Potential Role for Osteocalcin in the Development of Atherosclerosis and Blood Vessel Disease

Abstract
:1. Introduction
2. Atherosclerosis
3. Association between Osteocalcin and Atherosclerosis Outcomes
3.1. Measurement of Osteocalcin in Humans
3.2. In Vivo Osteocalcin Treatment and Cardiovascular Function in Animal Models
3.3. In Vivo Osteocalcin Treatment and Markers of Atherosclerosis Risk in Animal Models
4. Osteocalcin and Endothelial Function
4.1. In Vitro Osteocalcin Treatment in Human Cells
4.2. In Vitro Osteocalcin Treatment and Markers of Atherosclerosis Risk in Animal Cells
5. Osteocalcin and Vascular Calcification
5.1. Osteocalcin and Calcified Human Vascular Tissue
5.2. Vascular Calcification and Osteocalcin in Animal Models
6. The Putative Osteocalcin Receptor: GPRC6A
7. Future Directions
8. Summary and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abubakar, I.; Tillmann, T.; Banerjee, A. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. heart disease and stroke statistics-2017 update: A report from the american heart association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.; Peterson, K.; Herbert, J.; Alston, L.; Allender, S. Australian Heart Disease Statistics 2015; National Heart Foundation of Australia: Melbourne, Australia, 2016. [Google Scholar]
- Sakakura, K.; Nakano, M.; Otsuka, F.; Ladich, E.; Kolodgie, F.D.; Virmani, R. Pathophysiology of atherosclerosis plaque progression. Heart Lung Circ. 2013, 22, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Glasser, S.P.; Selwyn, A.P.; Ganz, P. Atherosclerosis: Risk factors and the vascular endothelium. Am. Heart J. 1996, 131, 379–384. [Google Scholar] [CrossRef]
- Vogel, R.A. Coronary risk factors, endothelial function, and atherosclerosis: A review. Clin. Cardiol. 1997, 20, 426–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, G.L.; Park, K.; Li, Q. Selective insulin resistance and the development of cardiovascular diseases in diabetes: The 2015 Edwin Bierman award lecture. Diabetes 2016, 65, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. Cell Metab. 2013, 17, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P. The role of osteocalcin in the endocrine cross-talk between bone remodelling and energy metabolism. Diabetologia 2011, 54, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Hinoi, E.; Karsenty, G.; Ducy, P. Osteocalcin differentially regulates beta cell and adipocyte gene expression and affects the development of metabolic diseases in wild-type mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Wei, J.; Yoshizawa, T.; Del Fattore, A.; DePinho, R.A.; Teti, A.; Ducy, P.; Karsenty, G. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 2010, 142, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.K.; Sowa, H.; Hinoi, E.; Ferron, M.; Ahn, J.D.; Confavreux, C.; Dacquin, R.; Mee, P.J.; McKee, M.D.; Jung, D.Y.; et al. Endocrine regulation of energy metabolism by the skeleton. Cell 2007, 130, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, H.; Yang, C.; Li, Y.; Dai, Z. An overview of osteocalcin progress. J. Bone Miner. Metab. 2016, 34, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Hauschka, P.V.; Lian, J.B.; Cole, D.E.; Gundberg, C.M. Osteocalcin and matrix Gla protein: Vitamin K-dependent proteins in bone. Physiol. Rev. 1989, 69, 990–1047. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Williamson, M.K.; Lothringer, J.W. Origin of the vitamin K-dependent bone protein found in plasma and its clearance by kidney and bone. J. Biol Chem. 1981, 256, 12760–12766. [Google Scholar] [PubMed]
- Booth, S.L.; Centi, A.; Smith, S.R.; Gundberg, C. The role of osteocalcin in human glucose metabolism: Marker or mediator? Nat. Rev. Endocrinol. 2013, 9, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Levinger, I.; Jerums, G.; Stepto, N.K.; Parker, L.; Serpiello, F.R.; McConell, G.K.; Anderson, M.; Hare, D.L.; Byrnes, E.; Ebeling, P.R.; et al. The effect of acute exercise on undercarboxylated osteocalcin and insulin sensitivity in obese men. J. Bone Miner. Res. 2014, 29, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Levinger, I.; Seeman, E.; Jerums, G.; McConell, G.K.; Rybchyn, M.S.; Cassar, S.; Byrnes, E.; Selig, S.; Mason, R.S.; Ebeling, P.R.; et al. Glucose-loading reduces bone remodeling in women and osteoblast function in vitro. Physiol. Rep. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Yeap, B.B.; Alfonso, H.; Chubb, S.A.; Gauci, R.; Byrnes, E.; Beilby, J.P.; Ebeling, P.R.; Handelsman, D.J.; Allan, C.A.; Grossmann, M.; et al. Higher serum undercarboxylated osteocalcin and other bone turnover markers are associated with reduced diabetes risk and lower estradiol concentrations in older men. J. Clin. Endocrinol. Metab. 2015, 100, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Brennan-Speranza, T.C.; Conigrave, A.D. Osteocalcin: An osteoblast-derived polypeptide hormone that modulates whole body energy metabolism. Calcif. Tissue Int. 2015, 96, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Karsenty, G. An overview of the metabolic functions of osteocalcin. Curr. Osteoporos Rep. 2015, 13, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G. Update on the biology of osteocalcin. Endocr. Pract. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I. Osteocalcin as a hormone regulating glucose metabolism. World J. Diabetes 2015, 6, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Levinger, I.; Brennan-Speranza, T.C.; Zulli, A.; Parker, L.; Lin, X.; Lewis, J.R.; Yeap, B.B. Multifaceted interaction of bone, muscle, lifestyle interventions and metabolic and cardiovascular disease: Role of osteocalcin. Osteoporos Int. 2017, 28, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Zhou, M.; Lu, Z.; Li, H.; Wang, Y.; Sun, L.; Gao, M.; Wei, M.; Jia, W. Serum levels of osteocalcin are inversely associated with the metabolic syndrome and the severity of coronary artery disease in Chinese men. Clin. Endocrinol. 2011, 75, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Fahrleitner-Pammer, A.; Herberth, J.; Browning, S.R.; Obermayer-Pietsch, B.; Wirnsberger, G.; Holzer, H.; Dobnig, H.; Malluche, H.H. Bone markers predict cardiovascular events in chronic kidney disease. J. Bone Miner. Res. 2008, 23, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Yamaguchi, T.; Yamamoto, M.; Yamauchi, M.; Kurioka, S.; Yano, S.; Sugimoto, T. Serum osteocalcin level is associated with glucose metabolism and atherosclerosis parameters in type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2009, 94, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Alfadda, A.A.; Masood, A.; Shaik, S.A.; Dekhil, H.; Goran, M. Association between osteocalcin, metabolic syndrome, and cardiovascular risk factors: Role of total and undercarboxylated osteocalcin in patients with type 2 diabetes. Int. J. Endocrinol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Ni, Z.; Zhou, W.; Qian, J. Undercarboxylated osteocalcin as a biomarker of subclinical atherosclerosis in non-dialysis patients with chronic kidney disease. J. Biomed. Sci. 2015, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.C.; Leopold, J.A.; Loscalzo, J. Vascular calcification: Pathobiological mechanisms and clinical implications. Circ. Res. 2006, 99, 1044–1059. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, H.; Liu, J.; Xu, L.; Guo, Q.; Zang, W.; Sun, H.; Wu, S. Autophagic dysfunction is improved by intermittent administration of osteocalcin in obese mice. Int. J. Obes. (Lond) 2016, 40, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Corti, R.; Fuster, V.; Badimon, J.J. Pathogenetic concepts of acute coronary syndromes. J. Am. Coll. Cardiol. 2003, 41, 7S–14S. [Google Scholar] [CrossRef]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Composition and classification of human atherosclerotic lesions. Virchows Arch. A Pathol. Anat. Histopathol. 1992, 421, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Bornfeldt, K.E.; Tabas, I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell. Metab. 2011, 14, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Schachinger, V.; Zeiher, A.M. Atherosclerosis-associated endothelial dysfunction. Z. Kardiol. 2000, 89 (Suppl. 9), 70–74. [Google Scholar] [CrossRef]
- Rubanyi, G.M. The role of endothelium in cardiovascular homeostasis and diseases. J. Cardiovasc. Pharmacol. 1993, 22, S1–S14. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.; Haudenschild, C.C. Endothelium and atherogenesis: Endothelial therapy revisited. J. Cardiovasc. Pharmacol. 2001, 38, S23–S25. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Farzaneh-Far, A.; Proudfoot, D.; Shanahan, C.; Weissberg, P.L. Vascular and valvar calcification: Recent advances. Heart 2001, 85, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Vascular calcification: Pathobiology of a multifaceted disease. Circulation 2008, 117, 2938–2948. [Google Scholar] [CrossRef] [PubMed]
- Rennenberg, R.J.; Kessels, A.G.; Schurgers, L.J.; van Engelshoven, J.M.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.M.; Asotra, K.; Fitzpatrick, L.A.; Qiao, J.H.; Wilkin, D.J.; Detrano, R.C.; Dunstan, C.R.; Shah, P.K.; Rajavashisth, T.B. Calcification in atherosclerosis: Bone biology and chronic inflammation at the arterial crossroads. Proc. Natl. Acad. Sci. USA 2003, 100, 11201–11206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, F.; Sakakura, K.; Yahagi, K.; Joner, M.; Virmani, R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb. Vasc. Biol. 2014, 34, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.; Delanaye, P.; Kamel, S.; Cristol, J.P.; Cavalier, E.; calcifications, S. Vascular calcification: From pathophysiology to biomarkers. Clin. Chim. Acta. 2015, 438, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.N.; Golledge, J. Osteo-progenitors in vascular calcification: A circulating cell theory. J. Atheroscler. Thromb. 2011, 18, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.A.; Patel, H.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Osteocalcin, vascular calcification, and atherosclerosis: A systematic review and meta-analysis. Front. Endocrinol. (Lausanne) 2017, 8, 183. [Google Scholar] [CrossRef] [PubMed]
- Villafan-Bernal, J.R.; Sanchez-Enriquez, S.; Munoz-Valle, J.F. Molecular modulation of osteocalcin and its relevance in diabetes (Review). Int. J. Mol. Med. 2011, 28, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, J.; Li, H.; Ma, X.; Zhang, M.; Fang, Q.; Nie, M.; Bao, Y.; Jia, W. Osteocalcin attenuates high fat diet-induced impairment of endothelium-dependent relaxation through Akt/eNOS-dependent pathway. Cardiovasc. Diabetol. 2014, 13, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Yang, L.; Luo, L.; Wu, P.; Yan, S. Osteocalcin improves metabolic profiles, body composition and arterial stiffening in an induced diabetic rat model. Exp. Clin. Endocrinol. Diabetes 2017, 125, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Kawakubo-Yasukochi, T.; Mizokami, A.; Chishaki, S.; Takeuchi, H.; Hirata, M. Uncarboxylated osteocalcin increases serum nitric oxide levels and ameliorates hypercholesterolemia in mice fed an atherogenic diet. Elect. J. Biol. 2016, 13, 22–28. [Google Scholar]
- Zhou, B.; Li, H.; Liu, J.; Xu, L.; Zang, W.; Wu, S.; Sun, H. Intermittent injections of osteocalcin reverse autophagic dysfunction and endoplasmic reticulum stress resulting from diet-induced obesity in the vascular tissue via the NFkappaB-p65-dependent mechanism. Cell Cycle 2013, 12, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Barbato, J.E.; Tzeng, E. Nitric oxide and arterial disease. J. Vasc. Surg. 2004, 40, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.; Mille-Baker, B.; Laffan, M. Human umbilical vein endothelial cells differ from other endothelial cells in failing to express ABO blood group antigens. J. Vasc. Res. 2000, 37, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Lee, W.J.; Hwang, J.Y.; Lee, M.J.; Seol, S.M.; Kim, Y.M.; Lee, Y.L.; Park, J.Y. The preventive effect of uncarboxylated osteocalcin against free fatty acid-induced endothelial apoptosis through the activation of phosphatidylinositol 3-kinase/Akt signaling pathway. Metabolism 2013, 62, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Li, H.; Xu, L.; Wu, S.; Sun, H.; Zhou, B. Undercarboxylated osteocalcin reverts insulin resistance induced by endoplasmic reticulum stress in human umbilical vein endothelial cells. Sci. Rep. 2017, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Li, H.; Xu, L.; Zang, W.; Wu, S.; Sun, H. Osteocalcin reverses endoplasmic reticulum stress and improves impaired insulin sensitivity secondary to diet-induced obesity through nuclear factor-kappaB signaling pathway. Endocrinology 2013, 154, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Vanhoutte, P.M.; Leung, S.W. Vascular nitric oxide: Beyond eNOS. J. Pharmacol. Sci. 2015, 129, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byon, C.H.; Javed, A.; Dai, Q.; Kappes, J.C.; Clemens, T.L.; Darley-Usmar, V.M.; McDonald, J.M.; Chen, Y. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J. Biol. Chem. 2008, 283, 15319–15327. [Google Scholar] [CrossRef] [PubMed]
- Tyson, K.L.; Reynolds, J.L.; McNair, R.; Zhang, Q.; Weissberg, P.L.; Shanahan, C.M. Osteo/chondrocytic transcription factors and their target genes exhibit distinct patterns of expression in human arterial calcification. Arterioscler Thromb Vasc. Biol. 2003, 23, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. Vascular calcification mechanisms. J. Am. Soc. Nephrol. 2004, 15, 2959–2964. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.J.; Gundberg, C.; Scheinman, R. The identification of the vitamin K-dependent bone protein osteocalcin as one of the gamma-carboxyglutamic acid containing proteins present in calcified atherosclerotic plaque and mineralized heart valves. Atherosclerosis 1983, 46, 49–56. [Google Scholar] [CrossRef]
- Levy, R.J.; Zenker, J.A.; Lian, J.B. Vitamin K-dependent calcium binding proteins in aortic valve calcification. J. Clin. Invest. 1980, 65, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Fleet, J.C.; Hock, J.M. Identification of osteocalcin mRNA in nonosteoid tissue of rats and humans by reverse transcription-polymerase chain reaction. J. Bone Miner. Res. 1994, 9, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Severson, A.R.; Ingram, R.T.; Fitzpatrick, L.A. Matrix proteins associated with bone calcification are present in human vascular smooth muscle cells grown in vitro. Cell Dev. Biol. Anim. 1995, 31, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Davies, J.D.; Skepper, J.N.; Weissberg, P.L.; Shanahan, C.M. Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation 2002, 106, 3044–3050. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular matrix mineralization is regulated locally; different roles of two GLA-containing proteins. J. Cell. Biol. 2004, 165, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.N.; Rush, C.; Parr, A.; Van Campenhout, A.; Golledge, J. Osteocalcin positive mononuclear cells are associated with the severity of aortic calcification. Atherosclerosis 2010, 210, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morony, S.; Tintut, Y.; Zhang, Z.; Cattley, R.C.; Van, G.; Dwyer, D.; Stolina, M.; Kostenuik, P.J.; Demer, L.L. Osteoprotegerin inhibits vascular calcification without affecting atherosclerosis in Ldlr(-/-) mice. Circulation 2008, 117, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, T.; Ota, H.; Iijima, K.; Son, B.K.; Kahyo, T.; Setou, M.; Ogawa, S.; Ouchi, Y.; Akishita, M. A novel organ culture model of aorta for vascular calcification. Atherosclerosis 2016, 244, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idelevich, A.; Rais, Y.; Monsonego-Ornan, E. Bone Gla protein increases HIF-1alpha-dependent glucose metabolism and induces cartilage and vascular calcification. Arterioscler Thromb Vasc. Biol. 2011, 31, e55–e71. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.; Shanahan, C.M. Targeting vascular calcification: Softening-up a hard target. Curr. Opin. Pharmacol. 2009, 9, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Shanahan, C.M.; Proudfoot, D.; Farzaneh-Far, A.; Weissberg, P.L. The role of Gla proteins in vascular calcification. Crit. Rev. Eukaryot. Gene. Expr. 1998, 8, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pi, M.; Kapoor, K.; Ye, R.; Nishimoto, S.K.; Smith, J.C.; Baudry, J.; Quarles, L.D. Evidence for Osteocalcin Binding and Activation of GPRC6A in beta-Cells. Endocrinology 2016, 157, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Nishimoto, S.K.; Quarles, L.D. GPRC6A: Jack of all metabolism (or master of none). Mol. Metab. 2017, 6, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Pi, M.; Wu, Y.; Quarles, L.D. GPRC6A mediates responses to osteocalcin in beta-cells in vitro and pancreas in vivo. J. Bone Miner. Res. 2011, 26, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Oury, F.; Sumara, G.; Sumara, O.; Ferron, M.; Chang, H.; Smith, C.E.; Hermo, L.; Suarez, S.; Roth, B.L.; Ducy, P.; et al. Endocrine regulation of male fertility by the skeleton. Cell 2011, 144, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Hanna, T.; Suda, N.; Karsenty, G.; Ducy, P. Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a. Diabetes 2014, 63, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.; Laue, K.; Ferron, M.; Confavreux, C.; Wei, J.; Galan-Diez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 2016, 23, 1078–1092. [Google Scholar] [CrossRef] [PubMed]
- Mera, P.; Laue, K.; Wei, J.; Berger, J.M.; Karsenty, G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol. Metab. 2016, 5, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Oury, F.; Ferron, M.; Huizhen, W.; Confavreux, C.; Xu, L.; Lacombe, J.; Srinivas, P.; Chamouni, A.; Lugani, F.; Lejeune, H.; et al. Osteocalcin regulates murine and human fertility through a pancreas-bone-testis axis. J. Clin. Investig. 2013, 123, 2421–2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Toni, L.; Di Nisio, A.; Speltra, E.; Rocca, M.S.; Ghezzi, M.; Zuccarello, D.; Turiaco, N.; Ferlin, A.; Foresta, C. Polymorphism rs2274911 of GPRC6A as a novel risk factor for testis failure. J. Clin. Endocrinol. Metab. 2016, 101, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.E.; Norskov-Lauritsen, L.; Thomsen, A.R.; Smajilovic, S.; Wellendorph, P.; Larsson, N.H.; Lehmann, A.; Bhatia, V.K.; Brauner-Osborne, H. Delineation of the GPRC6A receptor signaling pathways using a mammalian cell line stably expressing the receptor. J. Pharmacol. Exp. Ther. 2013, 347, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Rueda, P.; Harley, E.; Lu, Y.; Stewart, G.D.; Fabb, S.; Diepenhorst, N.; Cremers, B.; Rouillon, M.H.; Wehrle, I.; Geant, A.; et al. Murine GPRC6A mediates cellular responses to l.-amino acids, but not osteocalcin variants. PLoS ONE 2016, 11, e0146846. [Google Scholar] [CrossRef] [PubMed]
- Harno, E.; Edwards, G.; Geraghty, A.R.; Ward, D.T.; Dodd, R.H.; Dauban, P.; Faure, H.; Ruat, M.; Weston, A.H. Evidence for the presence of GPRC6A receptors in rat mesenteric arteries. Cell. Calcium. 2008, 44, 210–219. [Google Scholar] [CrossRef] [PubMed]
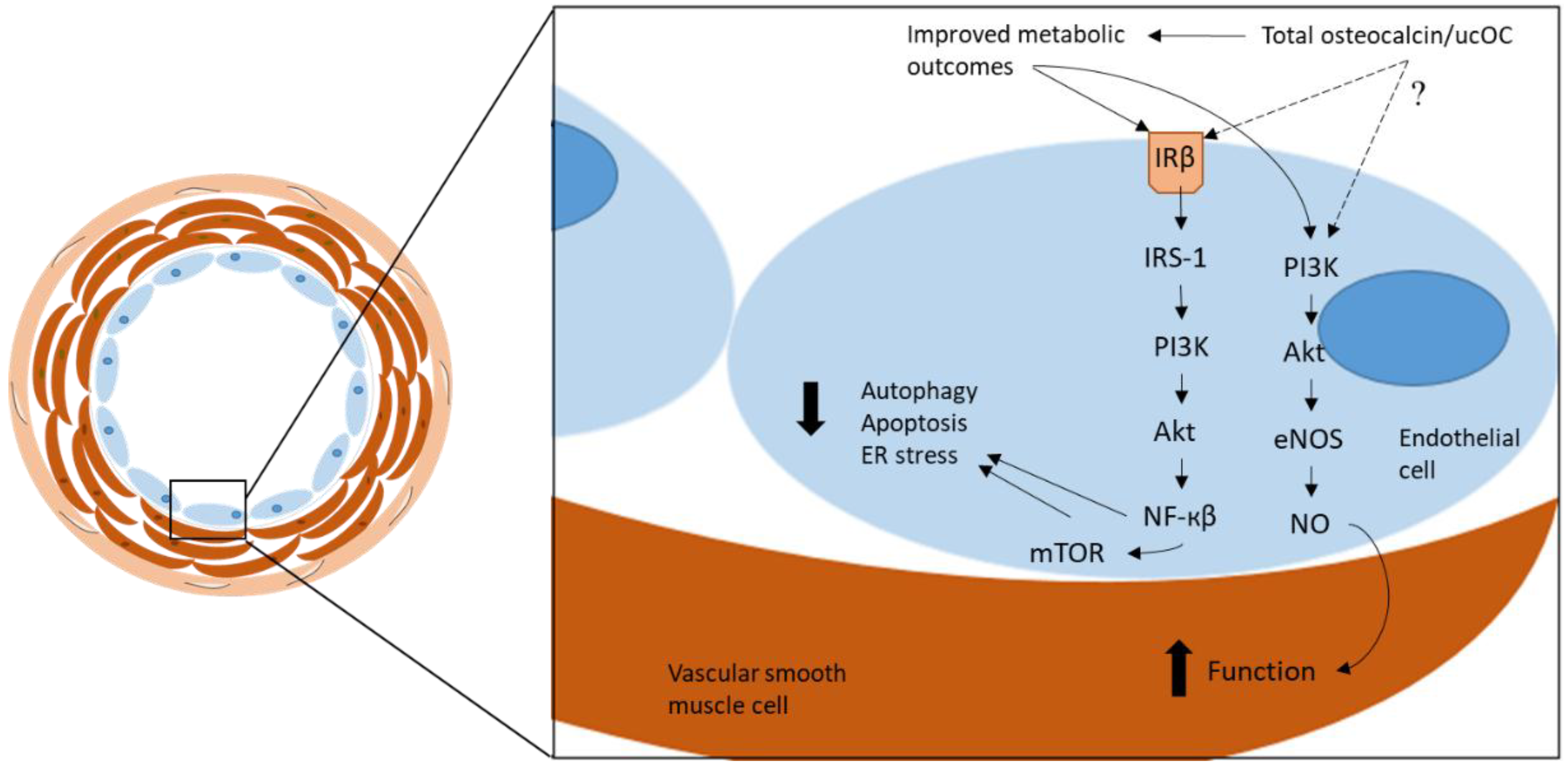

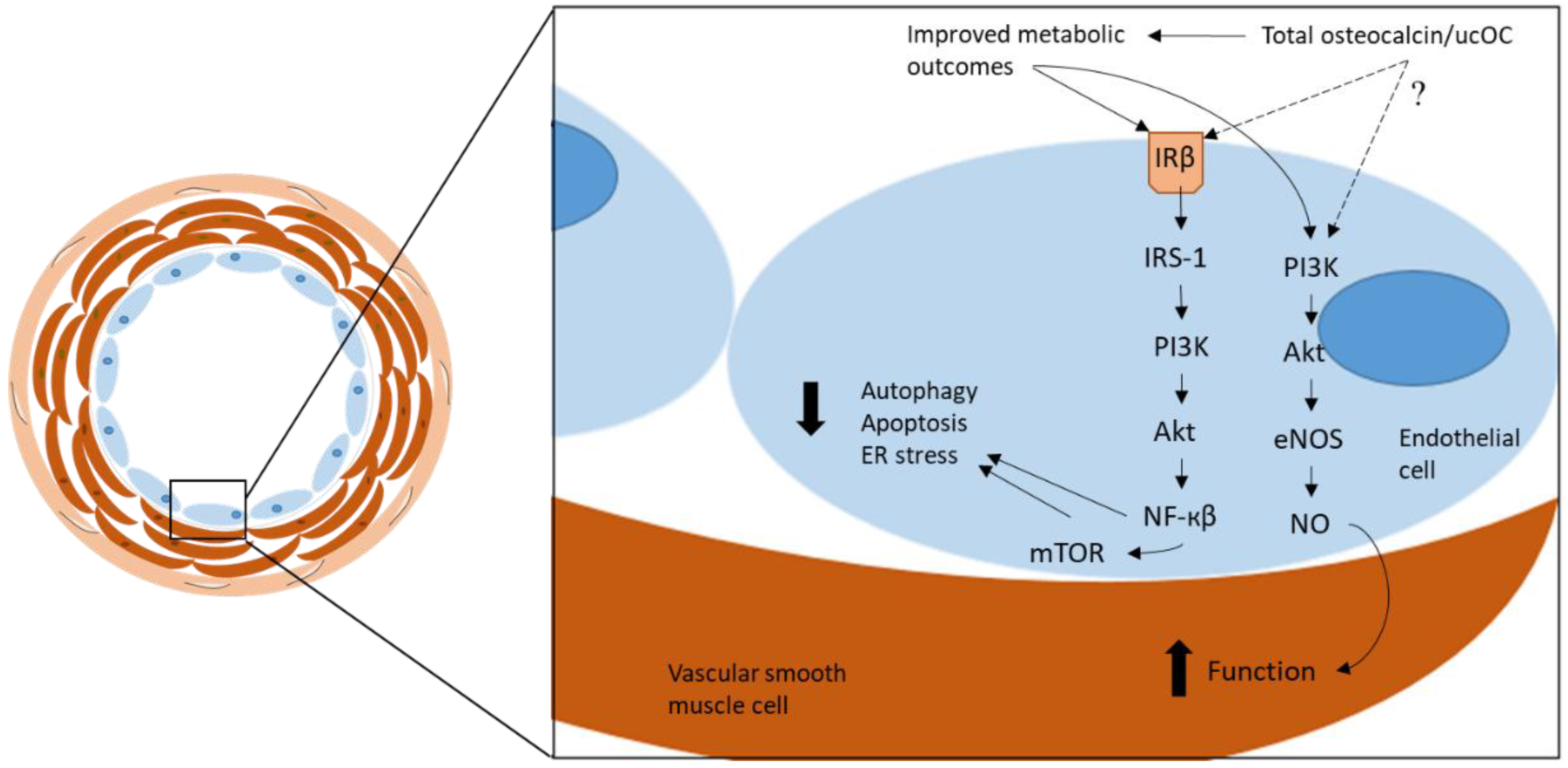{kind=link}
| First Author, Year [Ref.] | Experimental Overview | Measurement of Vascular Function | Results |
|---|---|---|---|
| Dou, 2014 [55] | ApoE-/- mice received ND or HFD and treatment daily for 12 weeks with vehicle or total osteocalcin (30 ng/g) | BP, heart rate, and isometric myography | In vivo: mean and diastolic BP normalized by osteocalcin treatment in HFD group, no change in systolic BP or heart rate. Ex vivo: 20% improvement in relaxation in osteocalcin-treated mice on HFD |
| Huang, 2017 [56] | Sprague Dawley rats induced with diabetes via STZ injection and received ND or HFD, daily treatment of vehicle or total osteocalcin (30 ng/g) for 12 weeks | BP, PWV, heart rate, pulse pressure, and mean arterial pressure | PWV normalized in osteocalcin-treated rats with diabetes compared to diabetic rats treated with vehicle, no change in BP, heart rate, mean arterial pressure, and pulse pressure |
| Kondo, 2016 [57] | Wild type C57BL/6 mice received HFD and treated 5 times a week for 10 weeks with vehicle or ucOC (30 ng/g) | Nitric oxide production | Increased nitric oxide concentration in ucOC-treated mice compared to vehicle-treated mice |
| Zhou, 2013 [58] | C57BL/6J mice received ND or HFD for 8 weeks with daily injections of vehicle or ucOC (30 ng/g) | Autophagy and ER stress | Autophagy and ER stress attenuated in mice receiving ucOC |
| First Author, Year [Ref.] | Experimental Overview | Outcomes | Results |
|---|---|---|---|
| Dou, 2014 [55] | HUVECs incubated with total osteocalcin (10–150 ng/mL) for 15 min–2 h. Descending aorta of ApoE-/- mice, previously treated with osteocalcin, incubated with LY294002 (10 µmol/L) and Akt inhibitor V (5 µmol/L) | eNOS, Akt, and PI3K phosphorylation and expression | Max phosphorylation of eNOS and Akt with 100 ng/mL of osteocalcin. Max phosphorylation of eNOS and Akt occurred after 1 h and 2 h, respectively. In aorta, PI3K, Akt, and eNOS phosphorylation and expression increased, inhibited with LY294002 and Akt inhibitor V |
| Kondo, 2016 [57] | HAECs incubated with ucOC (5, 25, and 100 ng/mL) and cOC (25 and 100 ng/mL) for 30 min | eNOS phosphorylation | Incubation of ucOC increased eNOS phosphorylation in a dose-dependent manner, cOC had no effect |
| Jung, 2013 [61] | HAECs incubated with ucOC (0.3–30 ng/mL), linoleic acid (100 µmol/L for 16 h), and wortmannin (100 nmol/L for 15 min) | Nitric oxide concentration, eNOS and Akt phosphorylation and apoptosis | UcOC increased eNOS and Akt phosphorylation and nitric oxide concentration, which was inhibited by wortmannin. UcOC attenuated linoleic acid-induced apoptosis |
| Guo, 2017 [62] | HUVECs incubated with ucOC (5 ng/mL for 4 h), tunicamycin (5 µg/mL for 4 h), insulin (10 nM for 10 min), wortmannin, and Akti-1/2 (10 µM for 4 h) | Insulin resistance, ER stress | UcOC blocked ER stress and insulin resistance, which was inhibited by wortmannin and Akti-1/2 |
| Zhou, 2013 [58] | Mouse VECs and VSMCs incubated with tunicamycin (5 µg/mL for 4 h), ucOC (5 ng/mL for 0, 2, 4, and 8 h), Akti-1/2 (10 µM for 4 h) and rapamycin (10 nM for 4 h) | Autophagy and ER stress | UcOC attenuated autophagy and ER stress in mouse VECs and VSMCs, which was inhibited by Akti-1/2 and rapamycin |
| First Author, Year [Ref.] | Experimental Overview | Outcomes | Results |
|---|---|---|---|
| Levy, 1983 [69] | Human aortic and valve tissue | Osteocalcin and Gla levels | Osteocalcin and Gla levels higher in calcified tissue than in non-calcified tissue |
| Levy, 1980 [70] | Human aortic and valve tissue | Gla levels | Higher Gla levels in calcified aorta and valves than non-calcified tissue |
| Fleet, 1994 [71] | Human aortic tissue | Osteocalcin mRNA levels | Osteocalcin mRNA increased in calcified aorta and plaque compared to non-calcified aorta |
| Tyson, 2003 [67] | Human aortic and carotid tissue | Osteocalcin expression | Calcified vessels had an increase in the expression of osteocalcin |
| Severson, 1995 [72] | Cultured human aortic VSMCs | Immunostaining for osteocalcin | Minimal immunostaining of human VSMCs |
| Proudfoot, 2002 [73] | Cultured human aortic VSMCs with lipid content modification | Osteocalcin expression | Osteocalcin expression increased in calcified cells compared to non-calcified cells, which was altered with the modification of lipid content |
| Murshed, 2004 [74] | MGP-/- mice inter-crossed with pSM22α-Osteocalcin | Mineralization of aorta | Osteocalcin gain of function model did not inhibit the mineralization of mouse aorta |
| Pal, 2010 [75] | OPG +/+ and OPG-/- mice | Calcification and mononuclear cells expressing osteocalcin | Increased calcification in OPG-/- mice, which was associated with an increased percentage of osteocalcin positive mononuclear cells |
| Morony, 2008 [76] | Ldlr -/- mice fed HFD for 5 months and treated with OPG | Calcification, osteocalcin mRNA and circulating levels | Osteocalcin mRNA levels were unchanged, circulating osteocalcin increased over the 5 months, which was associated with calcification |
| Akiyoshi, 2016 [77] | Thoracic aorta of C57BL/6 mice cultured to induced calcification | Osteocalcin expression | Osteocalcin expression increased in calcified thoracic aortas |
| Idelevich, 2011 [78] | Cultured MOVAS cells induced with calcification and overexpressed with osteocalcin. Sprague Dawley rats induced with calcification | Mineralization, osteocalcin mRNA, metabolic signaling pathways | In vitro: overexpression of osteocalcin in MOVAS cells associated with mineralization and upregulation of insulin signaling In vitro: osteocalcin mRNA is increased in calcified vasculature and associated with activation of metabolic signaling pathways |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tacey, A.; Qaradakhi, T.; Brennan-Speranza, T.; Hayes, A.; Zulli, A.; Levinger, I. Potential Role for Osteocalcin in the Development of Atherosclerosis and Blood Vessel Disease. Nutrients 2018, 10, 1426. https://doi.org/10.3390/nu10101426
Tacey A, Qaradakhi T, Brennan-Speranza T, Hayes A, Zulli A, Levinger I. Potential Role for Osteocalcin in the Development of Atherosclerosis and Blood Vessel Disease. Nutrients. 2018; 10(10):1426. https://doi.org/10.3390/nu10101426
Chicago/Turabian StyleTacey, Alexander, Tawar Qaradakhi, Tara Brennan-Speranza, Alan Hayes, Anthony Zulli, and Itamar Levinger. 2018. "Potential Role for Osteocalcin in the Development of Atherosclerosis and Blood Vessel Disease" Nutrients 10, no. 10: 1426. https://doi.org/10.3390/nu10101426