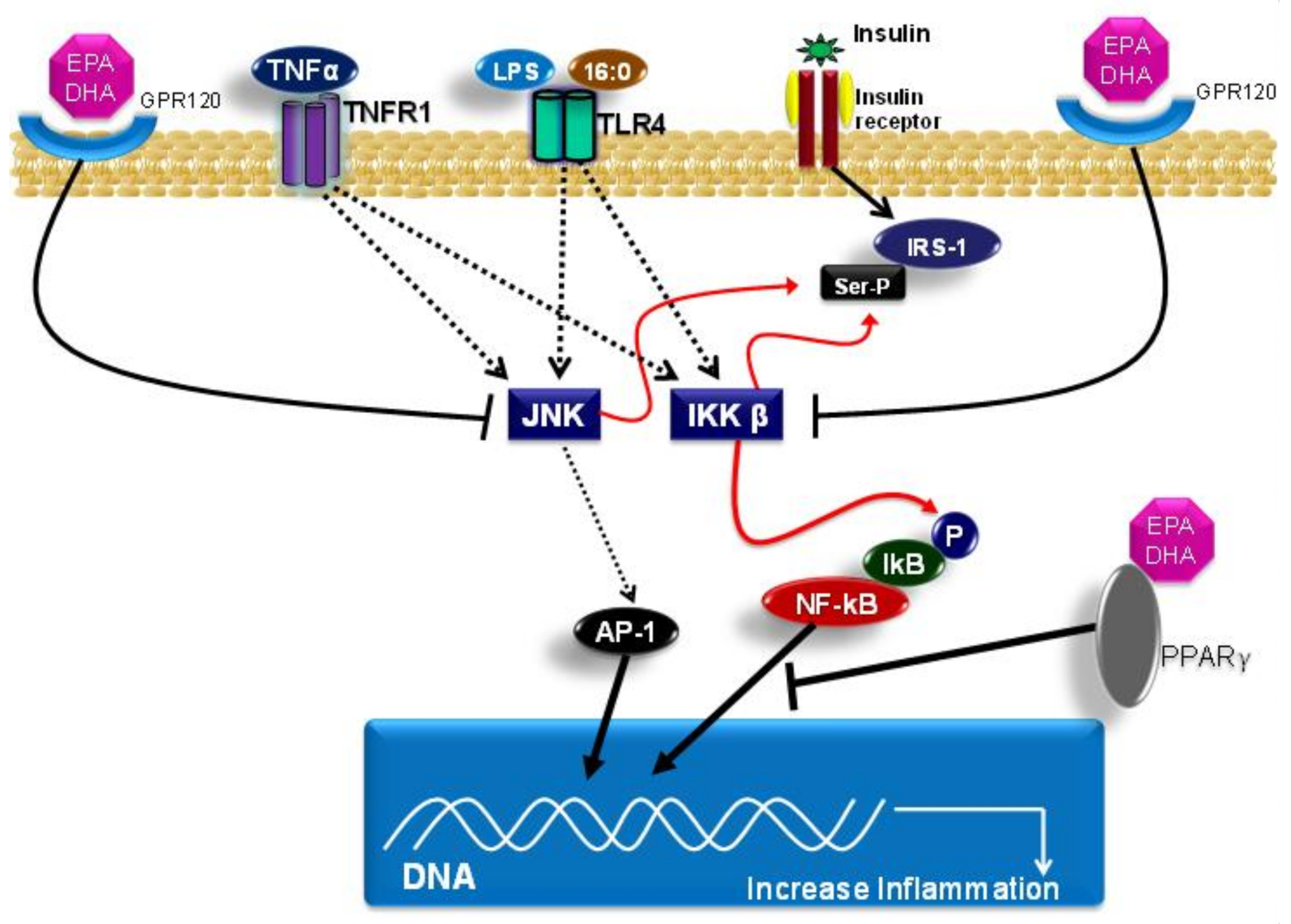
4. Fatty Acids, Toll-Like Receptors and Inflammation
4.1. Saturated Fatty Acids
4.2. Polyunsaturated Fatty Acids
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Flegal, K.M.; Carroll, M.D.; Kit, B.K.; Ogden, C.L. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 2012, 307, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Yach, D.; Stuckler, D.; Brownell, K.D. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat. Med. 2006, 12, 62–66. [Google Scholar] [CrossRef] [PubMed]
- WHO—World Health Organization. World Health Organization Obesity and overweight Fact Sheet (2016). Available online: http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 30 January 2018).
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F.; et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Alexandratos, N.; Bruinsma, J. World Agriculture Towards 2030/2050: The 2012 Revision; FAO: Rome, Italy, 2012. [Google Scholar]
- United Nations News Centre, 2015. United Nations News Centre. Available online: http://www.un.org/sustainabledevelopment/blog/2015/07/un-projects-world-population-to-reach-8-5-billion-by-2030-driven-by-growth-in-developing-countries/ (accessed on 17 January 2018).
- Ogden, C.L.; Carroll, M.D.; Curtin, L.R.; Lamb, M.M.; Flegal, K.M. Prevalence of high body mass index in US children and adolescents, 2007–2008. JAMA 2010, 303, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Ford, E.S.; Bowman, B.A.; Dietz, W.H.; Vinicor, F.; Bales, V.S.; Marks, J.S. Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA 2003, 289, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Daousi, C.; Casson, I.F.; Gill, G.V.; MacFarlane, I.A.; Wilding, J.P.; Pinkney, J.H. Prevalence of obesity in type 2 diabetes in secondary care: Association with cardiovascular risk factors. Postgrad. Med. J. 2006, 82, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Thun, M.J.; Petrelli, J.M.; Rodriguez, C.; Heath, C.W., Jr. Body-mass index and mortality in a prospective cohort of U.S. adults. N. Engl. J. Med. 1999, 341, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Prospective Studies Collaboration; Whitlock, G.; Lewington, S.; Sherliker, P.; Clarke, R.; Emberson, J.; Halsey, J.; Qizilbash, N.; Collins, R.; Peto, R. Body-mass index and cause-specific mortality in 900 000 adults: Collaborative analyses of 57 prospective studies. Lancet 2009, 373, 1083–1096. [Google Scholar] [CrossRef] [PubMed]
- Amuna, P.; Zotor, F.B. Epidemiological and nutrition transition in developing countries: Impact on human health and development. Proc. Nutr. Soc. 2008, 67, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Vandevijvere, S.; Chow, C.C.; Hall, K.D.; Umali, E.; Swinburn, B.A. Increased food energy supply as a major driver of the obesity epidemic: A global analysis. Bull. World Health Organ. 2015, 93, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.K.; Barnard, R.J. Effects of exercise and diet on chronic disease. J. Appl. Physiol. (1985) 2005, 98, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Simopoulos, A.P.; DiNicolantonio, J.J. The importance of a balanced ω-6 to ω-3 ratio in the prevention and management of obesity. Open Heart 2016, 3, e000385. [Google Scholar] [CrossRef] [PubMed]
- Galli, C.; Calder, P.C. Effects of fat and fatty acid intake on inflammatory and immune responses: A critical review. Ann. Nutr. Metab. 2009, 55, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Polyunsaturated fatty acids and inflammatory processes: New twists in an old tale. Biochimie 2009, 91, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Albers, R.; Bosco, N.; Bourdet-Sicard, R.; Haller, D.; Holgate, S.T.; Jönsson, L.S.; Latulippe, M.E.; Marcos, A.; et al. A consideration of biomarkers to be used for evaluation of inflammation in human nutritional studies. Br. J. Nutr. 2013, 109, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Molfino, A.; Amabile, M.I.; Monti, M.; Muscaritoli, M. Omega-3 Polyunsaturated Fatty Acids in Critical Illness: Anti-Inflammatory, Proresolving, or Both? Oxid. Med. Cell. Longev. 2017, 2017, 5987082. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Dalli, J. New pro-resolving n-3 mediators bridge resolution of infectious inflammation to tissue regeneration. Mol. Asp. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jönsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106, 5–78. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. The role of marine omega-3 (n-3) fatty acids in inflammatory processes, atherosclerosis and plaque stability. Mol. Nutr. Food Res. 2012, 56, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Marine omega-3 fatty acids and inflammatory processes: Effects, mechanisms and clinical relevance. Biochim. Biophys. Acta 2015, 1851, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Fatty acids and inflammation: The cutting edge between food and pharma. Eur. J. Pharmacol. 2011, 668, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Yaqoob, P. Marine omega-3 fatty acids and coronary heart disease. Curr. Opin. Cardiol. 2012, 27, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Thorleifsson, G.; Zhang, B.; Leonardson, A.S.; Zink, F.; Zhu, J.; Carlson, S.; Helgason, A.; Walters, G.B.; Gunnarsdottir, S.; et al. Genetics of gene expression and its effect on disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-alpha: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Stulnig, T.M. Adipose tissue macrophages. Immunol. Lett. 2007, 112, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Clement, K. Is obesity an inflammatory illness? Role of low-grade inflammation and macrophage infiltration in human white adipose tissue. BJOG 2006, 113, 1141–1147. [Google Scholar] [CrossRef] [PubMed]
- Cave, M.C.; Hurt, R.T.; Frazier, T.H.; Matheson, P.J.; Garrison, R.N.; McClain, C.J.; McClave, S.A. Obesity, inflammation, and the potential application of pharmaconutrition. Nutr. Clin. Pract. 2008, 23, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, A.W. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Bengtsson, T.; Cannon, B. Unexpected evidence for active brown adipose tissue in adult humans. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E444–E452. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Choi, E.Y.; Liu, X.; Martin, A.; Wang, C.; Xu, X.; During, M.J. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 2011, 14, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Lowell, B.B.; Susulic, V.; Hamann, A.; Lawitts, J.A.; Himms-Hagen, J.; Boyer, B.B.; Kozak, L.P.; Flier, J.S. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 1993, 366, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.; Morrisey, R.D.; Carnie, J.A. The effect of interscapular brown adipose tissue removal on body-weight and cold response in the mouse. Br. J. Nutr. 1982, 47, 653–658. [Google Scholar] [CrossRef] [PubMed]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Briscini, L.; Giordano, A.; Tonello, C.; Wiesbrock, S.M.; Uysal, K.T.; Cinti, S.; Carruba, M.O.; Hotamisligil, G.S. Tumor necrosis factor alpha mediates apoptosis of brown adipocytes and defective brown adipocyte function in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 8033–8038. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cell 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed]
- Guo, S. Insulin signaling, resistance, and the metabolic syndrome: Insights from mouse models into disease mechanisms. J. Endocrinol. 2014, 220, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, A.M.; Lenighan, Y.M.; O’Reilly, M.E.; McGillicuddy, F.C.; Roche, H.M. Nutritional modulation of metabolic inflammation. Biochem. Soc. Trans. 2017, 45, 979–985. [Google Scholar] [CrossRef] [PubMed]
- Egger, G.; Dixon, J. Obesity and chronic disease: Always offender or often just accomplice? Br. J. Nutr. 2009, 102, 1238–1242. [Google Scholar] [CrossRef] [PubMed]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Arruda, A.P.; Coope, A.; Ignacio-Souza, L.M.; Nunez, C.E.; Roman, E.A.; Romanatto, T.; Pascoal, L.B.; Caricilli, A.M.; Torsoni, M.A.; et al. Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 2012, 61, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Varma, V.; Yao-Borengasser, A.; Rasouli, N.; Nolen, G.T.; Phanavanh, B.; Starks, T.; Gurley, C.; Simpson, P.; McGehee, R.E., Jr.; Kern, P.A.; et al. Muscle inflammatory response and insulin resistance: Synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am. J. Physiol. Endocrinol. Metab. 2009, 296, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Basith, S.; Manavalan, B.; Lee, G.; Kim, S.G.; Choi, S. Toll-like receptor modulators: A patent review (2006–2010). Expert Opin. Ther. Pat. 2011, 21, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kawai, T.; Akira, S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2014, 7, a016246. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Akira, S. Toll-Like Receptor Signaling and Its Inducible Proteins. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Moresco, E.M.; LaVine, D.; Beutler, B. Toll-like receptors. Curr. Biol. 2011, 21, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Li, M.; Fang, D.; Fang, J.; Su, S.B. The essential roles of Toll-like receptor signaling pathways in sterile inflammatory diseases. Int. Immunopharmacol. 2011, 11, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, F.A.; Aitken, J.D.; Vijay-Kumar, M.; Gewirtz, A.T. Toll-like receptor-gut microbiota interactions: Perturb at your own risk! Annu. Rev. Physiol. 2012, 74, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Farfara, D.; Frenkel, D. Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: Towards future therapeutic application. Mediators Inflamm. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.J.; O’Neill, L.A. New developments in Toll-like receptor targeted therapeutics. Curr. Opin. Pharmacol. 2012, 12, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, M.; Triantafilou, K. The dynamics of LPS recognition: Complex orchestration of multiple receptors. J. Endotoxin Res. 2005, 11, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; Vogel, S.N. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002, 4, 903–914. [Google Scholar] [CrossRef]
- Triantafilou, M.; Triantafilou, K. Lipopolysaccharide recognition: CD14, TLRs and the LPS-activation cluster. Trends Immunol. 2002, 23, 301–304. [Google Scholar] [CrossRef]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.J.; Gangloff, M. Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 2007, 76, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Al-ofi, E.; Coffelt, S.B.; Anumba, D.O. Fibrinogen, an endogenous ligand of Toll-like receptor 4, activates monocytes in pre-eclamptic patients. J. Reprod. Immunol. 2014, 103, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Pathogen recognition with Toll-like receptors. Curr. Opin. Immunol. 2005, 17, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Caamaño, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Verma, I.M. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2002, 2, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.E.; Georgiou, E.; Monaco, C. The expression and functions of toll-like receptors in atherosclerosis. Mediat. Inflamm. 2010, 2010, 393946. [Google Scholar] [CrossRef] [PubMed]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.; Pham, M.; Luttrell, I.; Bannerman, D.D.; Tupper, J.; Thaler, J.; Hawn, T.R.; Raines, E.W.; Schwartz, M.W. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ. Res. 2007, 100, 1589–1596. [Google Scholar] [CrossRef] [PubMed]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Dasu, M.R.; Rockwood, J.; Winter, W.; Griffen, S.C.; Jialal, I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 2008, 93, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Al-Mass, A.; Atizado, V.; Al-Hubail, A.; Al-Ghimlas, F.; Al-Arouj, M.; Bennakhi, A.; Dermime, S.; Behbehani, K. Elevated expression of the toll like receptors 2 and 4 in obese individuals: Its significance for obesity-induced inflammation. J. Inflamm. (Lond). 2012, 9, 48. [Google Scholar] [CrossRef] [PubMed]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Haehnel, V.; Schwarzfischer, L.; Fenton, M.J.; Rehli, M. Transcriptional regulation of the human toll-like receptor 2 gene in monocytes and macrophages. J. Immunol. 2002, 168, 5629–5637. [Google Scholar] [CrossRef] [PubMed]
- Rehli, M.; Poltorak, A.; Schwarzfischer, L.; Krause, S.W.; Andreesen, R.; Beutler, B. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 gene. J. Biol. Chem. 2000, 275, 9773–9781. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Mohanty, P.; Deopurkar, R.; Sia, C.L.; Korzeniewski, K.; Abuaysheh, S.; Chaudhuri, A.; Dandona, P. Acute modulation of toll-like receptors by insulin. Diabetes Care 2008, 31, 1827–1831. [Google Scholar] [CrossRef] [PubMed]
- Wang, C. Obesity, inflammation, and lung injury (OILI): The good. Mediat. Inflamm. 2014, 2014, 978463. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Potempa, L.A.; El Kebir, D.; Filep, J.G. C-reactive protein and inflammation: Conformational changes affect function. Biol. Chem. 2015, 396, 1181–1197. [Google Scholar] [CrossRef] [PubMed]
- Aljada, A.; Mohanty, P.; Ghanim, H.; Abdo, T.; Tripathy, D.; Chaudhuri, A.; Dandona, P. Increase in intranuclear nuclear factor kappaB and decrease in inhibitor kappaB in mononuclear cells after a mixed meal: Evidence for a proinflammatory effect. Am. J. Clin. Nutr. 2004, 79, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Abuaysheh, S.; Sia, C.L.; Korzeniewski, K.; Chaudhuri, A.; Fernandez-Real, J.M.; Dandona, P. Increase in plasma endotoxin concentrations and the expression of Toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: Implications for insulin resistance. Diabetes Care 2009, 32, 2281–2287. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Calder, P.C. Plasma cytokine response during the postprandial period: A potential causal process in vascular disease? Br. J. Nutr. 2005, 93, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Van Oostrom, A.J.; Rabelink, T.J.; Verseyden, C.; Sijmonsma, T.P.; Plokker, H.W.; De Jaegere, P.P.; Cabezas, M.C. Activation of leukocytes by postprandial lipemia in healthy volunteers. Atherosclerosis 2004, 177, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.H.; Kim, J.A.; Lee, J.Y. Mechanisms for the activation of Toll-like receptor 2/4 by saturated fatty acids and inhibition by docosahexaenoic acid. Eur. J. Pharmacol. 2016, 785, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Sohn, K.H.; Rhee, S.H.; Hwang, D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J. Biol. Chem. 2001, 276, 16683–16689. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Takeuchi, O.; Kawai, T.; Sanjo, H.; Ogawa, T.; Takeda, Y.; Takeda, K.; Akira, S. Cutting edge: Toll-like receptor 4 (TLR4)-deficient mice are hyporesponsive to lipopolysaccharide: Evidence for TLR4 as the Lps gene product. J. Immunol. 1999, 162, 3749–3752. [Google Scholar] [PubMed]
- Mylona, E.E.; Mouktaroudi, M.; Crisan, T.O.; Makri, S.; Pistiki, A.; Georgitsi, M.; Savva, A.; Netea, M.G.; van der Meer, J.W.; Giamarellos-Bourboulis, E.J.; et al. Enhanced interleukin-1β production of PBMCs from patients with gout after stimulation with Toll-like receptor-2 ligands and urate crystals. Arthritis Res. Ther. 2012, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Snodgrass, R.G.; Huang, S.; Choi, I.W.; Rutledge, J.C.; Hwang, D.H. Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J. Immunol. 2013, 191, 4337–4347. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids: Preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef] [PubMed]
- Caricilli, A.M.; Nascimento, P.H.; Pauli, J.R.; Tsukumo, D.M.; Velloso, L.A.; Carvalheira, J.B.; Saad, M.J. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J. Endocrinol. 2008, 199, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Erridge, C.; Samani, N.J. Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1944–1949. [Google Scholar] [CrossRef] [PubMed]
- Capurso, C.; Capurso, A. From excess adiposity to insulin resistance: The role of free fatty acids. Vascul. Pharmacol. 2012, 57, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Holland, W.L.; Bikman, B.T.; Wang, L.P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.S.; Buras, E.D.; Balasubramanyam, A. The role of the immune system in obesity and insulin resistance. J. Obes. 2013, 2013, 616193. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. n-3 Fatty acids and cardiovascular disease: Evidence explained and mechanisms explored. Clin. Sci. (Lond). 2004, 107, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C.; Yaqoob, P. Understanding omega-3 polyunsaturated fatty acids. Postgrad. Med. 2009, 121, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, P.R.; Kemmerer, E.S.; Buck, B.J.; Osetek, A.J.; Yan, C.; Koch, L.G.; Britton, S.L.; Evans, S.J. Dietary n-3:n-6 fatty acid ratios differentially influence hormonal signature in a rodent model of metabolic syndrome relative to healthy controls. Nutr. Metab. (Lond). 2010, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.B.; Huang, L.L.; Rong, R.; Tan, R.; Wang, J.; Kang, J.X. Endogenously decreasing tissue n-6/n-3 fatty acid ratio reduces atherosclerotic lesions in apolipoprotein E-deficient mice by inhibiting systemic and vascular inflammation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Baker, E.J.; Miles, E.A.; Burdge, G.C.; Yaqoob, P.; Calder, P.C. Metabolism and functional effects of plant-derived omega-3 fatty acids in humans. Prog. Lipid Res. 2016, 64, 30–56. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Wootton, S.A. Conversion of alpha-linolenic acid to eicosapentaenoic, docosapentaenoic and docosahexaenoic acids in young women. Br. J. Nutr. 2002, 88, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Goyens, P.L.; Spilker, M.E.; Zock, P.L.; Katan, M.B.; Mensink, R.P. Conversion of alpha-linolenic acid in humans is influenced by the absolute amounts of alpha-linolenic acid and linoleic acid in the diet and not by their ratio. Am. J. Clin. Nutr. 2006, 84, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. The relationship between the fatty acid composition of immune cells and their function. Prostaglandins Leukot. Essent. Fatty Acids 2008, 79, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Casula, M.; Soranna, D.; Catapano, A.L.; Corrao, G. Long-term effect of high dose omega-3 fatty acid supplementation for secondary prevention of cardiovascular outcomes: A meta-analysis of randomized, placebo controlled trials [corrected]. Atheroscler. Suppl. 2013, 14, 243–251. [Google Scholar] [CrossRef]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.A.; Johnson, L.; Franco, O.H.; Butterworth, A.S.; Forouhi, N.G.; Thompson, S.G.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Marik, P.E.; Varon, J. Omega-3 dietary supplements and the risk of cardiovascular events: A systematic review. Clin. Cardiol. 2009, 32, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Briel, M.; Leimenstoll, B.; Glass, T.R.; Bucher, H.C. Effect of different antilipidemic agents and diets on mortality: A systematic review. Arch. Intern. Med. 2005, 165, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Buettner, R.; Parhofer, K.G.; Woenckhaus, M.; Wrede, C.E.; Kunz-Schughart, L.A.; Schölmerich, J.; Bollheimer, L.C. Defining high-fat-diet rat models: Metabolic and molecular effects of different fat types. J. Mol. Endocrinol. 2006, 36, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Hartweg, J.; Farmer, A.J.; Holman, R.R.; Neil, A. Potential impact of omega-3 treatment on cardiovascular disease in type 2 diabetes. Curr. Opin. Lipidol. 2009, 20, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Bayona, W.; Kallen, C.B.; Harding, H.P.; Ravera, C.P.; McMahon, G.; Brown, M.; Lazar, M.A. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J. Biol. Chem. 1995, 270, 23975–23983. [Google Scholar] [CrossRef] [PubMed]
- Kalupahana, N.S.; Claycombe, K.; Newman, S.J.; Stewart, T.; Siriwardhana, N.; Matthan, N.; Lichtenstein, A.H.; Moustaid-Moussa, N. Eicosapentaenoic acid prevents and reverses insulin resistance in high-fat diet-induced obese mice via modulation of adipose tissue inflammation. J. Nutr. 2010, 140, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Caughey, G.E.; Mantzioris, E.; Gibson, R.A.; Cleland, L.G.; James, M.J. The effect on human tumor necrosis factor alpha and interleukin 1 beta production of diets enriched in n-3 fatty acids from vegetable oil or fish oil. Am. J. Clin. Nutr. 1996, 63, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.; Serhan, C.N. Structural elucidation and physiologic functions of specialized pro-resolving mediators and their receptors. Mol. Asp. Med. 2017, 58, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Ntambi, J.M. Polyunsaturated fatty acid regulation of gene expression. Nutr. Rev. 2004, 62, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Stryjecki, C.; Mutch, D.M. Fatty acid-gene interactions, adipokines and obesity. Eur. J. Clin. Nutr. 2011, 65, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Endres, S.; Meydani, S.N.; Ghorbani, R.; Schindler, R.; Dinarello, C.A. Dietary supplementation with n-3 fatty acids suppresses interleukin-2 production and mononuclear cell proliferation. J. Leukoc. Biol. 1993, 54, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Talukdar, S.; Bae, E.J.; Imamura, T.; Morinaga, H.; Fan, W.; Li, P.; Lu, W.J.; Watkins, S.M.; Olefsky, J.M. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 2010, 142, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Olefsky, J.M. Omega 3 fatty acids and GPR120. Cell Metab. 2012, 15, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Li, A.C.; Glass, C.K. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J. Lipid Res. 2004, 45, 2161–2173. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Fernández, L.; Laiglesia, L.M.; Huerta, A.E.; Martínez, J.A.; Moreno-Aliaga, M.J. Omega-3 fatty acids and adipose tissue function in obesity and metabolic syndrome. Prostaglandins Other Lipid Mediat. 2015, 121, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Meital, L.T.; Sandow, S.L.; Calder, P.C.; Russell, F.D. Abdominal aortic aneurysm and omega-3 polyunsaturated fatty acids: Mechanisms, animal models, and potential treatment. Prostaglandins Leukot. Essent. Fatty Acids 2017, 118, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, M.J.; Hasty, A.H.; Saraswathi, V. The role of adipose tissue in mediating the beneficial effects of dietary fish oil. J. Nutr. Biochem. 2011, 22, 101–108. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogero, M.M.; Calder, P.C. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients 2018, 10, 432. https://doi.org/10.3390/nu10040432
Rogero MM, Calder PC. Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients. 2018; 10(4):432. https://doi.org/10.3390/nu10040432
Chicago/Turabian StyleRogero, Marcelo Macedo, and Philip C. Calder. 2018. "Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids" Nutrients 10, no. 4: 432. https://doi.org/10.3390/nu10040432
APA StyleRogero, M. M., & Calder, P. C. (2018). Obesity, Inflammation, Toll-Like Receptor 4 and Fatty Acids. Nutrients, 10(4), 432. https://doi.org/10.3390/nu10040432