Copper-Fructose Interactions: A Novel Mechanism in the Pathogenesis of NAFLD

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology of NAFLD, Fructose Consumption, and Dietary Copper Intake
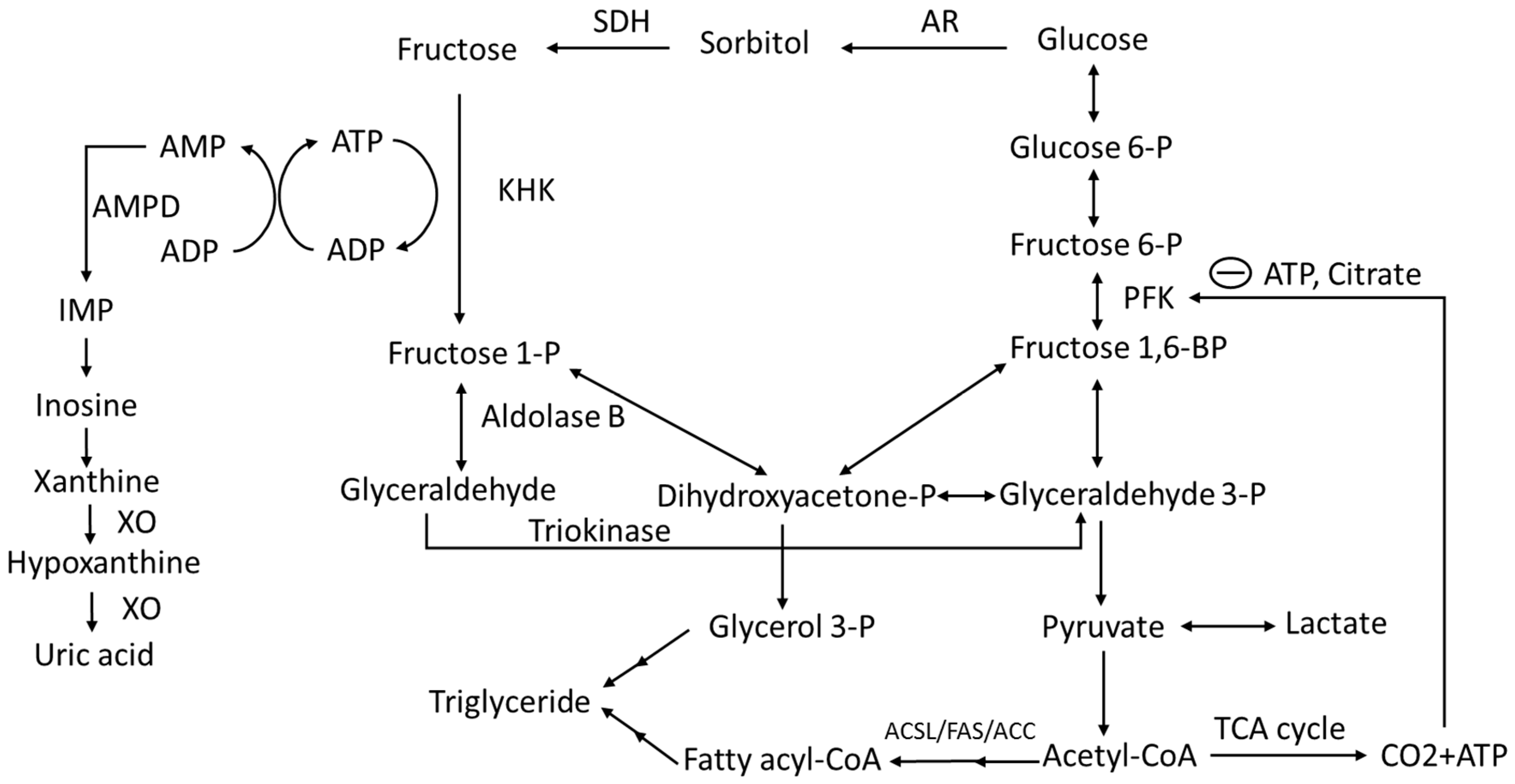
3. Fructose Absorption, Metabolism and Metabolic Fate
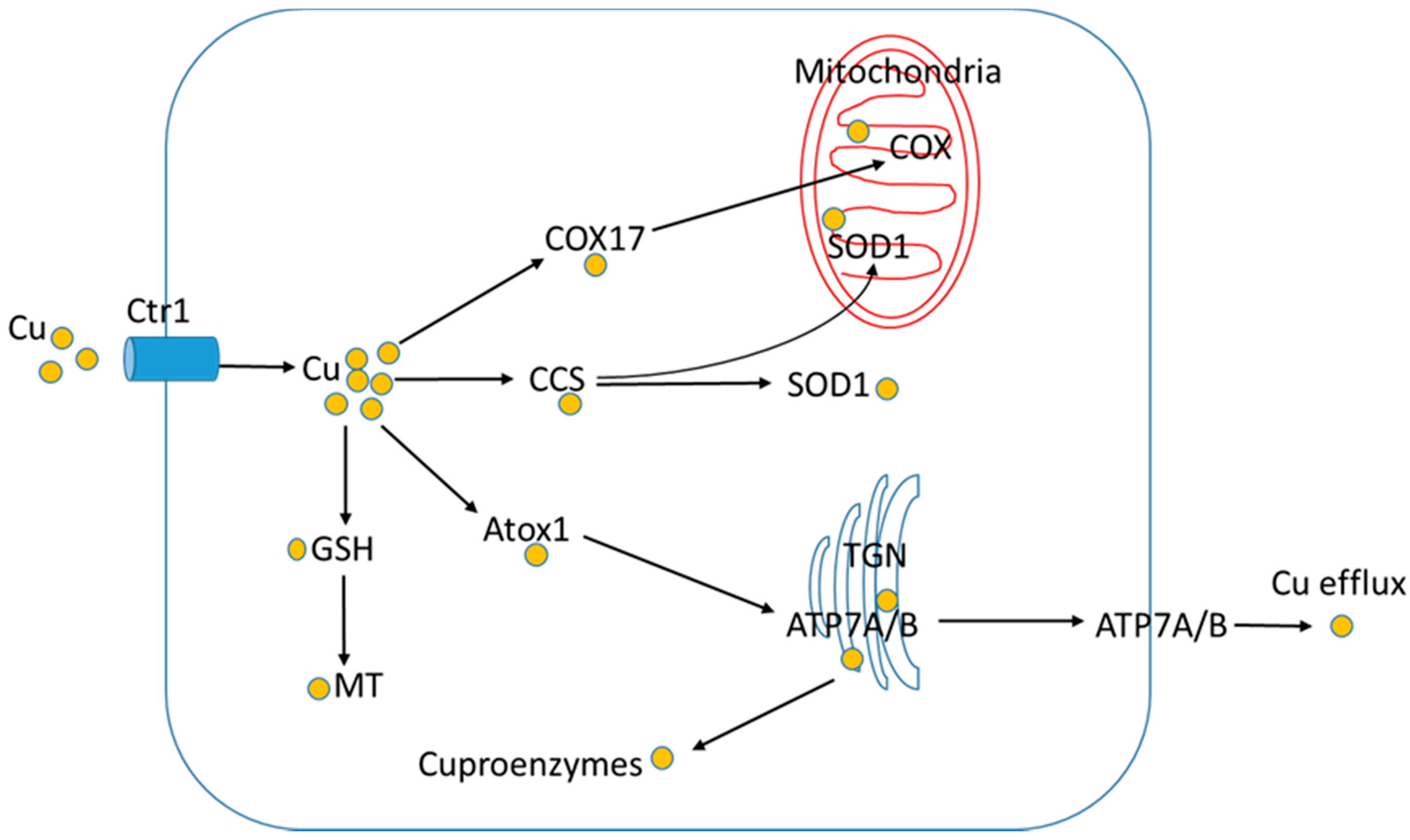
4. Copper Absorption, Distribution and Utilization
5. Copper Homeostasis and NAFLD
6. Copper-Fructose Interactions.
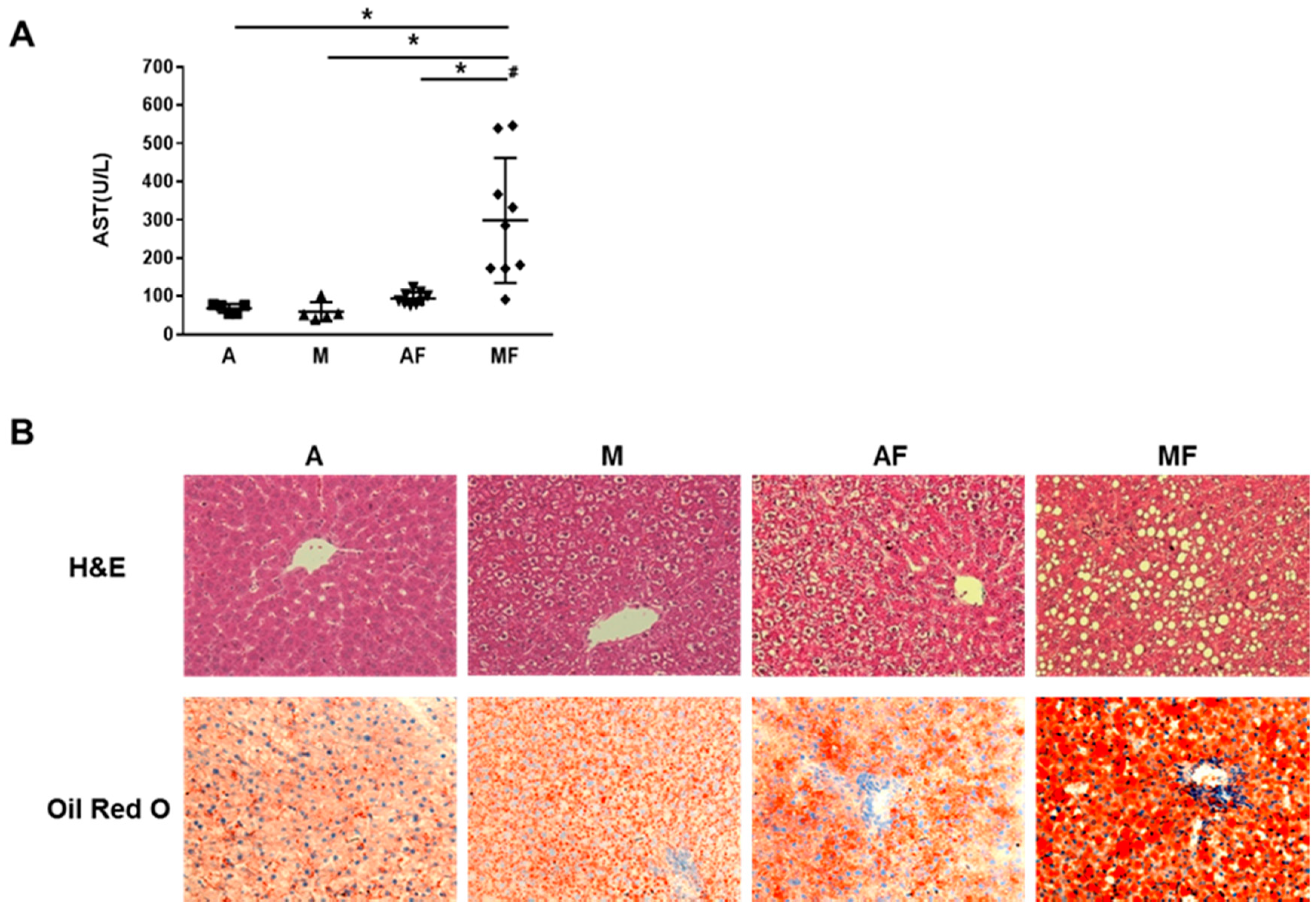
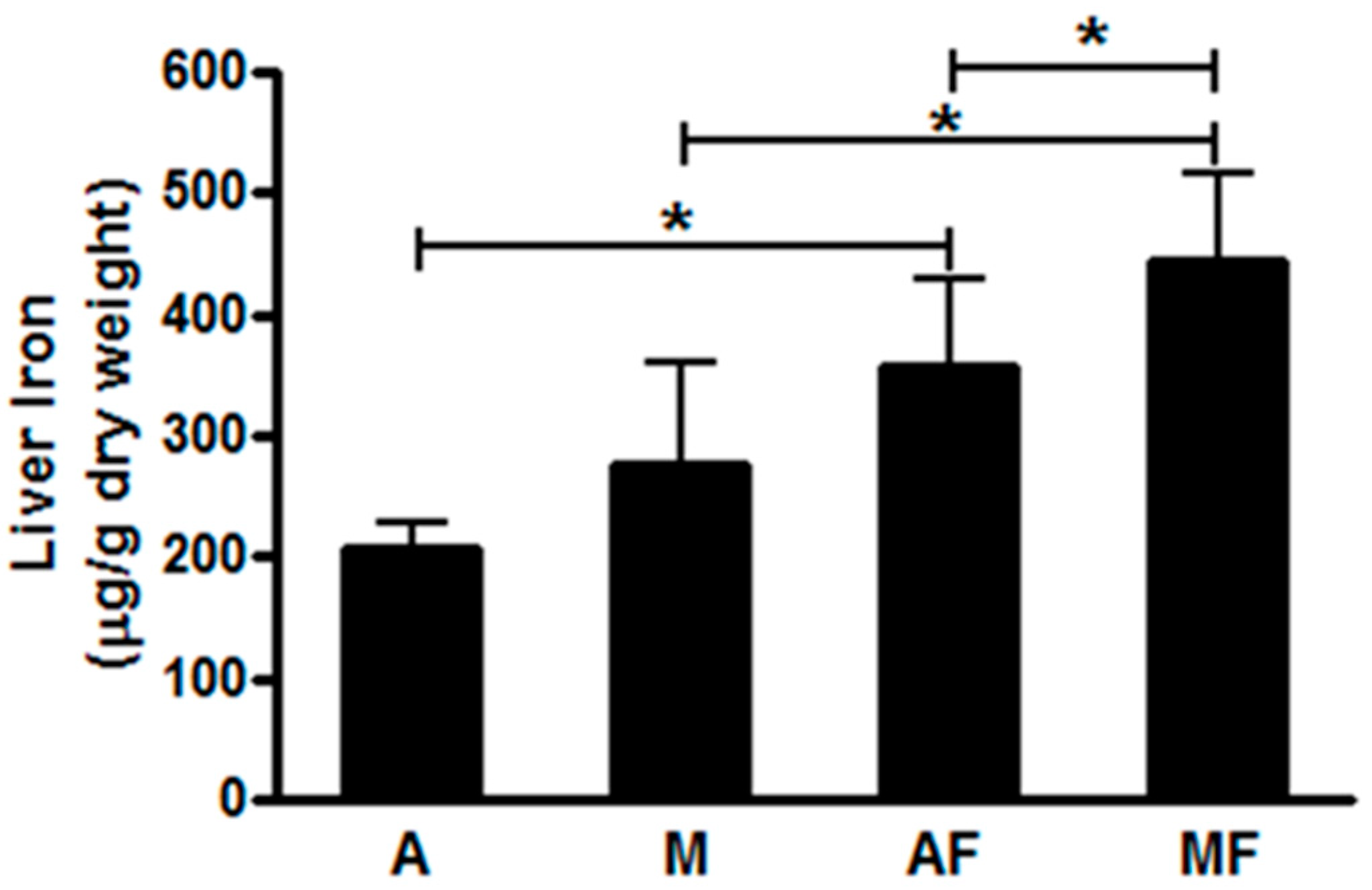
6.1. Copper-Fructose Interaction and NAFLD
6.2. Copper-Fructose Interaction and Hyperlipidemia
6.3. Copper-Fructose Interaction and Glucose Tolerance
6.4. Copper-Fructose Interaction and Gut Permeability
6.5. Copper-Fructose Interaction and Gut Microbiome
6.6. Sex Difference in the Copper-Fructose Interaction
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| NAFLD | nonalcoholic fatty liver disease |
| KC | Kupffer cell |
| HFCS | high-fructose corn syrup |
| KHK | ketohexokinase |
| TLR4 | toll like receptor 4 |
| MCD | methionine choline deficient |
| NASH | nonalcoholic steatohepatitis |
| T2D | type 2 diabetes |
| SSBs | sugar-sweetened beverages |
| CVDs | cardiovascular diseases |
| RDA | Recommended Dietary Allowance |
| EAR | Estimated Average Requirement |
| SOD1 | copper/zinc-superoxide dismutase |
| COX | cytochrome c oxidase |
| F1P | fructose 1-phosphate |
| DHAP | dihydroxyacetone phosphate |
| AMPD | adenosine monophosphate deaminase |
| IMP | inosine monophosphate |
| AMPK | adenosine monophosphate-activated protein kinase |
| XO | xanthine oxidase |
| AR | aldose reductase |
| CCS | copper chaperone for SOD1 |
| HIF-1 | hypoxia inducible factor-1 |
| Ctr1 | copper transporter 1 |
| MT | metallothionein |
| Atox1 | antioxidant protein 1 |
| TGN | trans-Golgi network |
| HCV | hepatitis C virus |
| FP-1 | ferroportin-1 |
| NAS | NAFLD Activity Score |
| WD | Wilson’s disease |
| FAS | fatty acid synthase |
| SREBP-1 | sterol regulatory element-binding protein-1 |
| BSO | L-buthionine sulfoximine |
| LPS | lipopolysaccharide |
| MCP-1 | monocyte chemoattractant protein-1 |
| LXR | liver X receptor |
| FXR | farnesoid X receptor |
| RXR | retinoid X receptor |
| SHP | small heterodimer partner |
| ER | endoplasmic reticulum |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| HMGCR | HMG-CoA reductase |
| GPx | glutathione peroxidase |
| SCFA | short chain fatty acid |
| Egfr | epidermal growth factor receptor |
| AQPs | aquaglyceroporins |
References
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sánchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Hu, F.B.; Malik, V.S. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: Epidemiologic evidence. Physiol. Behav. 2010, 100, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Kimmons, J.E.; Gillespie, C.; Welsh, J.; Blanck, H.M. Dietary fructose consumption among US children and adults: The third national health and nutrition examination survey. Medscape J. Med. 2008, 10, 160. [Google Scholar] [PubMed]
- Park, Y.K.; Yetley, E.A. Intakes and food sources of fructose in the United States. Am. J. Clin. Nutr. 1993, 58, 737S–747S. [Google Scholar] [CrossRef] [PubMed]
- Marriott, B.P.; Cole, N.; Lee, E. National estimates of dietary fructose intake increased from 1977 to 2004 in the United States. J. Nutr. 2009, 139, 1228S–1235S. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Karpen, S.; Vos, M.B. Increasing prevalence of nonalcoholic fatty liver disease among United States adolescents, 1988–1994 to 2007–2010. J. Pediatr. 2013, 162, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Assy, N.; Nasser, G.; Kamayse, I.; Nseir, W.; Beniashvili, Z.; Djibre, A.; Grosovski, M. Soft drink consumption linked with fatty liver in the absence of traditional risk factors. Can. J. Gastroenterol. 2008, 22, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Increased fructose consumption is associated with fibrosis severity in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L.; Le, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef] [PubMed]
- Heinz, F.; Lamprecht, W.; Kirsch, J. Enzymes of fructose metabolism in human liver. J. Clin. Investig. 1968, 47, 1826–1832. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, T.; Riby, J.; Kretchmer, N. Intestinal absorption of fructose in the rat. Gastroenterology 1991, 101, 360–367. [Google Scholar] [CrossRef]
- Riby, J.E.; Fujisawa, T.; Kretchmer, N. Fructose absorption. Am. J. Clin. Nutr. 1993, 58, 748S–753S. [Google Scholar] [CrossRef] [PubMed]
- Spruss, A.; Kanuri, G.; Wagnerberger, S.; Haub, S.; Bischoff, S.C.; Bergheim, I. Toll-like receptor 4 is involved in the development of fructose-induced hepatic steatosis in mice. Hepatology 2009, 50, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Kenmochi, H.; Miyashita, Y.; Sasaki, M.; Ojima, M.; Sasahara, M.; Koya, D.; Tsuneki, H.; Sasaoka, T. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology 2010, 151, 2040–2049. [Google Scholar] [CrossRef] [PubMed]
- Kelley, G.L.; Allan, G.; Azhar, S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology 2004, 145, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Park, O.J.; Cesar, D.; Faix, D.; Wu, K.; Shackleton, C.H.; Hellerstein, M.K. Mechanisms of fructose-induced hypertriglyceridaemia in the rat. Activation of hepatic pyruvate dehydrogenase through inhibition of pyruvate dehydrogenase kinase. Biochem. J. 1992, 282, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.; Hui, S.; Lu, W.; Cowan, A.J.; Morscher, R.J.; Lee, G.; Liu, W.; Tesz, G.J.; Birnbaum, M.J.; Rabinowitz, J.D. The Small intestine converts dietary fructose into glucose and organic acids. Cell MeTable 2018, 27, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Rivard, C.J.; Roncal-Jimenez, C.A.; Orlicky, D.J.; Cicerchi, C.; McMahan, R.H.; Abdelmalek, M.F.; Rosen, H.R.; Jackman, M.R.; et al. High-fat and high-sucrose (western) diet induces steatohepatitis that is dependent on fructokinase. Hepatology 2013, 58, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Ishimoto, T.; Lanaspa, M.A.; Le, M.T.; Garcia, G.E.; Diggle, C.P.; Maclean, P.S.; Jackman, M.R.; Asipu, A.; Roncal-Jimenez, C.A.; Kosugi, T.; et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 4320–4325. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Bergheim, I.; Weber, S.; Vos, M.; Kramer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Z.; Empie, M.W. Fructose metabolism in humans—What isotopic tracer studies tell us. Nutr. MeTable 2012, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Pan, A.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages and weight gain in children and adults: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2013, 98, 1084–1102. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Popkin, B.M.; Bray, G.A.; Despres, J.P.; Hu, F.B. Sugar-sweetened beverages, obesity, type 2 diabetes mellitus, and cardiovascular disease risk. Circulation 2010, 121, 1356–1364. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.S.; Schulze, M.B.; Hu, F.B. Intake of sugar-sweetened beverages and weight gain: A systematic review. Am. J. Clin. Nutr. 2006, 84, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L. Role of fructose-containing sugars in the epidemics of obesity and metabolic syndrome. Annu. Rev. Med. 2012, 63, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Maersk, M.; Belza, A.; Stodkilde-Jorgensen, H.; Ringgaard, S.; Chabanova, E.; Thomsen, H.; Pedersen, S.B.; Astrup, A.; Richelsen, B. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: A 6-mo randomized intervention study. Am. J. Clin. Nutr. 2012, 95, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Le, K.A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.D.; Stephenson, M.C.; Crossland, H.; Cordon, S.M.; Palcidi, E.; Cox, E.F.; Taylor, M.A.; Aithal, G.P.; Macdonald, I.A. No difference between high-fructose and high-glucose diets on liver triacylglycerol or biochemistry in healthy overweight men. Gastroenterology 2013, 145, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Kuzma, J.N.; Cromer, G.; Hagman, D.K.; Breymeyer, K.L.; Roth, C.L.; Foster-Schubert, K.E.; Holte, S.E.; Weigle, D.S.; Kratz, M. No differential effect of beverages sweetened with fructose, high-fructose corn syrup, or glucose on systemic or adipose tissue inflammation in normal-weight to obese adults: A randomized controlled trial. Am. J. Clin. Nutr. 2016, 104, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Schene, M.R.; Holman, R.; Romijn, J.A.; Serlie, M.J. Effect of fructose consumption on insulin sensitivity in nondiabetic subjects: A systematic review and meta-analysis of diet-intervention trials. Am. J. Clin. Nutr. 2016, 104, 1562–1576. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity (Silver Spring) 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Campos, V.; Despland, C.; Brandejsky, V.; Kreis, R.; Schneiter, P.; Chiolero, A.; Boesch, C.; Tappy, L. Sugar- and artificially sweetened beverages and intrahepatic fat: A randomized controlled trial. Obesity 2015, 23, 2335–2339. [Google Scholar] [CrossRef] [PubMed]
- Pickens, M.K.; Ogata, H.; Soon, R.K.; Grenert, J.P.; Maher, J.J. Dietary fructose exacerbates hepatocellular injury when incorporated into a methionine-choline-deficient diet. Liver Int. 2010, 30, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Pickens, M.K.; Yan, J.S.; Ng, R.K.; Ogata, H.; Grenert, J.P.; Beysen, C.; Turner, S.M.; Maher, J.J. Dietary sucrose is essential to the development of liver injury in the methionine-choline-deficient model of steatohepatitis. J. Lipid Res. 2009, 50, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Le, N.A.; Liu, S.; Farkas Epperson, M.; Ziegler, T.R.; Welsh, J.A.; Jones, D.P.; McClain, C.J.; Vos, M.B. Children with NAFLD are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J. Clin. Endocrinol. MeTable 2012, 97, E1088–E1098. [Google Scholar] [CrossRef] [PubMed]
- Teff, K.L.; Grudziak, J.; Townsend, R.R.; Dunn, T.N.; Grant, R.W.; Adams, S.H.; Keim, N.L.; Cummings, B.P.; Stanhope, K.L.; Havel, P.J. Endocrine and metabolic effects of consuming fructose- and glucose-sweetened beverages with meals in obese men and women: Influence of insulin resistance on plasma triglyceride responses. J. Clin. Endocrinol. MeTable 2009, 94, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M.; et al. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Li, X.; Zhang, X.; Shi, H.; Vos, M.B.; Wei, X.; Wang, Y.; Gao, H.; Rouchka, E.C.; Yin, X.; et al. Dietary copper-fructose interactions alter gut microbial activity in male rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G119–G130. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Chen, T.; Pierce, W.M., Jr.; Wang, R.; Johnson, W.T.; McClain, C.J. High fructose feeding induces copper deficiency in Sprague-Dawley rats: A novel mechanism for obesity related fatty liver. J. Hepatol 2012, 56, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Chen, T.; Shi, X.; Zhang, J.; Zhang, X.; Pierce, W.M., Jr.; Johnson, W.T.; Vos, M.B.; et al. Modest fructose beverage intake causes liver injury and fat accumulation in marginal copper deficient rats. Obesity (Silver Spring) 2013, 21, 1669–1675. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Ferretti, R.J.; Reiser, S.; Smith, J.C., Jr. The severity of copper deficiency in rats is determined by the type of dietary carbohydrate. Proc. Soc. Exp. Biol. Med. 1984, 175, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Ferretti, R.J.; Smith, J.C., Jr.; Reiser, S. The interaction of type of dietary carbohydrates with copper deficiency. Am. J. Clin. Nutr. 1984, 39, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Strasser, M.; Haufe, H.; Sonnweber, T.; Hohla, F.; Stadlmayr, A.; Solioz, M.; Tilg, H.; Patsch, W.; Weiss, G.; et al. A role for low hepatic copper concentrations in nonalcoholic Fatty liver disease. Am. J. Gastroenterol. 2010, 105, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Aigner, E.; Theurl, I.; Haufe, H.; Seifert, M.; Hohla, F.; Scharinger, L.; Stickel, F.; Mourlane, F.; Weiss, G.; Datz, C. Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease. Gastroenterology 2008, 135, 680–688. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.; Caltharp, S.; Song, M.; Collin, L.; Konomi, J.V.; McClain, C.J.; Vos, M.B. Low Hepatic Tissue Copper in Pediatric Nonalcoholic Fatty Liver Disease. J. Pediatr. Gastroenterol. Nutr. 2017, 65, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Stattermayer, A.F.; Traussnigg, S.; Aigner, E.; Kienbacher, C.; Huber-Schonauer, U.; Steindl-Munda, P.; Stadlmayr, A.; Wrba, F.; Trauner, M.; Datz, C.; et al. Low hepatic copper content and PNPLA3 polymorphism in non-alcoholic fatty liver disease in patients without metabolic syndrome. J. Trace Elem. Med. Biol. 2017, 39, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Siotto, M.; Bedogni, G.; Rava, L.; Pietrobattista, A.; Panera, N.; Alisi, A.; Squitti, R. Levels of serum ceruloplasmin associate with pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2013, 56, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.M.; Wolf, W.R.; Mertz, W. Zinc and copper in self-selected diets. J. Am. Diet. Assoc. 1979, 75, 23–28. [Google Scholar] [PubMed]
- Klevay, L.M.; Reck, S.J.; Jacob, R.A.; Logan, G.M., Jr.; Munoz, J.M.; Sandstead, H.H. The human requirement for copper. I. Healthy men fed conventional, American diets. Am. J. Clin. Nutr. 1980, 33, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Klevay, L.M. Is the Western diet adequate in copper? J. Trace Elem. Med. Biol. 2011, 25, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Reiser, S.; Smith, J.C., Jr.; Mertz, W.; Holbrook, J.T.; Scholfield, D.J.; Powell, A.S.; Canfield, W.K.; Canary, J.J. Indices of copper status in humans consuming a typical American diet containing either fructose or starch. Am. J. Clin. Nutr. 1985, 42, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Holbrook, J.; Scholfield, D.; Smith, J.C., Jr.; Reiser, S. Effect of fructose or starch on copper-67 absorption and excretion by the rat. J. Nutr. 1986, 116, 625–632. [Google Scholar] [PubMed]
- Holbrook, J.; Fields, M.; Smith, J.C., Jr.; Reiser, S. Tissue distribution and excretion of copper-67 intraperitoneally administered to rats fed fructose or starch. J. Nutr. 1986, 116, 831–838. [Google Scholar] [PubMed]
- Ernst, B.; Thurnheer, M.; Schultes, B. Copper deficiency after gastric bypass surgery. Obesity. (Silver Spring) 2009, 17, 1980–1981. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Hamza, N.; Madhok, B.; De Alwis, N.; Sharma, M.; Miras, A.D.; Mahawar, K.K. Copper Deficiency after Gastric Bypass for Morbid Obesity: A Systematic Review. Obes. Surg. 2016, 26, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Hedera, P.; Peltier, A.; Fink, J.K.; Wilcock, S.; London, Z.; Brewer, G.J. Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin II. The denture cream is a primary source of excessive zinc. Neurotoxicology 2009, 30, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Spain, R.I.; Leist, T.P.; De Sousa, E.A. When metals compete: A case of copper-deficiency myeloneuropathy and anemia. Nat. Clin. Pract. Neurol. 2009, 5, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Nanashima, A.; Hiyoshi, M.; Imamura, N.; Yano, K.; Hamada, T. Risk factors for development of nonalcoholic fatty liver disease after pancreatoduodenectomy. Ann. Gastroenterol. Surg. 2017, 1, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Ferretti, R.J.; Judge, J.M.; Smith, J.C.; Reiser, S. Effects of different dietary carbohydrates on hepatic enzymes of copper-deficient rats. Proc. Soc. Exp. Biol. Med. 1985, 178, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Ferretti, R.J.; Smith, J.C., Jr.; Reiser, S. Effect of copper deficiency on metabolism and mortality in rats fed sucrose or starch diets. J. Nutr. 1983, 113, 1335–1345. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Ferretti, R.J.; Smith, J.C., Jr.; Reiser, S. Effect of dietary carbohydrates and copper status on blood pressure of rats. Life Sci. 1984, 34, 763–769. [Google Scholar] [CrossRef]
- Fields, M.; Ferretti, R.J.; Smith, J.C., Jr.; Reiser, S. Impairment of glucose tolerance in copper-deficient rats: Dependency on the type of dietary carbohydrate. J. Nutr. 1984, 114, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G. Dietary fructose but not starch is responsible for hyperlipidemia associated with copper deficiency in rats: Effect of high-fat diet. J. Am. Coll. Nutr. 1999, 18, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524.e1–530.e1. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.R.; Burns, J.M.; Pedersen, R.A.; Watt, K.D.; Heimbach, J.K.; Dierkhising, R.A. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology 2011, 141, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Longnecker, M.P.; Lawlor, D.A. Prevalence of elevated alanine aminotransferase among US adolescents and associated factors: NHANES 1999–2004. Gastroenterology 2007, 133, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, E.; Tokushige, K. Prevalence, gender, ethnic variations, and prognosis of NASH. J. Gastroenterol. 2011, 46 (Suppl. 1), 63–69. [Google Scholar] [CrossRef] [PubMed]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-sweetened Beverage Consumption Among, U.S. Adults, 2011–2014. NCHS Data Briefs 2017, 270, 1–8. [Google Scholar]
- Rosinger, A.; Herrick, K.; Gahche, J.; Park, S. Sugar-sweetened Beverage Consumption Among, U.S.; Youth, 2011–2014. NCHS Data Briefs 2017, 271, 1–8. [Google Scholar]
- Bleich, S.N.; Vercammen, K.A. The negative impact of sugar-sweetened beverages on children’s health: An update of the literature. BMC Obes. 2018, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Fryar, C.D.; Flegal, K.M. Prevalence of Obesity Among Adults and Youth: United States, 2011–2014. NCHS Data Briefs 2015, 219, 1–8. [Google Scholar]
- Malik, V.S.; Hu, F.B. Fructose and Cardiometabolic HealthWhat the Evidence from Sugar-Sweetened Beverages Tells US. J. Am. Coll. Cardiol. 2015, 66, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.M.; Micha, R.; Khatibzadeh, S.; Lim, S.; Ezzati, M.; Mozaffarian, D. Estimated Global, Regional, and National Disease Burdens Related to Sugar-Sweetened Beverage Consumption in 2010. Circulation 2015, 132, 639–666. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.P.; Clegg, D.J. Dietary Guidelines for Americans—Eat Less Sugar. JAMA 2016, 315, 1196. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2001; Copyright 2001 by the National Academy of Sciences. All rights reserved., 2001. [Google Scholar]
- Klevay, L.M. Lack of a recommended dietary allowance for copper may be hazardous to your health. J. Am. Coll. Nutr. 1998, 17, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; MacIntosh, D.L.; Ryan, P.B. A longitudinal investigation of aggregate oral intake of copper. J. Nutr. 2001, 131, 2171–2176. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R. Impact of copper limitation on expression and function of multicopper oxidases (ferroxidases). Adv. Nutr. 2011, 2, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R. Impact of copper deficiency in humans. Ann. N. Y. Acad. Sci. 2014, 1314, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Milne, D.B.; Johnson, P.E. Assessment of copper status: Effect of age and gender on reference ranges in healthy adults. Clin. Chem. 1993, 39, 883–887. [Google Scholar] [PubMed]
- Milne, D.B. Assessment of copper nutritional status. Clin. Chem. 1994, 40, 1479–1484. [Google Scholar] [PubMed]
- Heffern, M.C.; Park, H.M.; Au-Yeung, H.Y.; Van de Bittner, G.C.; Ackerman, C.M.; Stahl, A.; Chang, C.J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2016, 113, 14219–14224. [Google Scholar] [CrossRef] [PubMed]
- David, E.S.; Cingari, D.S.; Ferraris, R.P. Dietary induction of intestinal fructose absorption in weaning rats. Pediatr. Res. 1995, 37, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. The role of fructose transporters in diseases linked to excessive fructose intake. J. Physiol. 2013, 591, 401–414. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.J.; Chen, Z.; Finck, B.N.; Chi, M.; Moley, K.H. Glucose transporter-8 (GLUT8) mediates glucose intolerance and dyslipidemia in high-fructose diet-fed male mice. Mol. Endocrinol. 2013, 27, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Debosch, B.J.; Chen, Z.; Saben, J.L.; Finck, B.N.; Moley, K.H. Glucose transporter 8 (GLUT8) mediates fructose-induced de novo lipogenesis and macrosteatosis. J. Biol. Chem. 2014, 289, 10989–10998. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.J.; Chi, M.; Moley, K.H. Glucose transporter 8 (GLUT8) regulates enterocyte fructose transport and global mammalian fructose utilization. Endocrinology 2012, 153, 4181–4191. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Ferraris, R.P. Regulation of the fructose transporter GLUT5 in health and disease. Am. J. Physiol. Endocrinol. MeTable 2008, 295, E227–E237. [Google Scholar] [CrossRef] [PubMed]
- Barone, S.; Fussell, S.L.; Singh, A.K.; Lucas, F.; Xu, J.; Kim, C.; Wu, X.; Yu, Y.; Amlal, H.; Seidler, U.; et al. Slc2a5 (Glut5) is essential for the absorption of fructose in the intestine and generation of fructose-induced hypertension. J. Biol. Chem. 2009, 284, 5056–5066. [Google Scholar] [CrossRef] [PubMed]
- Douard, V.; Choi, H.I.; Elshenawy, S.; Lagunoff, D.; Ferraris, R.P. Developmental reprogramming of rat GLUT5 requires glucocorticoid receptor translocation to the nucleus. J. Physiol. 2008, 586, 3657–3673. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; Leitch, D.; Brooke, D.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Ketohexokinase: Expression and localization of the principal fructose-metabolizing enzyme. J. Histochem. Cytochem. 2009, 57, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Shires, M.; McRae, C.; Crellin, D.; Fisher, J.; Carr, I.M.; Markham, A.F.; Hayward, B.E.; Asipu, A.; Bonthron, D.T. Both isoforms of ketohexokinase are dispensable for normal growth and development. Physiol. Genomics 2010, 42A, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Raivio, K.O.; Kekomaki, M.P.; Maenpaa, P.H. Depletion of liver adenine nucleotides induced by D-fructose. Dose-dependence and specificity of the fructose effect. Biochem. Pharmacol. 1969, 18, 2615–2624. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Andres-Hernando, A.; Orlicky, D.J.; Cicerchi, C.; Jang, C.; Li, N.; Milagres, T.; Kuwabara, M.; Wempe, M.F.; Rabinowitz, J.D.; et al. Ketohexokinase C blockade ameliorates fructose-induced metabolic dysfunction in fructose-sensitive mice. J. Clin. Investig. 2018, 128, 2226–2238. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Ishimoto, T.; Li, N.; Cicerchi, C.; Orlicky, D.J.; Ruzycki, P.; Rivard, C.; Inaba, S.; Roncal-Jimenez, C.A.; Bales, E.S.; et al. Endogenous fructose production and metabolism in the liver contributes to the development of metabolic syndrome. Nat. Commun. 2013, 4, 2434. [Google Scholar] [CrossRef] [PubMed]
- Maenpaa, P.H.; Raivio, K.O.; Kekomaki, M.P. Liver adenine nucleotides: Fructose-induced depletion and its effect on protein synthesis. Science 1968, 161, 1253–1254. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, T.W.; Kabra, P.M.; Booth, B.E.; Al-Bander, H.A.; Portale, A.A.; Serena, B.G.; Tsai, H.C.; Morris, R.C., Jr. Liquid-chromatographic measurements of inosine, hypoxanthine, and xanthine in studies of fructose-induced degradation of adenine nucleotides in humans and rats. Clin. Chem. 1986, 32, 782–786. [Google Scholar] [PubMed]
- van den Berghe, G.; Bronfman, M.; Vanneste, R.; Hers, H.G. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem. J. 1977, 162, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wan, X.; Xu, L.; Weng, H.; Yan, M.; Miao, M.; Sun, Y.; Xu, G.; Dooley, S.; Li, Y.; et al. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: One stone hits two birds. J. Hepatol. 2015, 62, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, R.G.; Barnett, P.; Aguayo, J.; Cheng, H.M.; Chylack, L.T., Jr. Direct measurement of polyol pathway activity in the ocular lens. Diabetes 1984, 33, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.D.; Clements, R.S., Jr.; Travis, S.B.; Oski, F.; Winegrad, A.I. Glucose utilization by the polyol pathway in human erythrocytes. Biochem. Biophys. Res. Commun. 1970, 40, 199–205. [Google Scholar] [CrossRef]
- Sapp, V.; Gaffney, L.; EauClaire, S.F.; Matthews, R.P. Fructose leads to hepatic steatosis in zebrafish that is reversed by mechanistic target of rapamycin (mTOR) inhibition. Hepatology 2014, 60, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Williams, J.R.; Muckett, P.J.; Mayer, F.V.; Liljevald, M.; Bohlooly, Y.M.; Carling, D. Liver-Specific Activation of AMPK Prevents Steatosis on a High-Fructose Diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef] [PubMed]
- Tappy, L. Fructose-containing caloric sweeteners as a cause of obesity and metabolic disorders. J. Exp. Biol. 2018, 221, 164202. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, K.W.; Serlie, M.J. Fructose Consumption, Lipogenesis, and Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 981. [Google Scholar] [CrossRef] [PubMed]
- Bonham, M.; O’Connor, J.M.; Hannigan, B.M.; Strain, J.J. The immune system as a physiological indicator of marginal copper status? Br. J. Nutr. 2002, 87, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Bertinato, J.; L’Abbe, M.R. Copper modulates the degradation of copper chaperone for Cu,Zn superoxide dismutase by the 26 S. proteosome. J. Biol. Chem. 2003, 278, 35071–35078. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Linden, T.; Katschinski, D.M.; Oehme, F.; Flamme, I.; Mukhopadhyay, C.K.; Eckhardt, K.; Tröger, J.; Barth, S.; Camenisch, G.; et al. Copper-dependent activation of hypoxia-inducible factor (HIF)-1: Implications for ceruloplasmin regulation. Blood 2005, 105, 4613–4619. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Ivy, K.; Yu, H.; Kaplan, J.H. Rate and regulation of copper transport by human copper transporter 1 (hCTR1). J. Biol. Chem. 2013, 288, 18035–18046. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Thiele, D.J. How copper traverses cellular membranes through the mammalian copper transporter 1, Ctr1. Ann. N. Y. Acad. Sci. 2014, 1314, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Nose, Y.; Kim, B.E.; Thiele, D.J. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell MeTable 2006, 4, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Turski, M.L.; Nose, Y.; Casad, M.; Rockman, H.A.; Thiele, D.J. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell MeTable 2010, 11, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Logeman, B.L.; Wood, L.K.; Lee, J.; Thiele, D.J. Gene duplication and neo-functionalization in the evolutionary and functional divergence of the metazoan copper transporters Ctr1 and Ctr2. J. Biol. Chem. 2017, 292, 11531–11546. [Google Scholar] [CrossRef] [PubMed]
- Ohrvik, H.; Nose, Y.; Wood, L.K.; Kim, B.E.; Gleber, S.C.; Ralle, M.; Thiele, D.J. Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain. Proc. Natl. Acad. Sci. USA 2013, 110, E4279–E4288. [Google Scholar] [CrossRef] [PubMed]
- van den Berghe, P.V.; Klomp, L.W. New developments in the regulation of intestinal copper absorption. Nutr. Rev. 2009, 67, 658–672. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V.; Muruais, G.; Tsuchiya, Y.; Pulido, D.; Sandoval, I.V. Molecular mechanisms of copper homeostasis. Front. Biosci. (Landmark Ed.) 2009, 14, 4878–4903. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, M.J.; Kim, Y.S.; Chun, H.; Won, B.Y.; Lee, J.H.; Han, K.; Rim, K.-S.; Park, K.-C. Low hair copper concentration is related to a high risk of nonalcoholic fatty liver disease in adults. J. Trace Elem. Med. Biol. 2018, 50, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Stattermayer, A.F.; Traussnigg, S.; Dienes, H.P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease—Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef]
- Huster, D. Structural and metabolic changes in Atp7b-/- mouse liver and potential for new interventions in Wilson’s disease. Ann. N. Y. Acad. Sci. 2014, 1315, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Medici, V.; Shibata, N.M.; Kharbanda, K.K.; LaSalle, J.M.; Woods, R.; Liu, S.; Engelberg, J.A.; Devaraj, S.; Török, N.J.; Jiang, J.X.; et al. Wilson’s disease: Changes in methionine metabolism and inflammation affect global DNA methylation in early liver disease. Hepatology 2013, 57, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Wooton-Kee, C.R.; Jain, A.K.; Wagner, M.; Grusak, M.A.; Finegold, M.J.; Lutsenko, S.; Moore, D.D. Elevated copper impairs hepatic nuclear receptor function in Wilson’s disease. J. Clin. Investig. 2015, 125, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Porcu, C.; Antonucci, L.; Barbaro, B.; Illi, B.; Nasi, S.; Martini, M.; Licata, A.; Miele, L.; Grieco, A.; Balsano, C. Copper/MYC/CTR1 interplay: A dangerous relationship in hepatocellular carcinoma. Oncotarget 2018, 9, 9325–9343. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Song, Z.; Barve, S.; Zhang, J.; Chen, T.; Liu, M.; Arteel, G.E.; Brewer, G.J.; McClain, C.J. Tetrathiomolybdate protects against bile duct ligation-induced cholestatic liver injury and fibrosis. J. Pharmacol. Exp. Ther. 2008, 325, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Zhou, Z.; Chen, T.; Zhang, J.; McClain, C.J. Copper deficiency exacerbates bile duct ligation-induced liver injury and fibrosis in rats. J. Pharmacol. Exp. Ther. 2011, 339, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, L.; Porcu, C.; Iannucci, G.; Balsano, C.; Barbaro, B. Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper. Nutrients 2017, 9, 1137. [Google Scholar] [CrossRef] [PubMed]
- Morrell, A.; Tallino, S.; Yu, L.; Burkhead, J.L. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life 2017, 69, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Tallino, S.; Duffy, M.; Ralle, M.; Cortes, M.P.; Latorre, M.; Burkhead, J.L. Nutrigenomics analysis reveals that copper deficiency and dietary sucrose up-regulate inflammation, fibrosis and lipogenic pathways in a mature rat model of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2015, 26, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Schuschke, D.A.; Zhou, Z.; Zhong, W.; Zhang, J.; Zhang, X.; Wang, Y.; McClain, C.J. Kupffer cell depletion protects against the steatosis, but not the liver damage, induced by marginal-copper, high-fructose diet in male rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G934–G945. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chao, P.Y.; Allen, K.G. Inhibition of elevated hepatic glutathione abolishes copper deficiency cholesterolemia. FASEB J. 1992, 6, 2467–2471. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Gasperkova, D.; Xu, J.; Baillie, R.; Lee, J.H.; Clarke, S.D. Copper deficiency induces hepatic fatty acid synthase gene transcription in rats by increasing the nuclear content of mature sterol regulatory element binding protein 1. J. Nutr. 2000, 130, 2915–2921. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.; Kim, S.; Allen, K.G.; Baillie, R.; Clarke, S.D. Hepatic fatty acid synthase gene transcription is induced by a dietary copper deficiency. Am. J. Physiol. 1997, 272, E1124–E1129. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Juge-Aubry, C.; Sgroi, A.; Conzelmann, S.; Pazienza, V.; Pittet-Cuenod, B.; Meier, C.A.; Negro, F. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology 2008, 48, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Mandrekar, P.; Ambade, A.; Lim, A.; Szabo, G.; Catalano, D. An essential role for monocyte chemoattractant protein-1 in alcoholic liver injury: Regulation of proinflammatory cytokines and hepatic steatosis in mice. Hepatology 2011, 54, 2185–2197. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G. Hepatic iron overload may contribute to hypertriglyceridemia and hypercholesterolemia in copper-deficient rats. Metabolism 1997, 46, 377–381. [Google Scholar] [CrossRef]
- Fields, M.; Lewis, C.G. Level of dietary iron, not type of dietary fat, is hyperlipidemic in copper-deficient rats. J. Am. Coll. Nutr. 1999, 18, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Lure, M.D.; Burns, W.A. Dietary ferric vs. ferrous iron in copper-deficient rats fed fructose-based diets. J. Am. Coll. Nutr. 1995, 14, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Lure, M.D.; Burns, W.A.; Antholine, W.E. The severity of copper deficiency can be ameliorated by deferoxamine. Metabolism 1991, 40, 105–109. [Google Scholar] [CrossRef]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell MeTable 2005, 2, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, J.R. Changes in Cu,Zn-superoxide dismutase, cytochrome c oxidase, glutathione peroxidase and glutathione transferase activities in copper-deficient mice and rats. J. Nutr. 1991, 121, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Dallman, P.R. Cytochrome oxidase repair during treatment of copper deficiency: Relation to mitochondrial turnover. J. Clin. Investig. 1967, 46, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.H.; Reeve, V.E.; Wright, R. Copper deficiency in the rat. Effect on the ultrastructure of hepatocytes. Aust. J. Exp. Biol. Med. Sci. 1973, 51, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Andreux, P.; Poitry-Yamate, C.; Auwerx, J.; Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 19507–19512. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.; Scholfield, D.J.; Powell, A.S.; Rose, A.J.; Reiser, S.; Smith, J.C. Female rats are protected against the fructose induced mortality of copper deficiency. Proc. Soc. Exp. Biol. Med. 1986, 183, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Xue, Q.; Aliabadi, H.; Hallfrisch, J. Effects of dietary galactose and fructose on rats fed diets marginal or adequate in copper for 9–21 months. Nutr. Res. 2001, 21, 1078–1087. [Google Scholar] [CrossRef]
- Fields, M.; Lewis, C.G.; Lure, M.D. Allopurinol, an inhibitor of xanthine oxidase, reduces uric acid levels and modifies the signs associated with copper deficiency in rats fed fructose. Free Radic. Biol. Med. 1996, 20, 595–600. [Google Scholar] [CrossRef]
- Rossi, L.; Lippe, G.; Marchese, E.; De Martino, A.; Mavelli, I.; Rotilio, G.; Ciriolo, M.R. Decrease of cytochrome c oxidase protein in heart mitochondria of copper-deficient rats. Biometals 1998, 11, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Metallomics 2017, 9, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.C. Redox regulation of SCO protein function: Controlling copper at a mitochondrial crossroad. Antioxid. Redox Signal. 2010, 13, 1403–1416. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.; Deepa, S.S.; Sataranatarajan, K.; Premkumar, P.; Pulliam, D.; Liu, Y.; Soto, V.Y.; Fischer, K.E.; Van Remmen, H. Sco2 deficient mice develop increased adiposity and insulin resistance. Mol. Cell. Endocrinol. 2017, 455, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Paret, C.; Stucka, R.; Horn, N.; Muller-Hocker, J.; Horvath, R.; Trepesch, N.; Stecker, G.; Freisinger, P.; Thirion, C.; et al. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum. Mol. Genet. 2001, 10, 3025–3035. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Pharaoh, G.; Kinter, M.; Diaz, V.; Fok, W.C.; Riddle, K.; Pulliam, D.; Hill, S.; Fischer, K.E.; Soto, V.; et al. Lifelong reduction in complex IV induces tissue-specific metabolic effects but does not reduce lifespan or healthspan in mice. Aging Cell 2018, e12769. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.S.; Pulliam, D.; Hill, S.; Shi, Y.; Walsh, M.E.; Salmon, A.; Sloane, L.; Zhang, N.; Zeviani, M.; Viscomi, C.; et al. Improved insulin sensitivity associated with reduced mitochondrial complex IV assembly and activity. FASEB J. 2013, 27, 1371–1380. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, D.A.; Deepa, S.S.; Liu, Y.; Hill, S.; Lin, A.L.; Bhattacharya, A.; Shi, Y.; Sloane, L.; Viscomi, C.; Zeviani, M.; et al. Complex IV-deficient Surf1(-/-) mice initiate mitochondrial stress responses. Biochem. J. 2014, 462, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Koganti, L.; Muchenditsi, A.; Pendyala, V.S.; Huso, D.; Hankin, J.; Murphy, R.C.; Huster, D.; Merle, U.; Mangels, C.; et al. Activation of liver X receptor/retinoid X receptor pathway ameliorates liver disease in Atp7B-/- (Wilson disease) mice. Hepatology 2016, 63, 1828–1841. [Google Scholar] [CrossRef] [PubMed]
- Bo, S.; Durazzo, M.; Gambino, R.; Berutti, C.; Milanesio, N.; Caropreso, A.; Gentile, L.; Cassader, M.; Cavallo-Perin, P.; Pagano, G. Associations of dietary and serum copper with inflammation, oxidative stress, and metabolic variables in adults. J. Nutr. 2008, 138, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lure, M.D.; Lewis, C.G. Effect of saturated versus unsaturated fat on the pathogenesis of copper deficiency in rats. J. Nutr. Biochem. 1996, 7, 246–251. [Google Scholar] [CrossRef]
- Allen, K.G.; Klevay, L.M. Copper deficiency and cholesterol metabolism in the rat. Atherosclerosis 1978, 31, 259–271. [Google Scholar] [CrossRef]
- Al-Othman, A.A.; Rosenstein, F.; Lei, K.Y. Pool size and concentration of plasma cholesterol are increased and tissue copper levels are reduced during early stages of copper deficiency in rats. J. Nutr. 1994, 124, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Lure, M.D.; Burns, W.A.; Antholine, W.E. Low dietary iron prevents free radical formation and heart pathology of copper-deficient rats fed fructose. Proc. Soc. Exp. Biol. Med. 1993, 202, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Chao, P.Y.; Allen, K.G. Glutathione production in copper-deficient isolated rat hepatocytes. Free Radic. Biol. Med. 1992, 12, 145–150. [Google Scholar] [PubMed]
- Hassel, C.A.; Marchello, J.A.; Lei, K.Y. Impaired glucose tolerance in copper-deficient rats. J. Nutr. 1983, 113, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Lure, M.D. Responses of insulin to oral glucose and fructose loads in marginally copper-deficient rats fed starch or fructose. Nutrition 1996, 12, 524–528. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Karhausen, J.; Furuta, G.T.; Tomaszewski, J.E.; Johnson, R.S.; Colgan, S.P.; Haase, V.H. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Investig. 2004, 114, 1098–1106. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, B.J.; Kao, D.J.; Kitzenberg, D.A.; Dobrinskikh, E.; Schwisow, K.D.; Masterson, J.C.; Kendrick, A.A.; Kelly, C.J.; Bayless, A.J.; Kominsky, D.J.; et al. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol. Biol. Cell 2015, 26, 2252–2262. [Google Scholar] [CrossRef] [PubMed]
- Fluck, K.; Breves, G.; Fandrey, J.; Winning, S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. 2016, 9, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Clambey, E.T.; McNamee, E.N.; Westrich, J.A.; Glover, L.E.; Campbell, E.L.; Jedlicka, P.; de Zoeten, E.F.; Cambier, J.C.; Stenmark, K.R.; Colgan, S.P.; et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc. Natl. Acad. Sci. USA 2012, 109, E2784–E2793. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Ye, F.; Xue, W.; Zhou, Z.; Kang, Y.J. Copper regulation of hypoxia-inducible factor-1 activity. Mol. Pharmacol. 2009, 75, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Song, M.; Yin, X.; Schuschke, D.A.; Koo, I.; McClain, C.J.; Zhang, X. Effects of Dietary Different Doses of Copper and High Fructose Feeding on Rat Fecal Metabolome. J. Proteome Res. 2015, 14, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Ferruzza, S.; Scacchi, M.; Scarino, M.L.; Sambuy, Y. Iron and copper alter tight junction permeability in human intestinal Caco-2 cells by distinct mechanisms. Toxicol. In Vitro 2002, 16, 399–404. [Google Scholar] [CrossRef]
- Rossi, A.; Poverini, R.; Di Lullo, G.; Modesti, A.; Modica, A.; Scarino, M.L. Heavy metal toxicity following apical and basolateral exposure in the human intestinal cell line Caco-2. Toxicol. In Vitro 1996, 10, 27–36. [Google Scholar] [CrossRef]
- Santos, S.; Silva, A.M.; Matos, M.; Monteiro, S.M.; Alvaro, A.R. Copper induced apoptosis in Caco-2 and Hep-G2 cells: Expression of caspases 3, 8 and 9, AIF and p53. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2016, 185–186, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, V.; Petris, M.J. Copper homeostasis at the host-pathogen interface. J. Biol. Chem. 2012, 287, 13549–13555. [Google Scholar] [CrossRef] [PubMed]
- Pontel, L.B.; Soncini, F.C. Alternative periplasmic copper-resistance mechanisms in Gram negative bacteria. Mol. Microbiol. 2009, 73, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Solioz, M.; Abicht, H.K.; Mermod, M.; Mancini, S. Response of gram-positive bacteria to copper stress. J. Biol. Inorg. Chem. 2010, 15, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, C.A.; Faughnan, M.S.; Gilmore, W.S.; Coulter, J.S.; Howard, A.N.; Strain, J.J. Plasma diamine oxidase activity is greater in copper-adequate than copper-marginal or copper-deficient rats. J. Nutr. 2000, 130, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Legleiter, L.R.; Spears, J.W. Plasma diamine oxidase: A biomarker of copper deficiency in the bovine. J. Anim. Sci. 2007, 85, 2198–2204. [Google Scholar] [CrossRef] [PubMed]
- Luk, G.D.; Bayless, T.M.; Baylin, S.B. Diamine oxidase (histaminase). A circulating marker for rat intestinal mucosal maturation and integrity. J. Clin. Investig. 1980, 66, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Galipeau, D.; Verma, S.; McNeill, J.H. Female rats are protected against fructose-induced changes in metabolism and blood pressure. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2478–H2484. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Beal, T.; Scholfield, D.; Patterson, K.; Smith, J.C.; Reiser, S. Sexual differences in the expression of copper deficiency in rats. Proc. Soc. Exp. Biol. Med. 1987, 186, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.; Lewis, C.G.; Lure, M.; Antholine, W.E. The influence of gender on developing copper deficiency and on free radical generation of rats fed a fructose diet. Metabolism 1992, 41, 989–994. [Google Scholar] [CrossRef]
- Bantle, J.P.; Raatz, S.K.; Thomas, W.; Georgopoulos, A. Effects of dietary fructose on plasma lipids in healthy subjects. Am. J. Clin. Nutr. 2000, 72, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Couchepin, C.; Le, K.A.; Bortolotti, M.; da Encarnacao, J.A.; Oboni, J.B.; Tran, C.; Schneiter, P.; Tappy, L. Markedly blunted metabolic effects of fructose in healthy young female subjects compared with male subjects. Diabetes Care 2008, 31, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Latour, C.; Kautz, L.; Besson-Fournier, C.; Island, M.L.; Canonne-Hergaux, F.; Loreal, O.; Ganz, T.; Coppin, H.; Roth, M.P. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology 2014, 59, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Werman, M.J.; Bhathena, S.J. Fructose metabolizing enzymes in the rat liver and metabolic parameters: Interactions between dietary copper, type of carbohydrates, and gender. J. Nutr. Biochem. 1995, 6, 373–379. [Google Scholar] [CrossRef]
- Millo, H.; Werman, M.J. Hepatic fructose-metabolizing enzymes and related metabolites: Role of dietary copper and gender. J. Nutr. Biochem. 2000, 11, 374–381. [Google Scholar] [CrossRef]
- Rodriguez, A.; Marinelli, R.A.; Tesse, A.; Fruhbeck, G.; Calamita, G. Sexual Dimorphism of Adipose and Hepatic Aquaglyceroporins in Health and Metabolic Disorders. Front. Endocrinol. (Lausanne) 2015, 6, 171. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.; Vos, M.B.; McClain, C.J. Copper-Fructose Interactions: A Novel Mechanism in the Pathogenesis of NAFLD. Nutrients 2018, 10, 1815. https://doi.org/10.3390/nu10111815
Song M, Vos MB, McClain CJ. Copper-Fructose Interactions: A Novel Mechanism in the Pathogenesis of NAFLD. Nutrients. 2018; 10(11):1815. https://doi.org/10.3390/nu10111815
Chicago/Turabian StyleSong, Ming, Miriam B. Vos, and Craig J. McClain. 2018. "Copper-Fructose Interactions: A Novel Mechanism in the Pathogenesis of NAFLD" Nutrients 10, no. 11: 1815. https://doi.org/10.3390/nu10111815
APA StyleSong, M., Vos, M. B., & McClain, C. J. (2018). Copper-Fructose Interactions: A Novel Mechanism in the Pathogenesis of NAFLD. Nutrients, 10(11), 1815. https://doi.org/10.3390/nu10111815