Orosensory Detection of Dietary Fatty Acids Is Altered in CB1R−/− Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Behavioral Experiments
2.2.1. Two-Bottle Preference Tests
2.2.2. Licking Tests
2.3. Papillae and Taste Buds Isolation
2.4. Real-Time qPCR
2.5. Western Blotting
2.6. Tissue Culture of TBC and GLP-1 Release
2.7. Measurement of Ca2+ Signaling
2.8. Statistics
3. Results
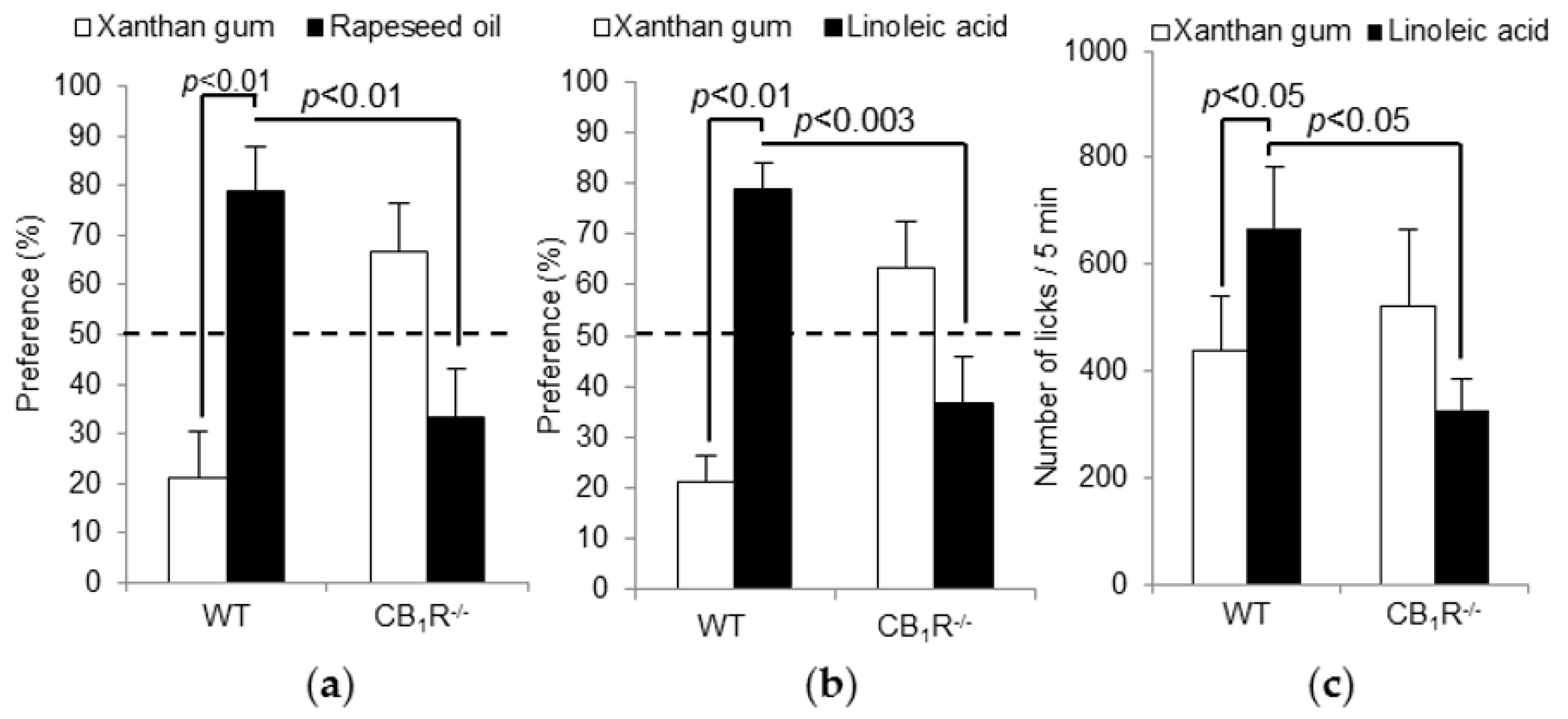
3.1. The Absence of CB1R Gene Induces a Low Preference for Fatty Solutions Independently of Postprandial Factors
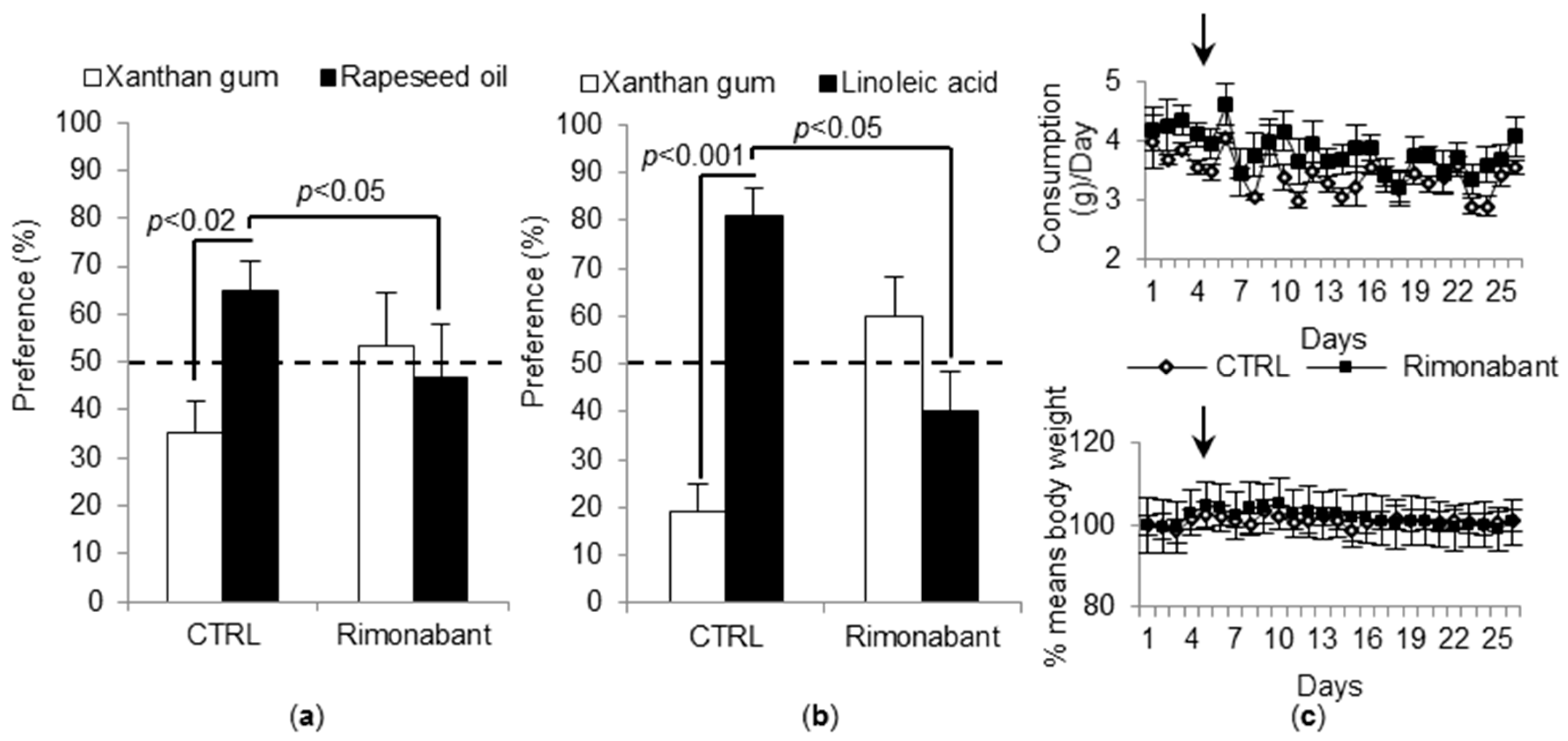
3.2. Treatment with Rimonabant Induces a Low Preference for Fat Solutions and Does Not Alter Feeding Behavior
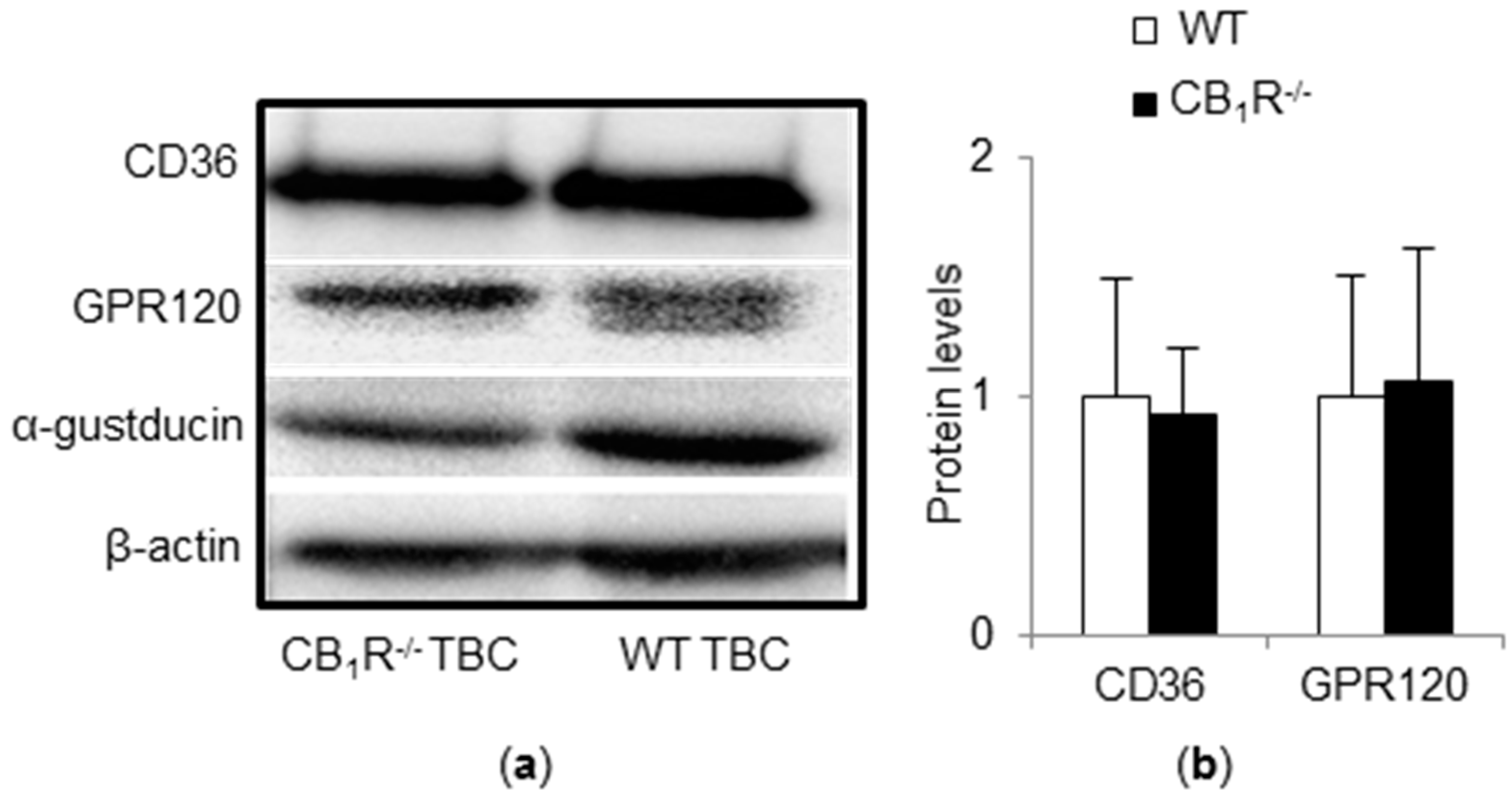
3.3. CD36 and GPR120 Protein Expressions Are Not Altered in TBC of CB1R−/− Mice
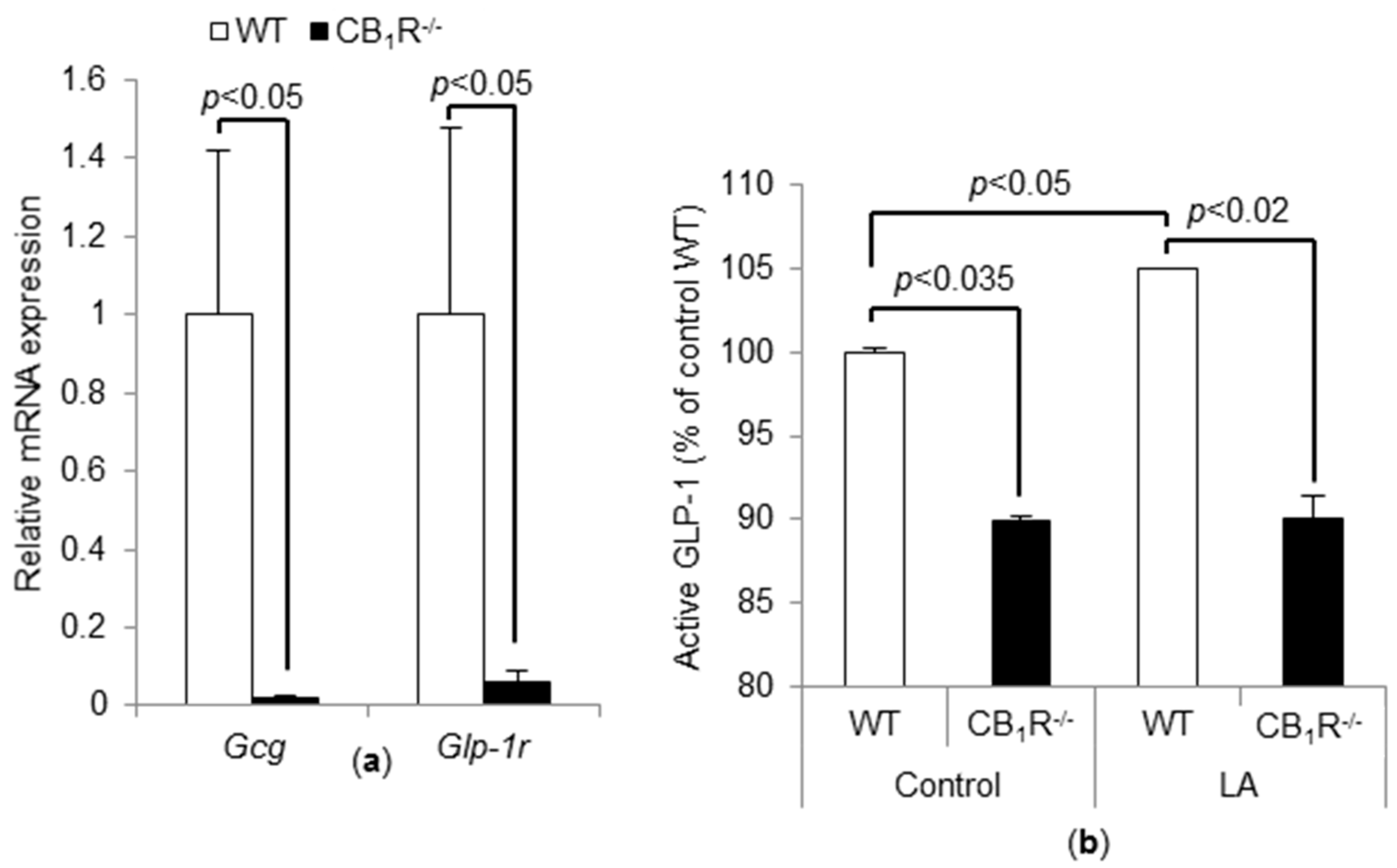
3.4. CB1R Gene Invalidation Induces a Decrease in Proglucagon and GLP-1r mRNA and Basal GLP-1 Level
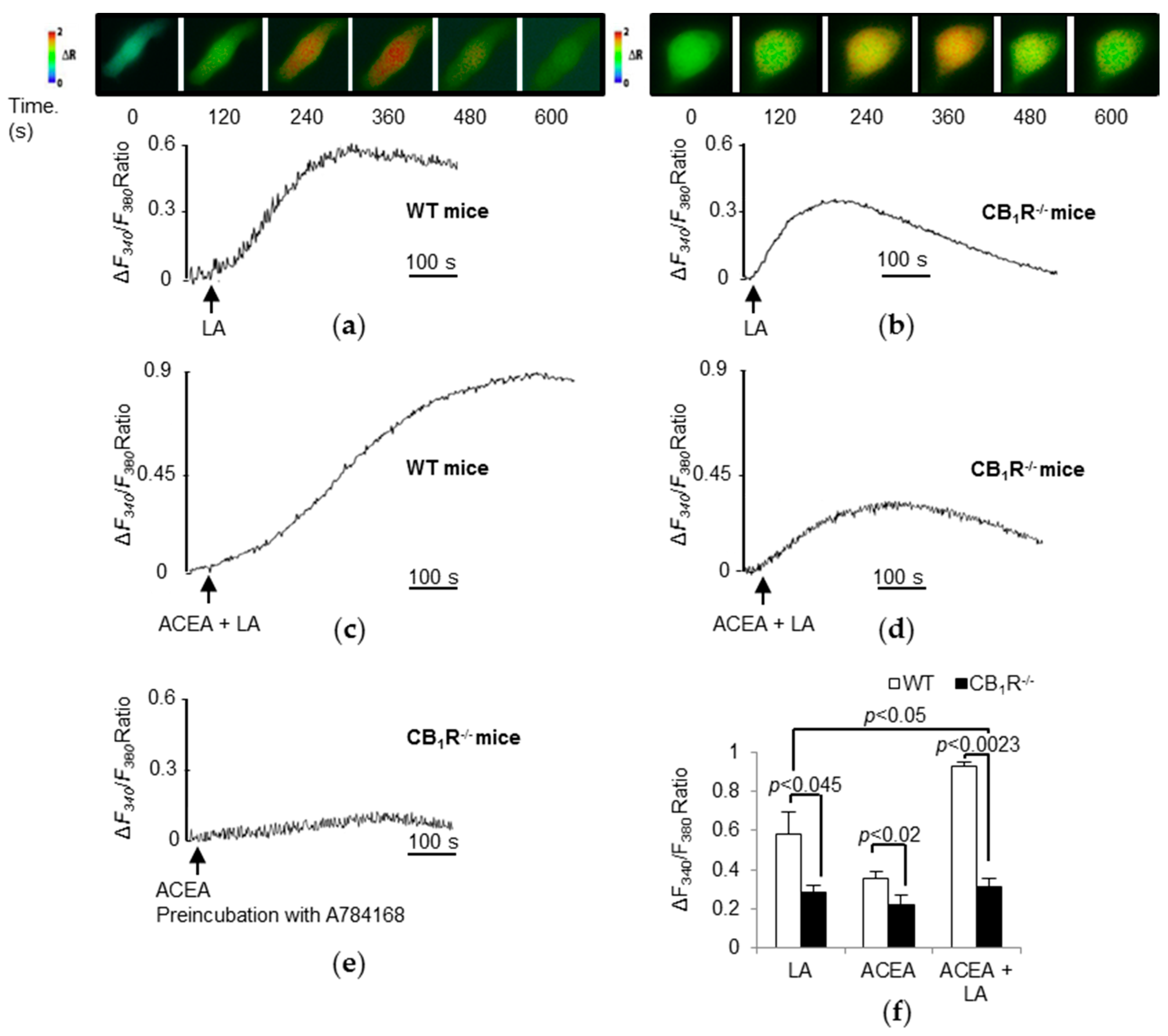
3.5. Both LA and Cannabinoids Induce CB1R-Dependent Ca2+ Responses in TBC
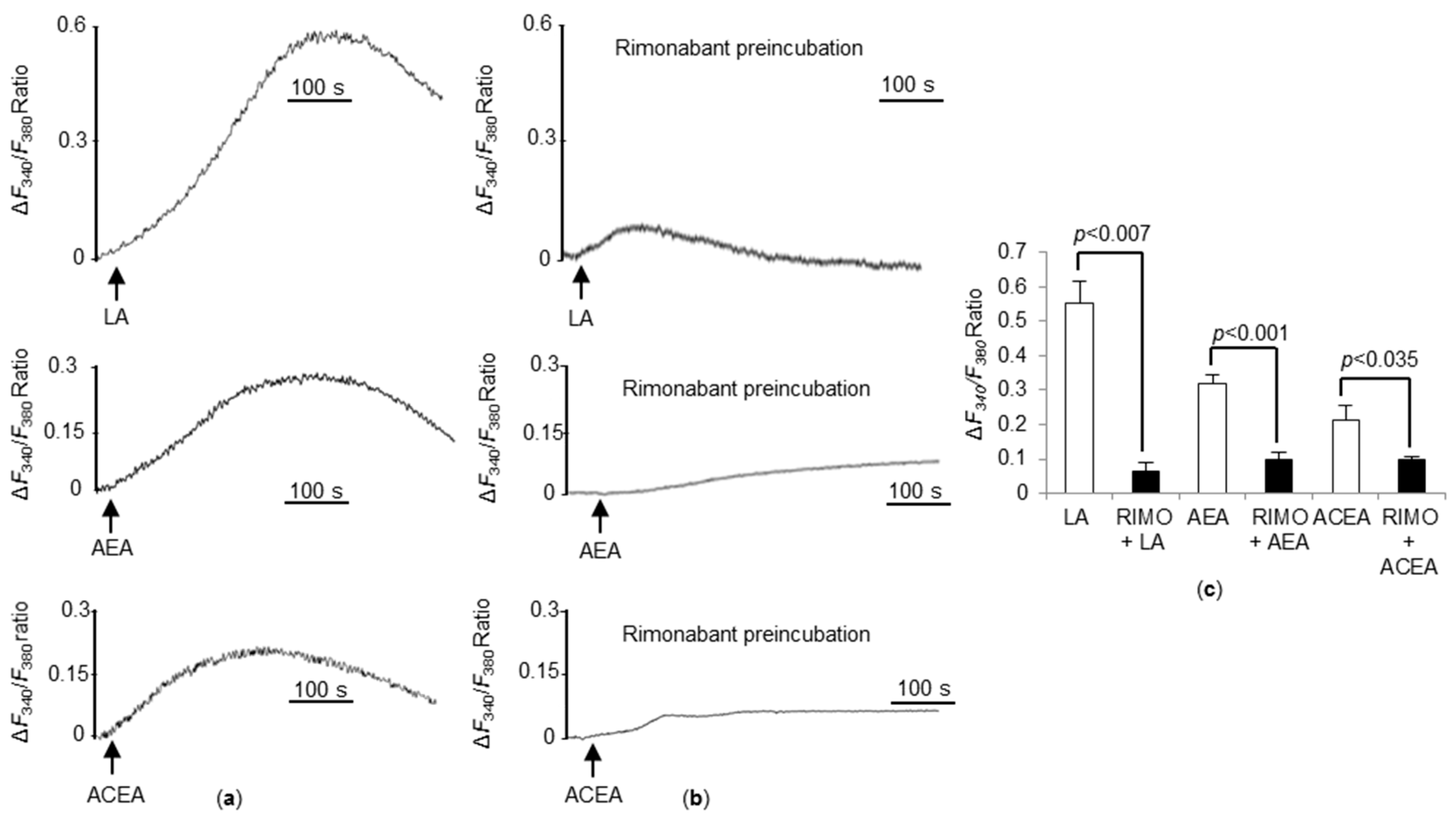
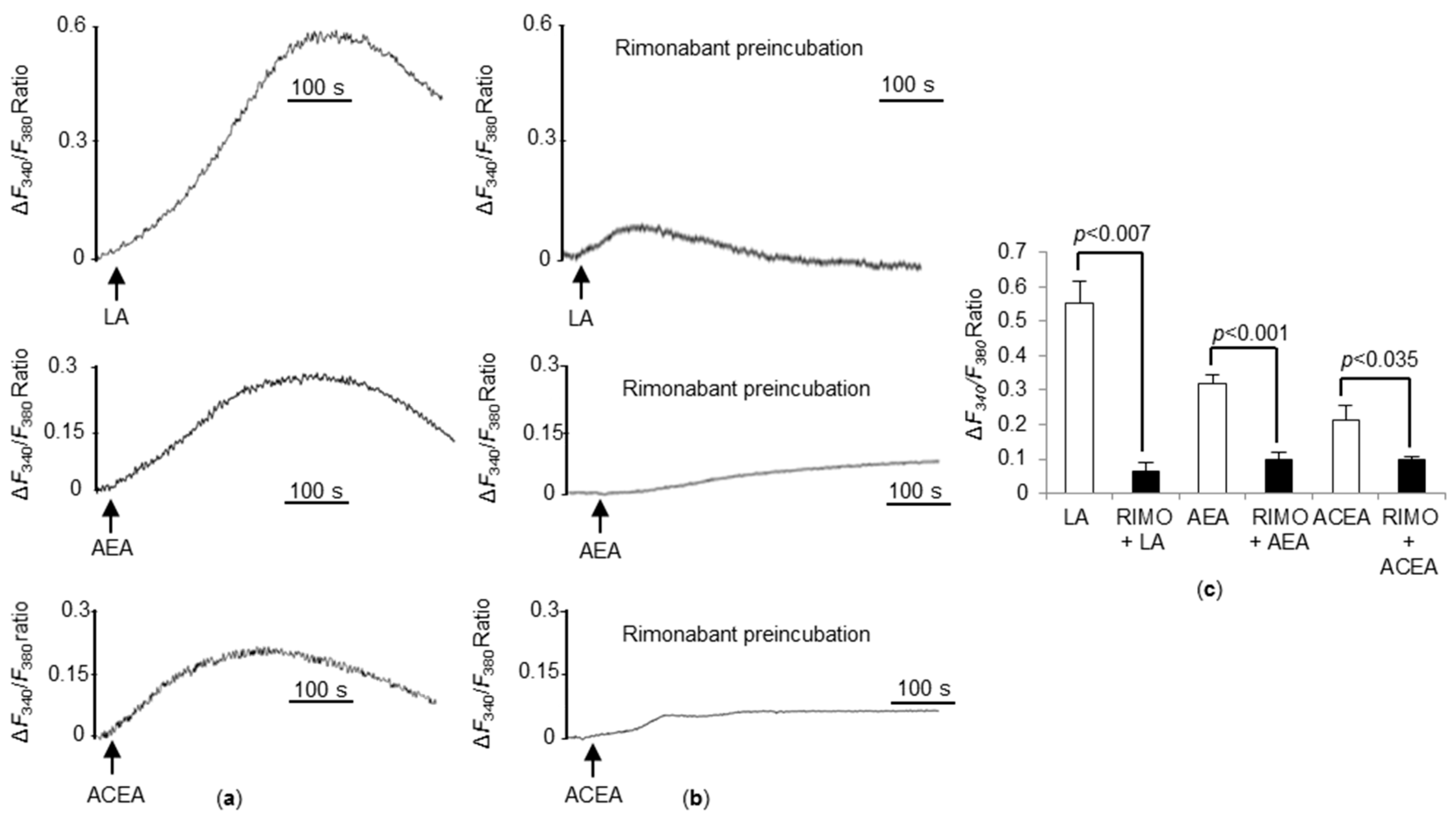
3.6. CB1R Blockade Significantly Decreases Ca2+ Responses Triggered by LA, AEA, and ACEA in WT TBC
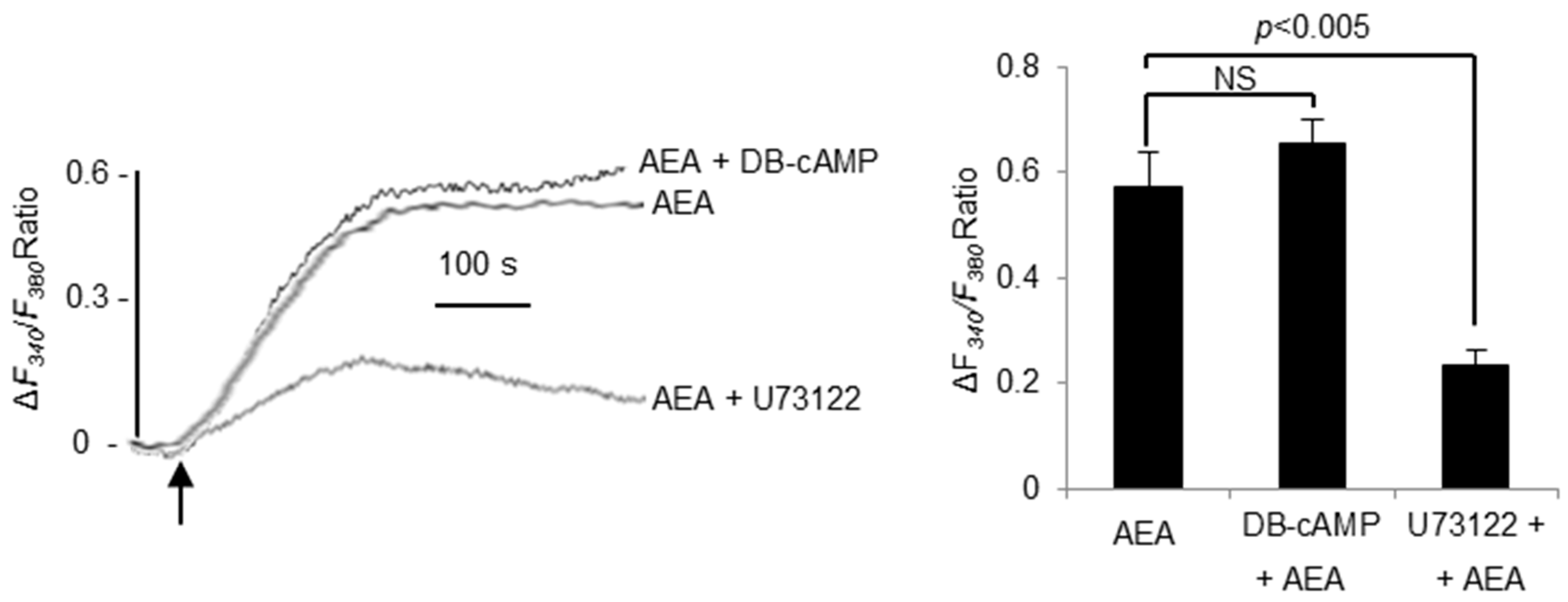
3.7. AEA-Induced Ca2+-Signaling Is PLC Dependent
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Drewnowski, A.; Brunzell, J.D.; Sande, K.; Iverius, P.H.; Greenwood, M.R. Sweet tooth reconsidered: Taste responsiveness in human obesity. Physiol. Behav. 1985, 35, 617–622. [Google Scholar] [CrossRef]
- Mela, D.J.; Sacchetti, D.A. Sensory preferences for fats: Relationships with diet and body composition. Am. J. Clin. Nutr. 1991, 53, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Laugerette, F.; Passilly-Degrace, P.; Patris, B.; Niot, I.; Febbraio, M.; Montmayeur, J.-P.; Besnard, P. CD36 involvement in orosensory detection of dietary lipids, spontaneous fat preference, and digestive secretions. J. Clin. Investig. 2005, 115, 3177–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Yassimi, A.; Hichami, A.; Besnard, P.; Khan, N.A. Linoleic acid induces calcium signaling, Src kinase phosphorylation, and neurotransmitter release in mouse CD36-positive gustatory cells. J. Biol. Chem. 2008, 283, 12949–12959. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, T.A.; Khan, N.A. Cell signaling mechanisms of oro-gustatory detection of dietary fat: Advances and challenges. Prog. Lipid Res. 2014, 53, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, D.; Laugerette, F.; Darcel, N.; El-Yassimi, A.; Passilly-Degrace, P.; Hichami, A.; Khan, N.A.; Montmayeur, J.-P.; Besnard, P. The gustatory pathway is involved in CD36-mediated orosensory perception of long-chain fatty acids in the mouse. FASEB J. 2008, 22, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Ohkuri, T.; Jyotaki, M.; Yasuo, T.; Horio, N.; Yasumatsu, K.; Sanematsu, K.; Shigemura, N.; Yamamoto, T.; Margolskee, R.F.; et al. Endocannabinoids selectively enhance sweet taste. Proc. Natl. Acad. Sci. USA 2010, 107, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Besnard, P.; Passilly-Degrace, P.; Khan, N.A. Taste of fat: A Sixth Taste Modality? Physiol. Rev. 2016, 96, 151–176. [Google Scholar] [CrossRef] [PubMed]
- Ozdener, M.H.; Subramaniam, S.; Sundaresan, S.; Sery, O.; Hashimoto, T.; Asakawa, Y.; Besnard, P.; Abumrad, N.A.; Khan, N.A. CD36- and GPR120-mediated Ca2+ signaling in human taste bud cells mediates differential responses to fatty acids and is altered in obese mice. Gastroenterology 2014, 146, 995–1005. [Google Scholar] [CrossRef] [PubMed]
- Cartoni, C.; Yasumatsu, K.; Ohkuri, T.; Shigemura, N.; Yoshida, R.; Godinot, N.; le Coutre, J.; Ninomiya, Y.; Damak, S. Taste preference for fatty acids is mediated by GPR40 and GPR120. J. Neurosci. 2010, 30, 8376–8382. [Google Scholar] [CrossRef] [PubMed]
- Sclafani, A.; Zukerman, S.; Ackroff, K. GPR40 and GPR120 fatty acid sensors are critical for postoral but not oral mediation of fat preferences in the mouse. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R1490–R1497. [Google Scholar] [CrossRef] [PubMed]
- Ancel, D.; Bernard, A.; Subramaniam, S.; Hirasawa, A.; Tsujimoto, G.; Hashimoto, T.; Passilly-Degrace, P.; Khan, N.-A.; Besnard, P. The oral lipid sensor GPR120 is not indispensable for the orosensory detection of dietary lipids in mice. J. Lipid Res. 2015, 56, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Passilly-Degrace, P.; Chevrot, M.; Ancel, D.; Sparks, S.M.; Drucker, D.J.; Besnard, P. Lipid-mediated release of GLP-1 by mouse taste buds from circumvallate papillae: Putative involvement of GPR120 and impact on taste sensitivity. J. Lipid Res. 2012, 53, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Baggio, L.L.; Drucker, D.J. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007, 132, 2131–2157. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.-K.; Martin, B.; Golden, E.; Doston, C.D.; Maudsley, S.; Kim, W.; Jang, H.-J.; Mattson, M.P.; Drucker, D.J.; Egan, J.M.; et al. Modulation of taste sensitivity by GLP-1 signaling. J. Neurochem. 2008, 106, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cota, D.; Marsicano, G.; Lutz, B.; Vicennati, V.; Stalla, G.K.; Pasquali, R.; Pagotto, U. Endogenous cannabinoid system as a modulator of food intake. Int. J. Obes. 2003, 27, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Engeli, S.; Böhnke, J.; Feldpausch, M.; Gorzelniak, K.; Janke, J.; Bátkai, S.; Pacher, P.; Harvey-White, J.; Luft, F.C.; Sharma, A.M.; Jordan, J. Activation of the peripheral endocannabinoid system in human obesity. Diabetes 2005, 54, 2838–2843. [Google Scholar] [CrossRef] [PubMed]
- Jamshidi, N.; Taylor, D.A. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br. J. Pharmacol. 2001, 134, 1151–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.M.; Kirkham, T.C. Anandamide induces overeating: Mediation by central cannabinoid (CB1) receptors. Psychopharmacology 1999, 143, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Kirkham, T.C. Observational analysis of feeding induced by delta 9-THC and anandamide. Physiol. Behav. 2002, 76, 241–250. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; Kunos, G. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Després, J.-P.; Golay, A.; Sjöström, L. Rimonabant in obesity-lipids study group effects of rimonabant on metabolic risk factors in overweight patients with dyslipidemia. N. Engl. J. Med. 2005, 353, 2121–2134. [Google Scholar]
- Van Gaal, L.; Pi-Sunyer, X.; Després, J.-P.; McCarthy, C.; Scheen, A. Efficacy and safety of rimonabant for improvement of multiple cardiometabolic risk factors in overweight/obese patients: Pooled 1-year data from the rimonabant in obesity (RIO) program. Diabetes Care 2008, 31 (Suppl. 2), S229–S240. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Rissanen, A.M.; Scheen, A.J.; Ziegler, O.; Rössner, S. RIO-Europe study group effects of the cannabinoid-1 receptor blocker rimonabant on weight reduction and cardiovascular risk factors in overweight patients: 1-year experience from the RIO-Europe study. Lancet Lond. Engl. 2005, 365, 1389–1397. [Google Scholar] [CrossRef]
- Zhang, X.; Fitzsimmons, R.L.; Cleland, L.G.; Ey, P.L.; Zannettino, A.C.W.; Farmer, E.-A.; Sincock, P.; Mayrhofer, G. CD36/fatty acid translocase in rats: Distribution, isolation from hepatocytes, and comparison with the scavenger receptor SR-B1. Lab. Investig. J. Tech. Methods Pathol. 2003, 83, 317–332. [Google Scholar] [CrossRef]
- Dramane, G.; Abdoul-Azize, S.; Hichami, A.; Vögtle, T.; Akpona, S.; Chouabe, C.; Sadou, H.; Nieswandt, B.; Besnard, P.; Khan, N.A. STIM1 regulates calcium signaling in taste bud cells and preference for fat in mice. J. Clin. Investig. 2012, 122, 2267–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, H.E.; Gribble, F.M.; Reimann, F. The role of gut endocrine cells in control of metabolism and appetite. Exp. Physiol. 2014, 99, 1116–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melis, M.; Scheggi, S.; Carta, G.; Madeddu, C.; Lecca, S.; Luchicchi, A.; Cadeddu, F.; Frau, R.; Fattore, L.; Fadda, P.; et al. PPARα regulates cholinergic-driven activity of midbrain dopamine neurons via a novel mechanism involving α7 nicotinic acetylcholine receptors. J. Neurosci. 2013, 33, 6203–6211. [Google Scholar] [CrossRef] [PubMed]
- Galindo, M.M.; Voigt, N.; Stein, J.; van Lengerich, J.; Raguse, J.-D.; Hofmann, T.; Meyerhof, W.; Behrens, M. G Protein—Coupled receptors in human fat taste perception. Chem. Sens. 2012, 37, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Kentish, S.J.; Page, A.J. The role of gastrointestinal vagal afferent fibres in obesity. J. Physiol. 2015, 593, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Ryskamp, D.A.; Redmon, S.; Jo, A.O.; Križaj, D. TRPV1 and endocannabinoids: Emerging molecular Signals that modulate mammalian vision. Cells 2014, 3, 914–938. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Marini, P.; Matias, I.; Moriello, A.S.; Starowicz, K.; Cristino, L.; Nigam, S.; Di Marzo, V. Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta-cells. Exp. Cell Res. 2007, 313, 2993–3004. [Google Scholar] [CrossRef] [PubMed]
- Takai, S.; Yasumatsu, K.; Inoue, M.; Iwata, S.; Yoshida, R.; Shigemura, N.; Yanagawa, Y.; Drucker, D.J.; Margolskee, R.F.; Ninomiya, Y. Glucagon-like peptide-1 is specifically involved in sweet taste transmission. FASEB J. 2015, 29, 2268–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brissard, L.; Leemput, J.; Hichami, A.; Passilly-Degrace, P.; Maquart, G.; Demizieux, L.; Degrace, P.; Khan, N.A. Orosensory Detection of Dietary Fatty Acids Is Altered in CB1R−/− Mice. Nutrients 2018, 10, 1347. https://doi.org/10.3390/nu10101347
Brissard L, Leemput J, Hichami A, Passilly-Degrace P, Maquart G, Demizieux L, Degrace P, Khan NA. Orosensory Detection of Dietary Fatty Acids Is Altered in CB1R−/− Mice. Nutrients. 2018; 10(10):1347. https://doi.org/10.3390/nu10101347
Chicago/Turabian StyleBrissard, Léa, Julia Leemput, Aziz Hichami, Patricia Passilly-Degrace, Guillaume Maquart, Laurent Demizieux, Pascal Degrace, and Naim Akhtar Khan. 2018. "Orosensory Detection of Dietary Fatty Acids Is Altered in CB1R−/− Mice" Nutrients 10, no. 10: 1347. https://doi.org/10.3390/nu10101347
APA StyleBrissard, L., Leemput, J., Hichami, A., Passilly-Degrace, P., Maquart, G., Demizieux, L., Degrace, P., & Khan, N. A. (2018). Orosensory Detection of Dietary Fatty Acids Is Altered in CB1R−/− Mice. Nutrients, 10(10), 1347. https://doi.org/10.3390/nu10101347