
One-Dimensional Numerical Simulation of Pt-Co Alloy Catalyst Aging for Proton Exchange Membrane Fuel Cells


Abstract
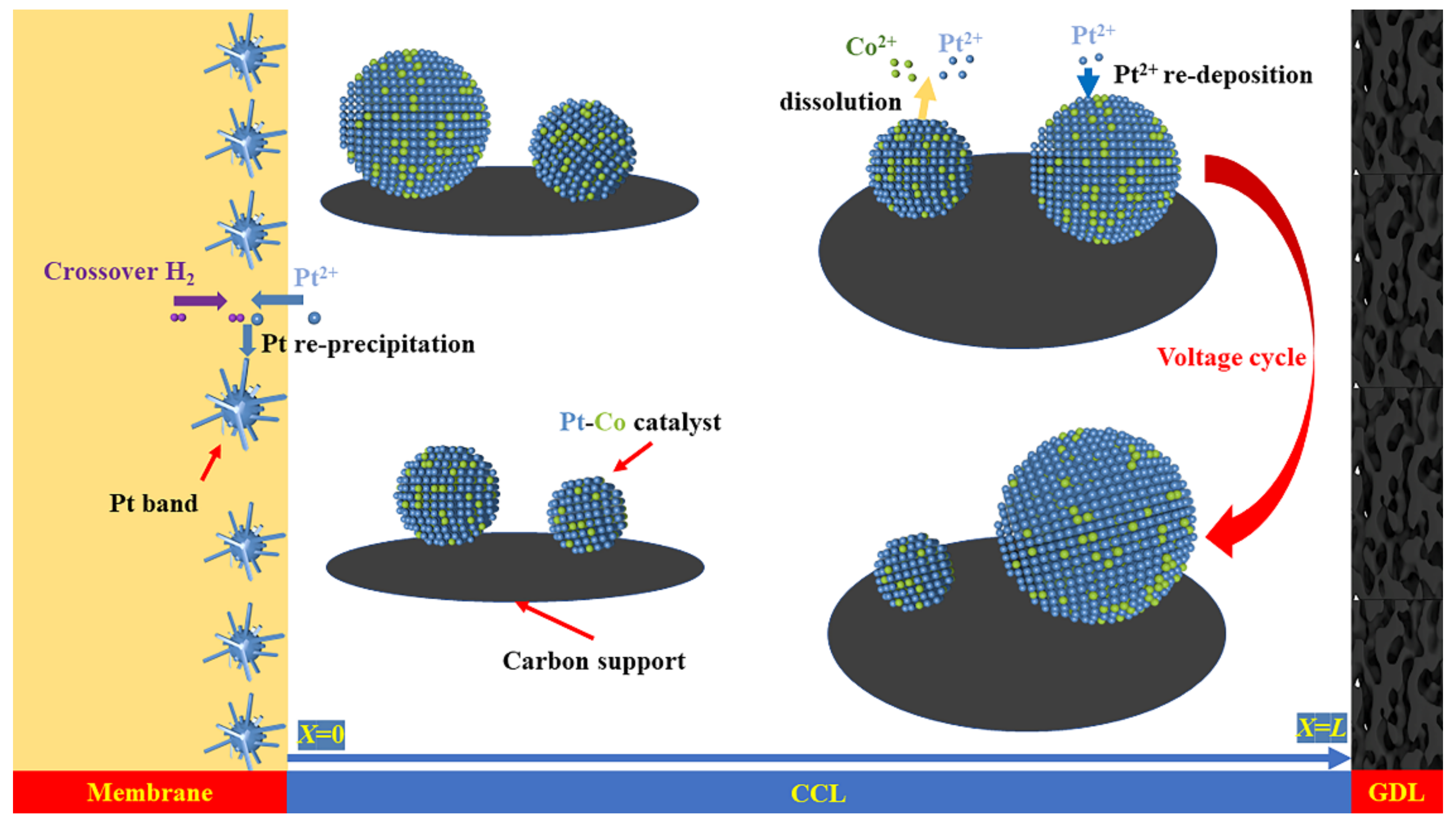
:1. Introduction
2. Model Description
3. Results and Discussion
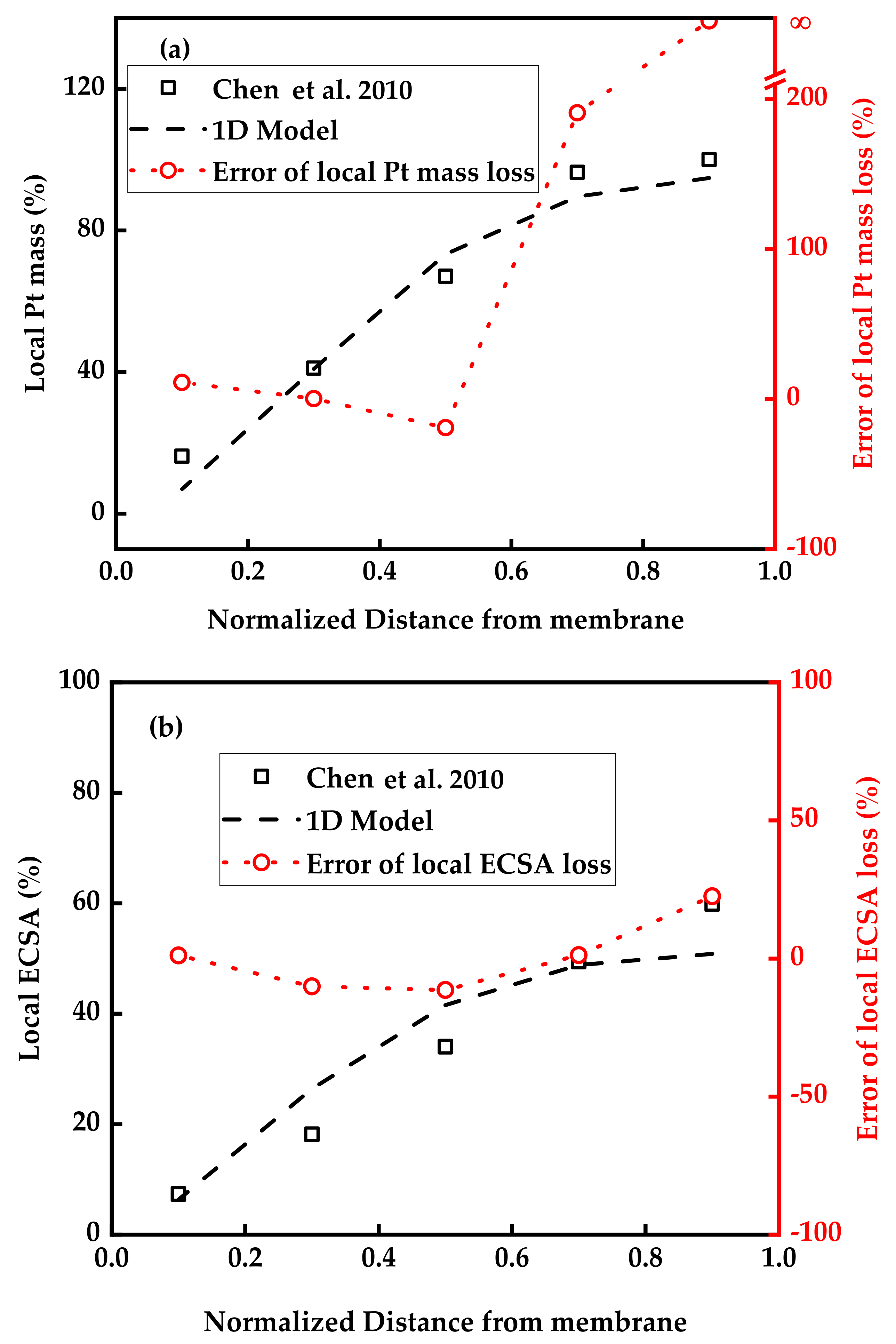
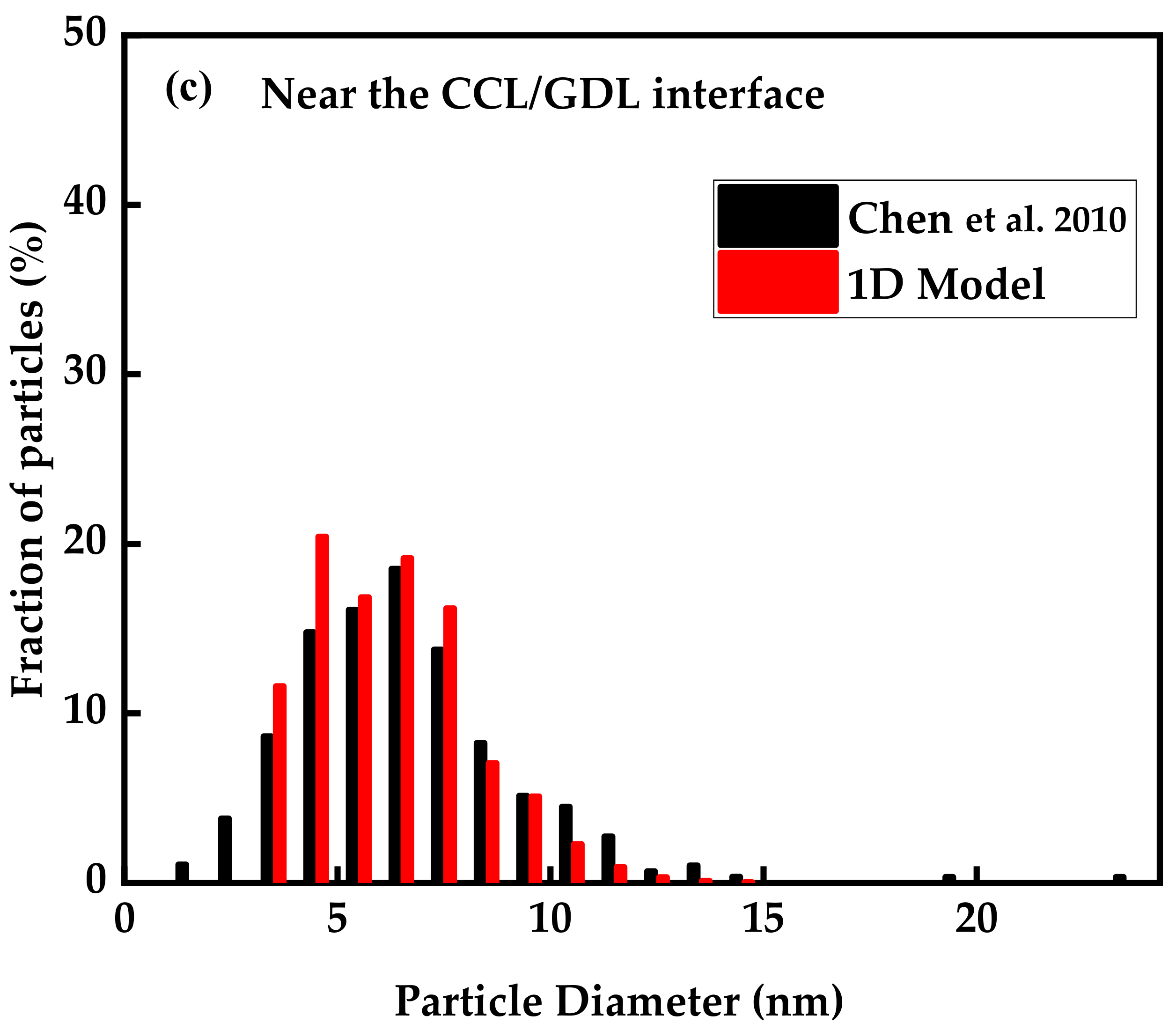
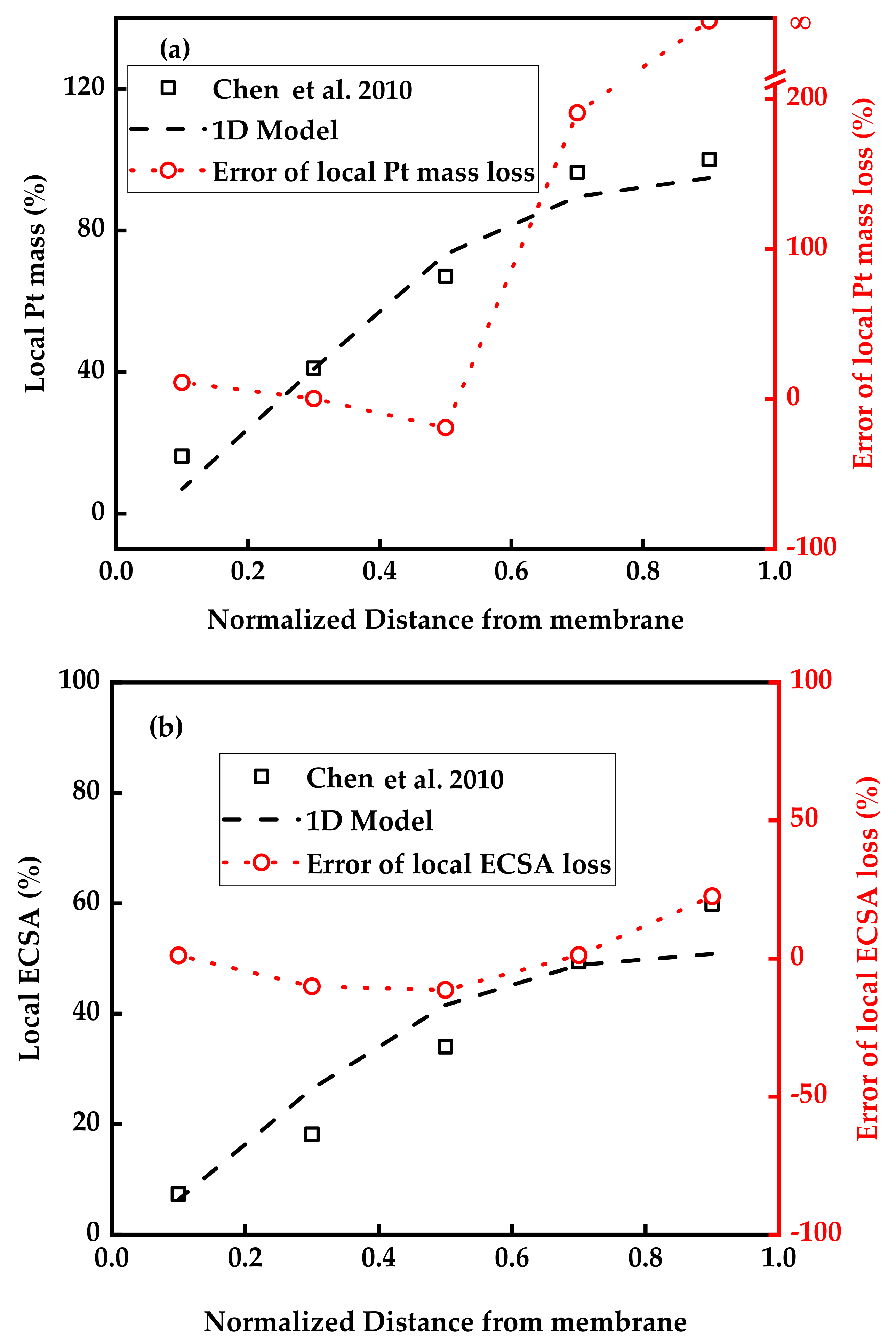
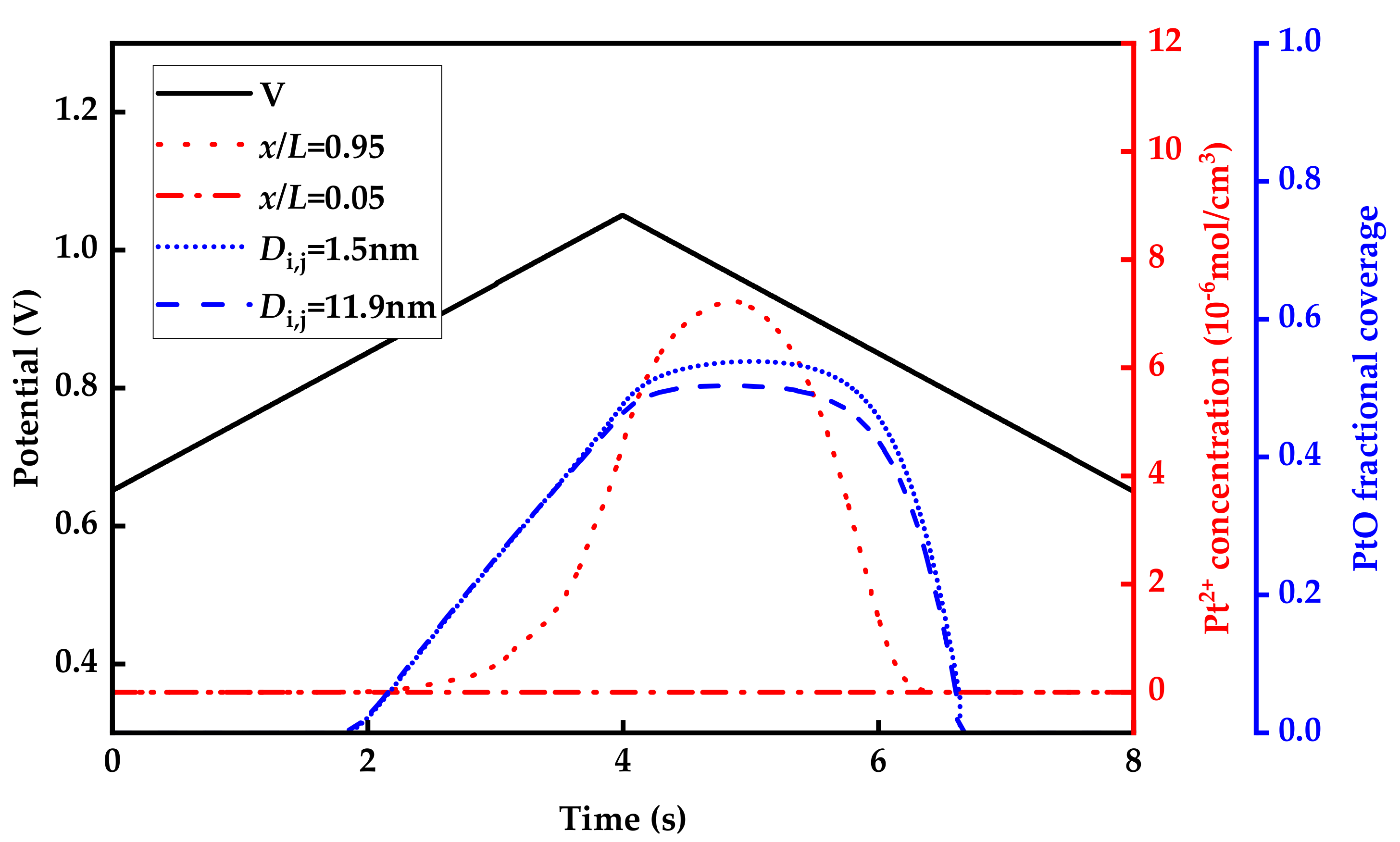
3.1. Model Validation
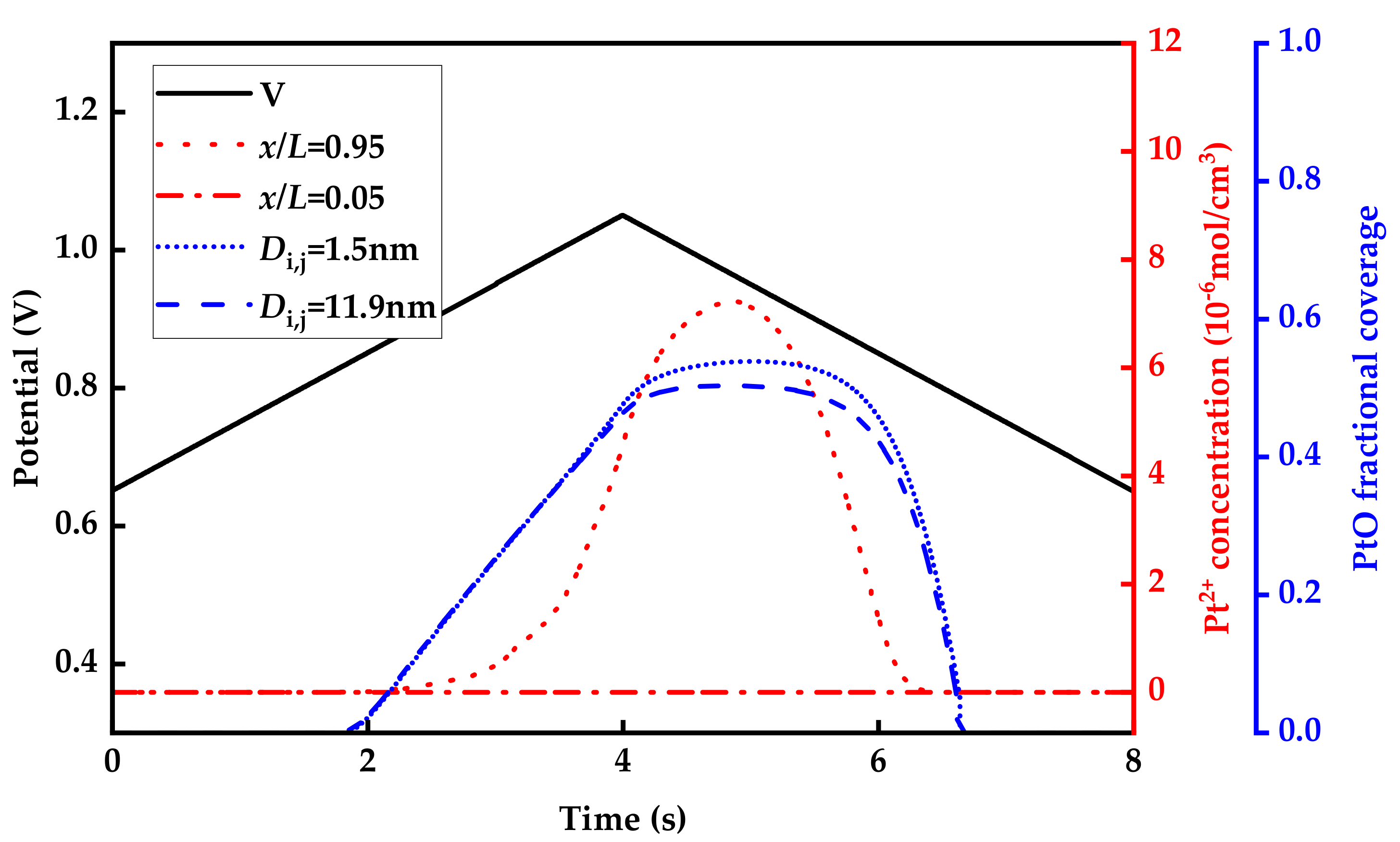
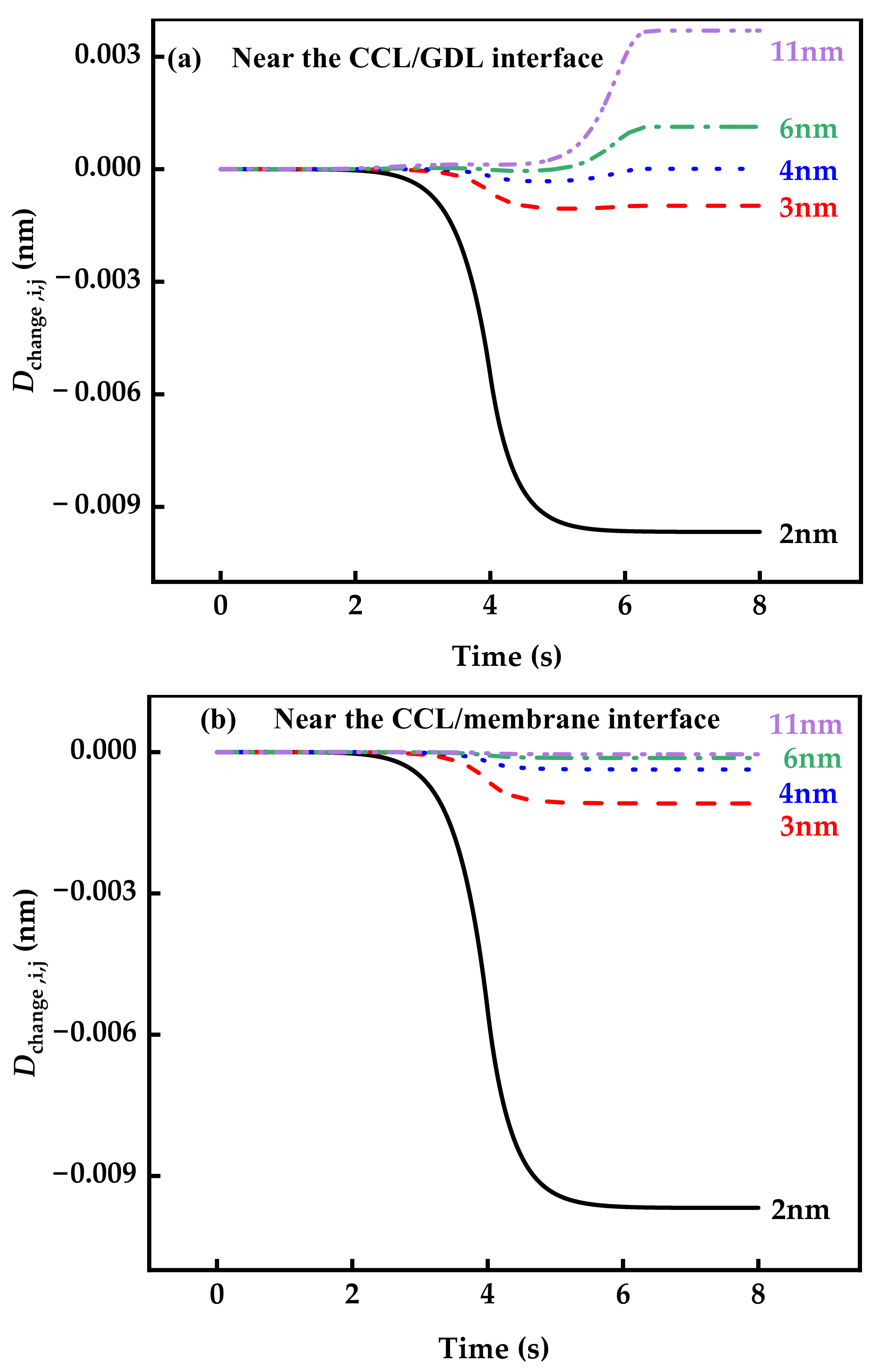
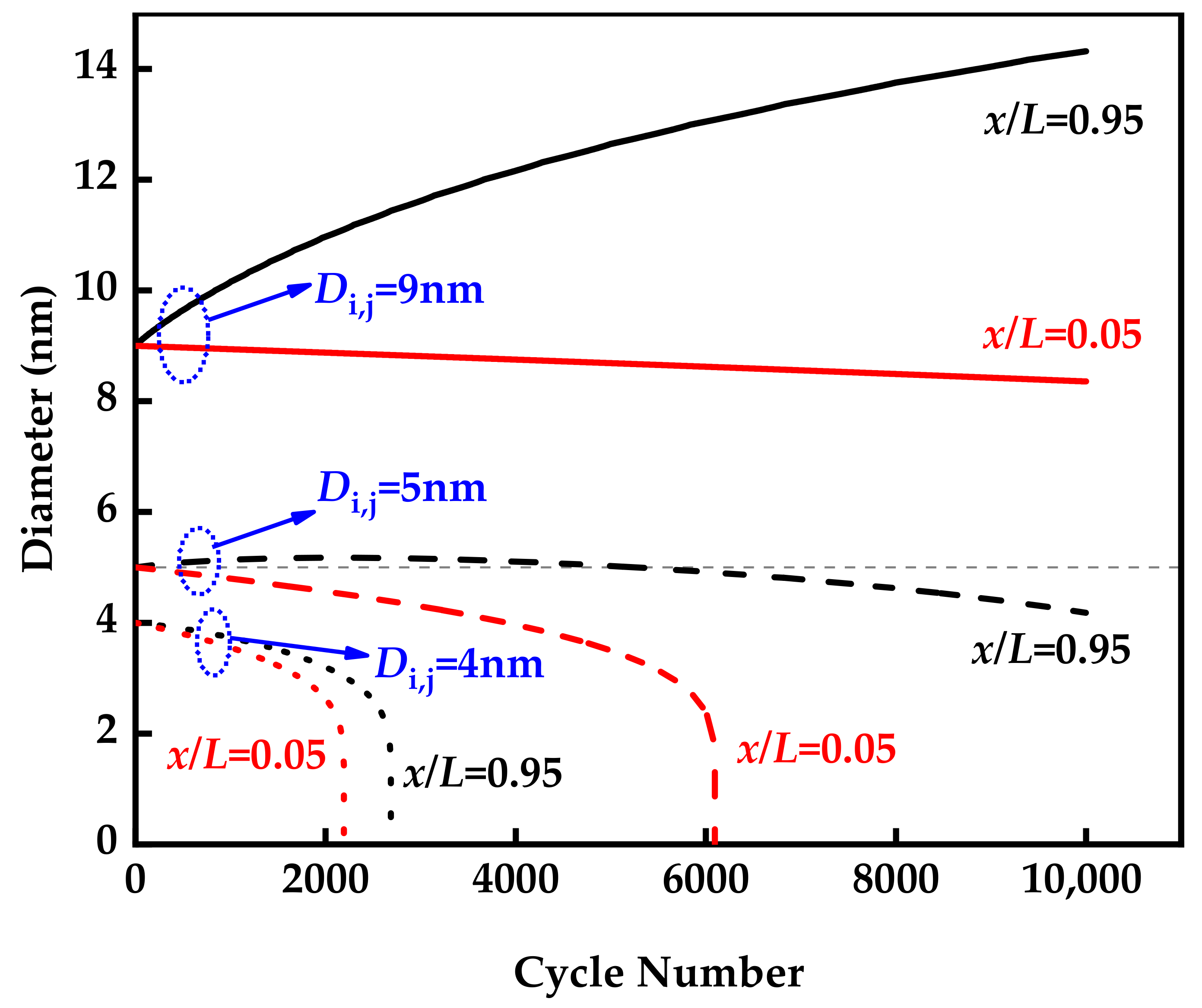
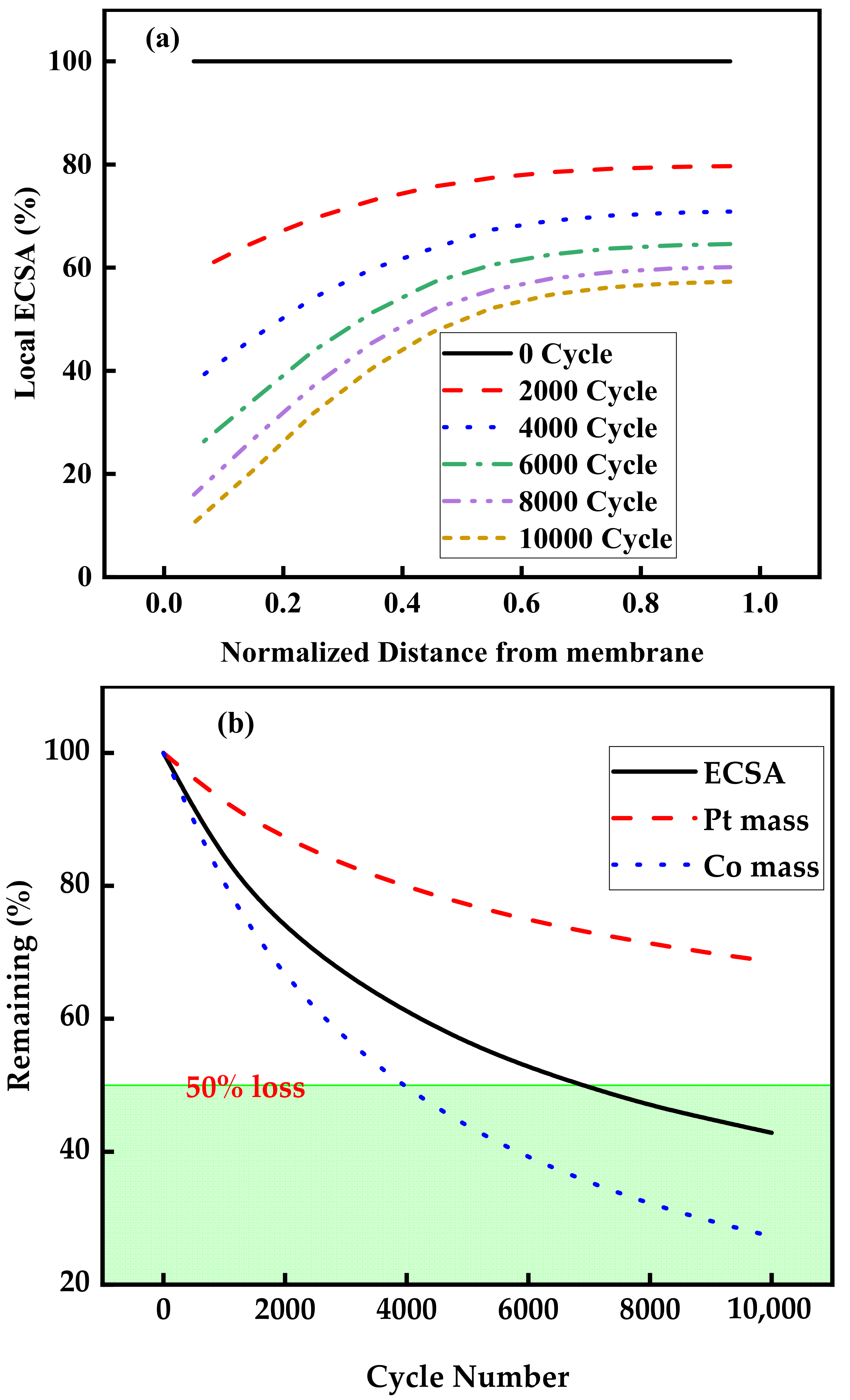
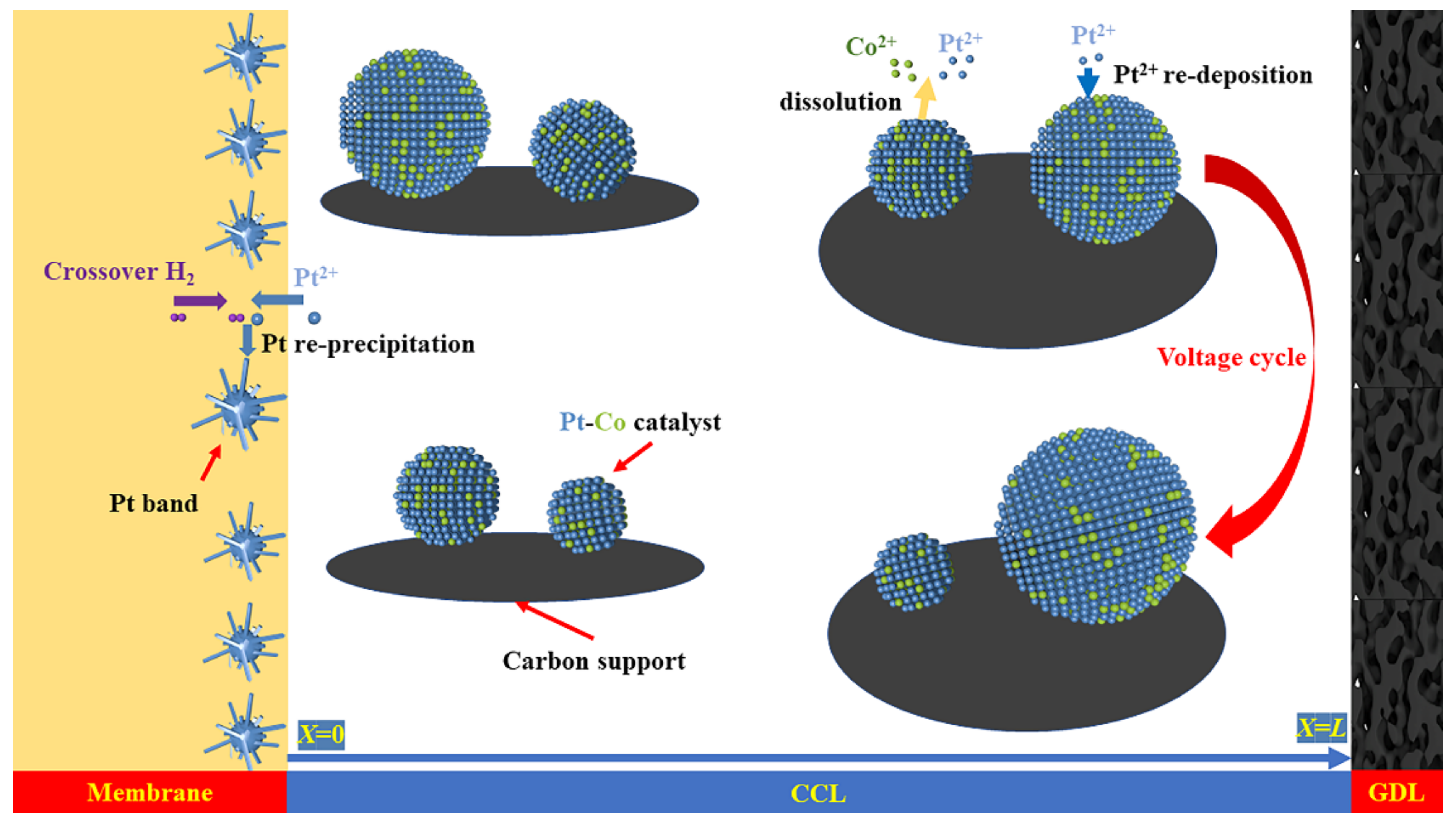
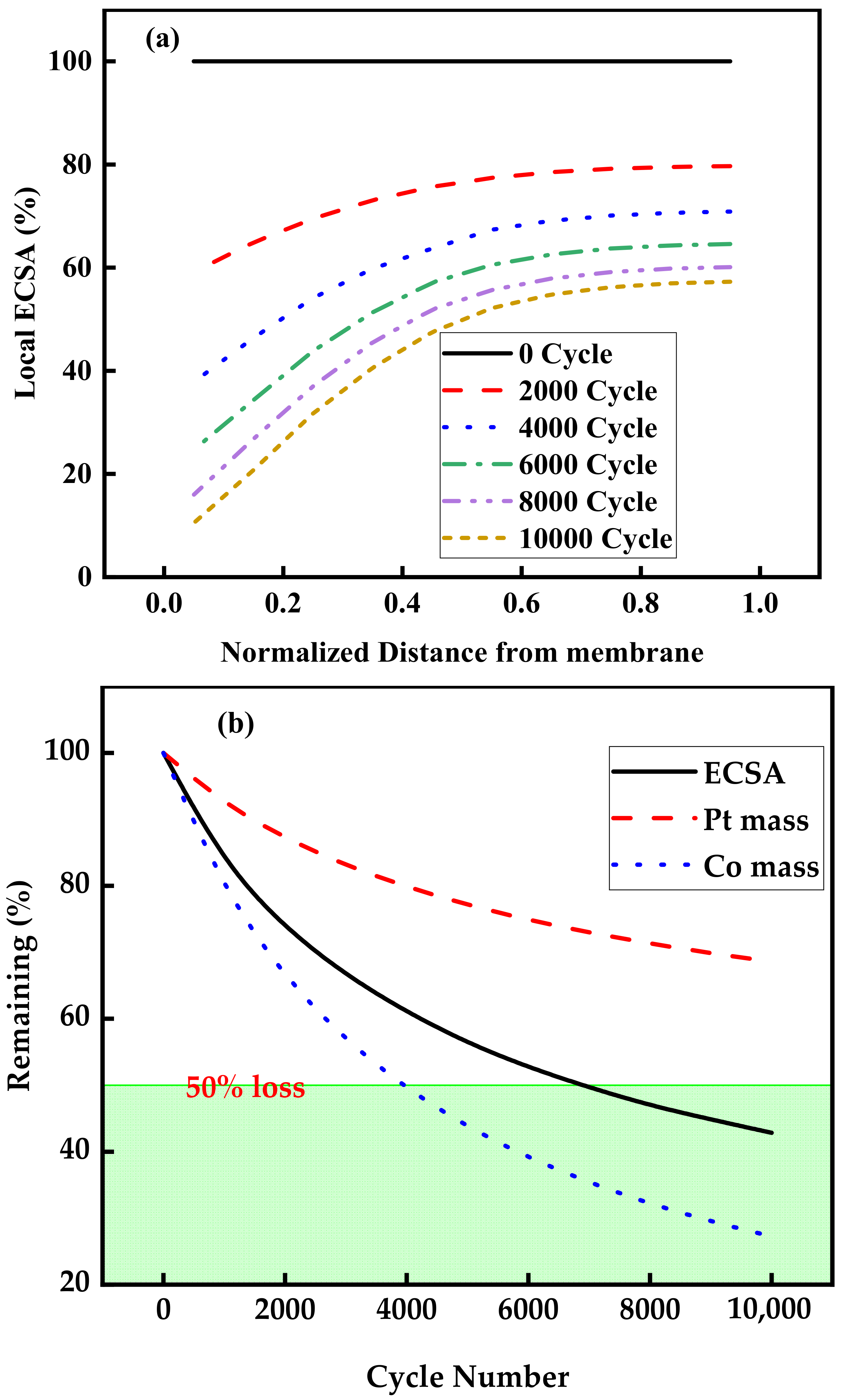
3.2. Aging of Catalysts during Voltage Cycle
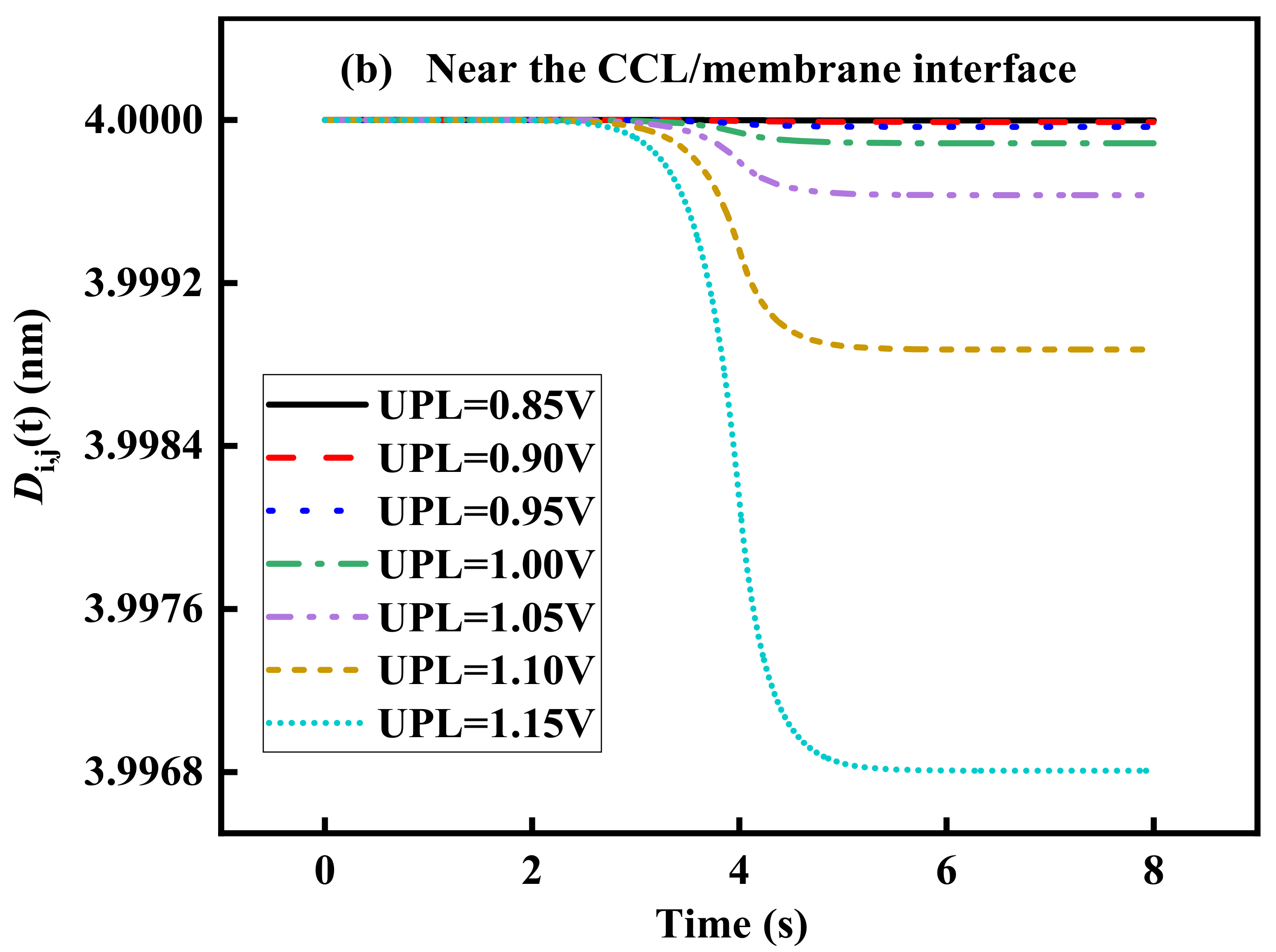
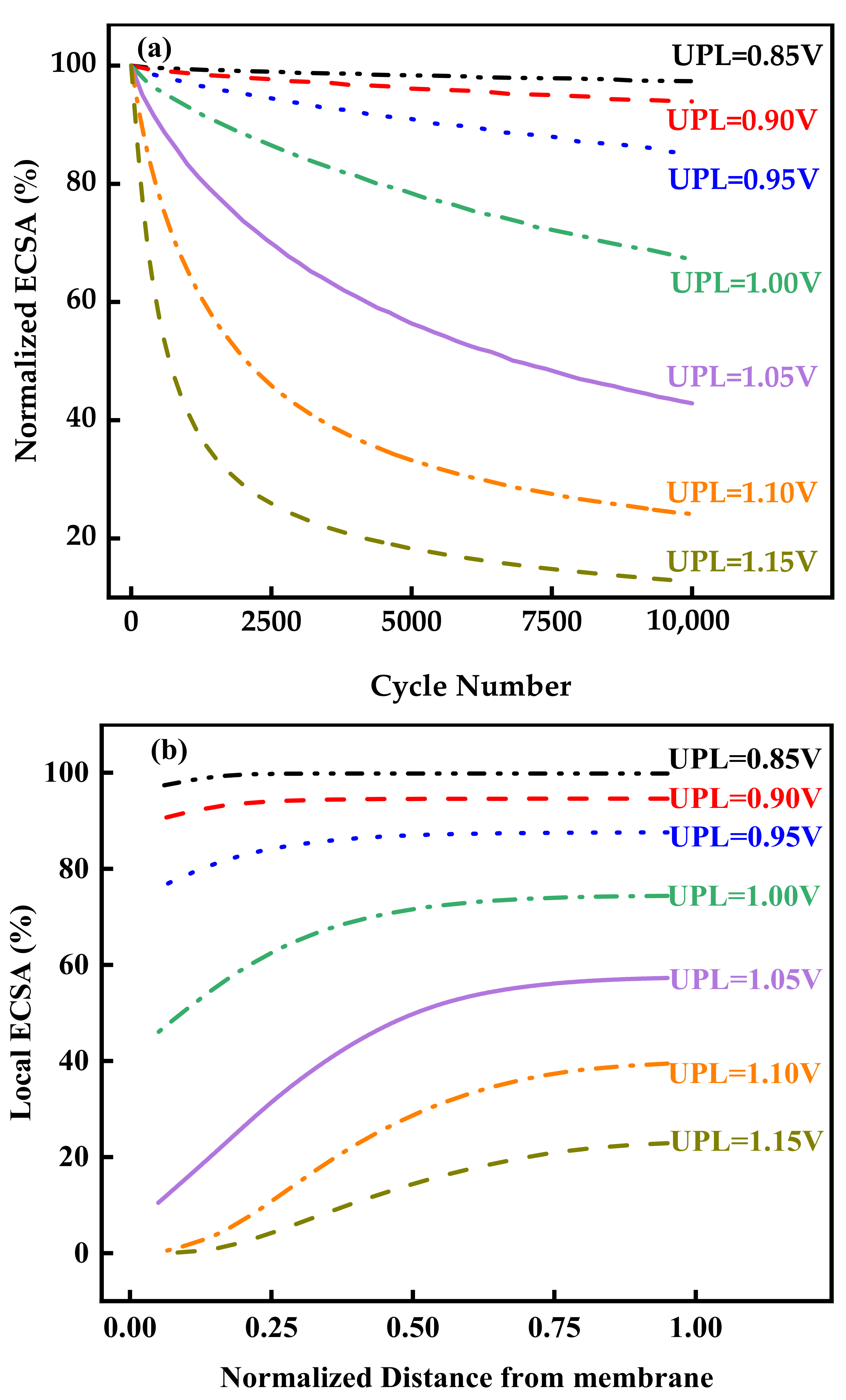
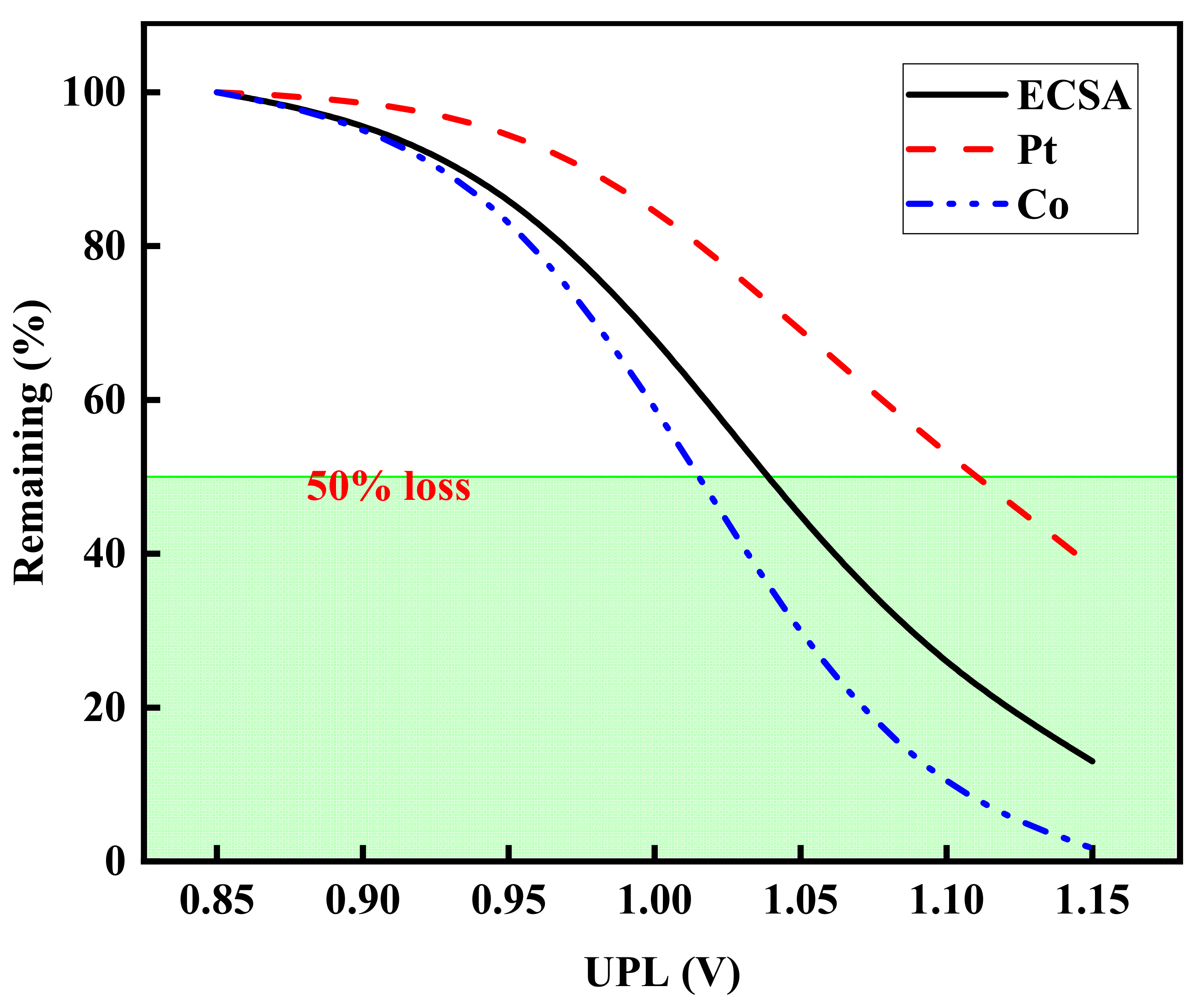
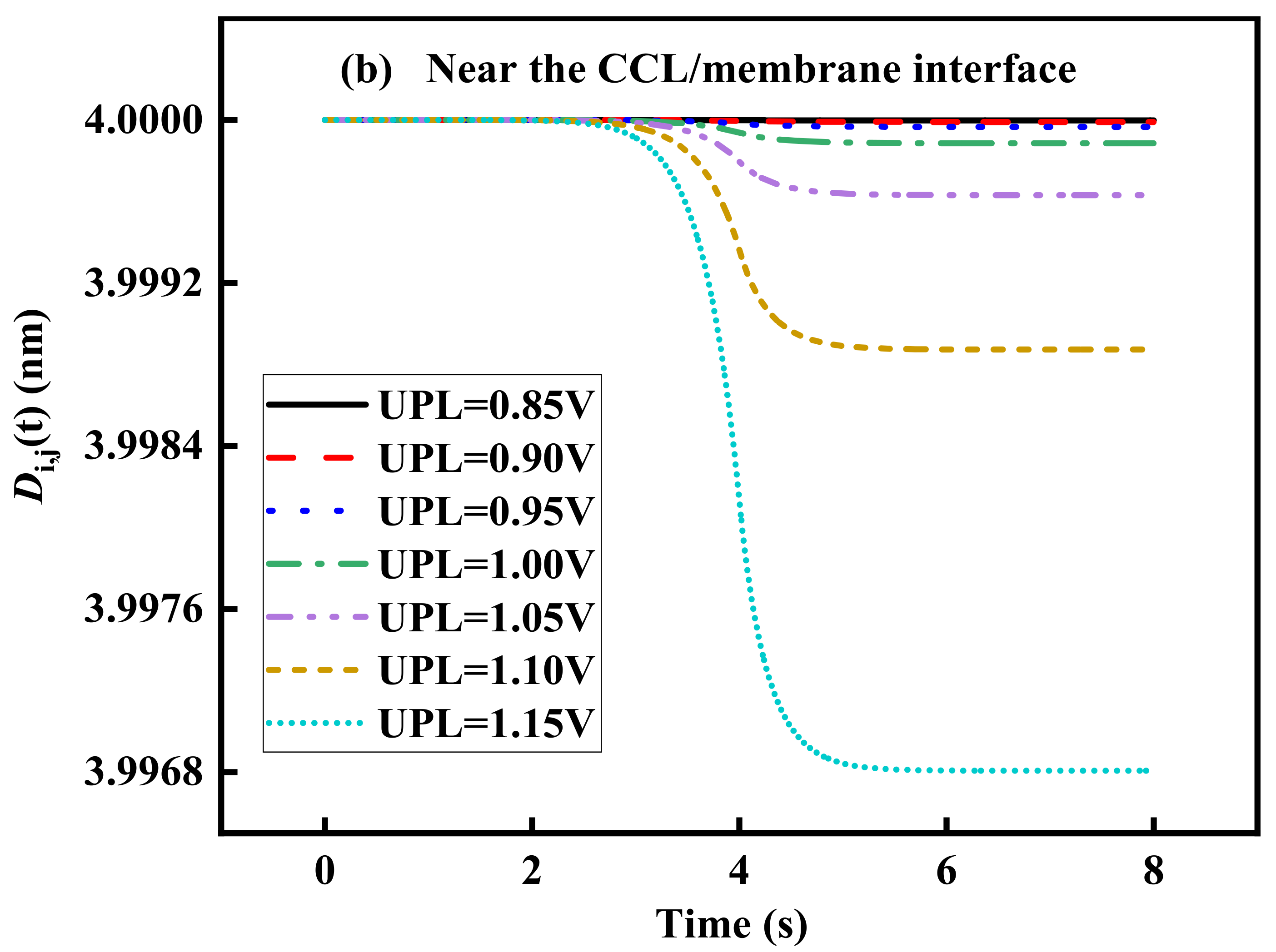
3.3. Effect of UPL
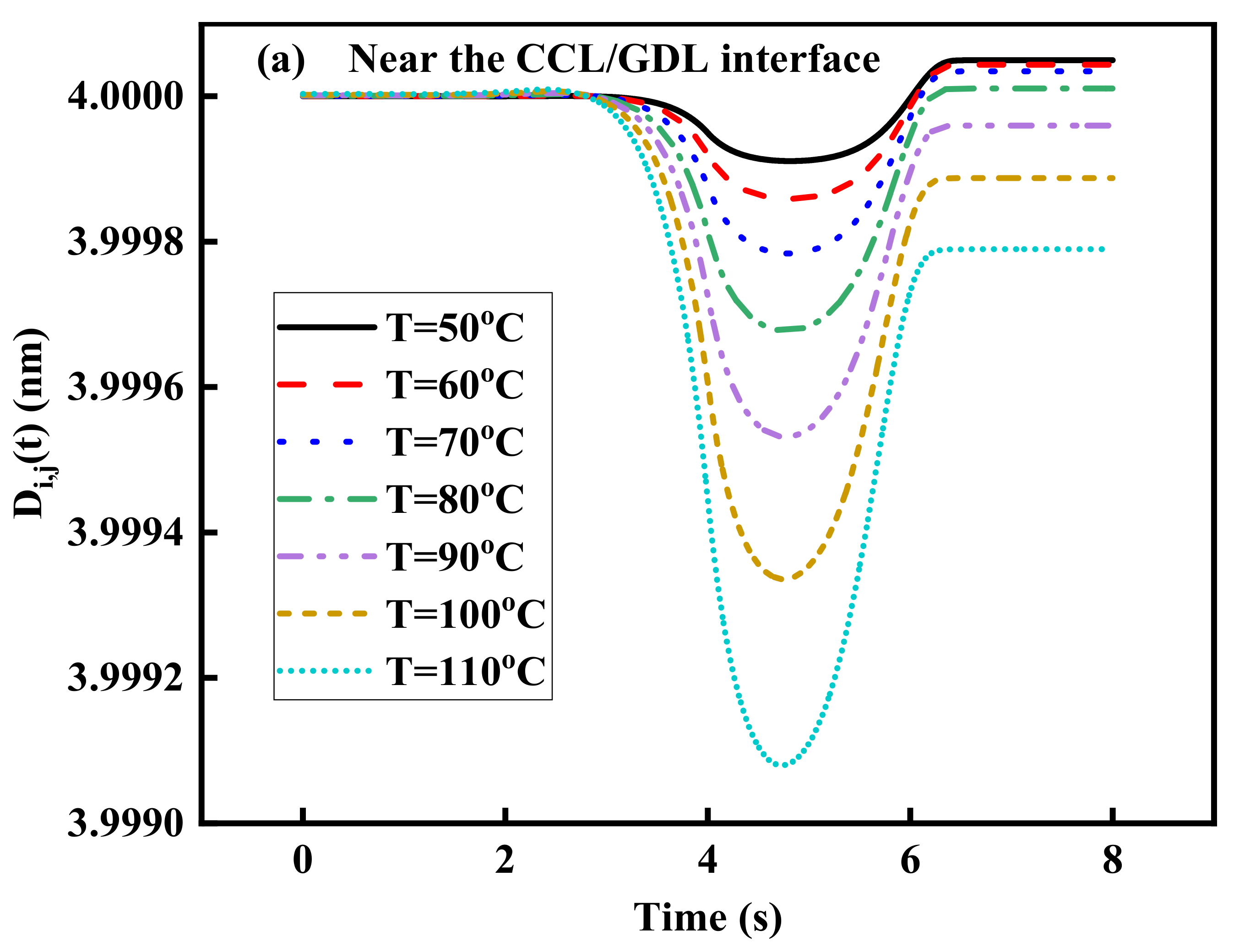
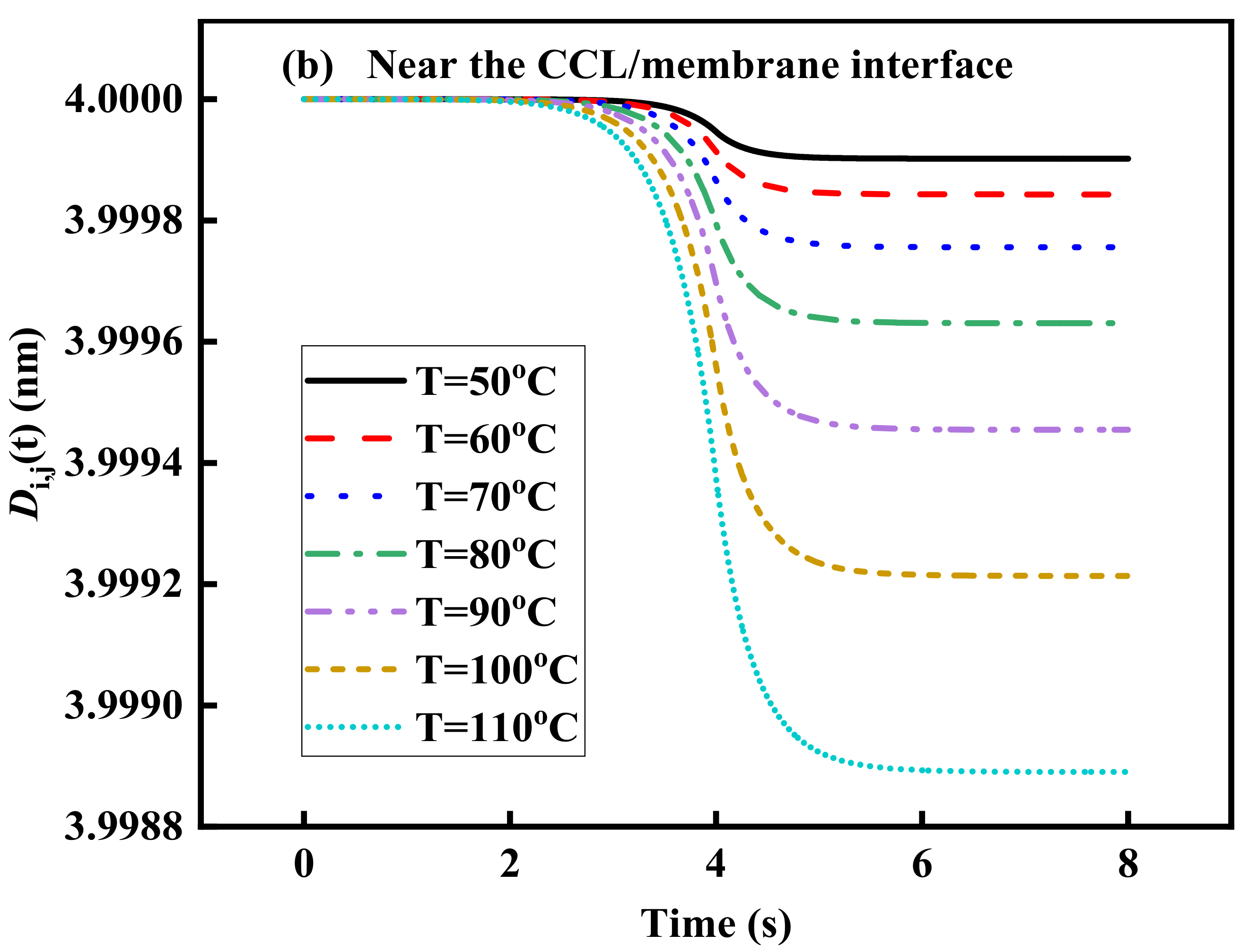
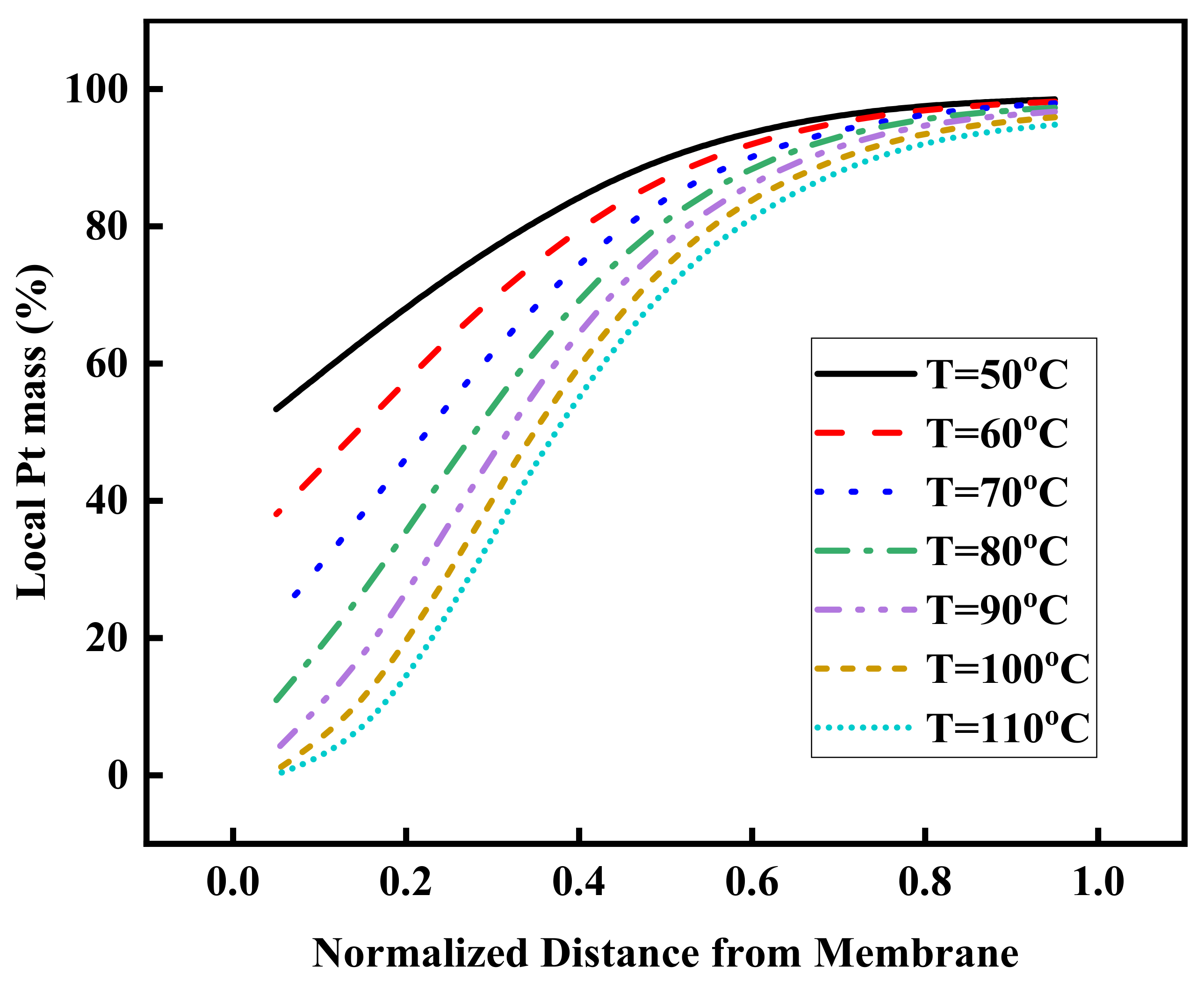
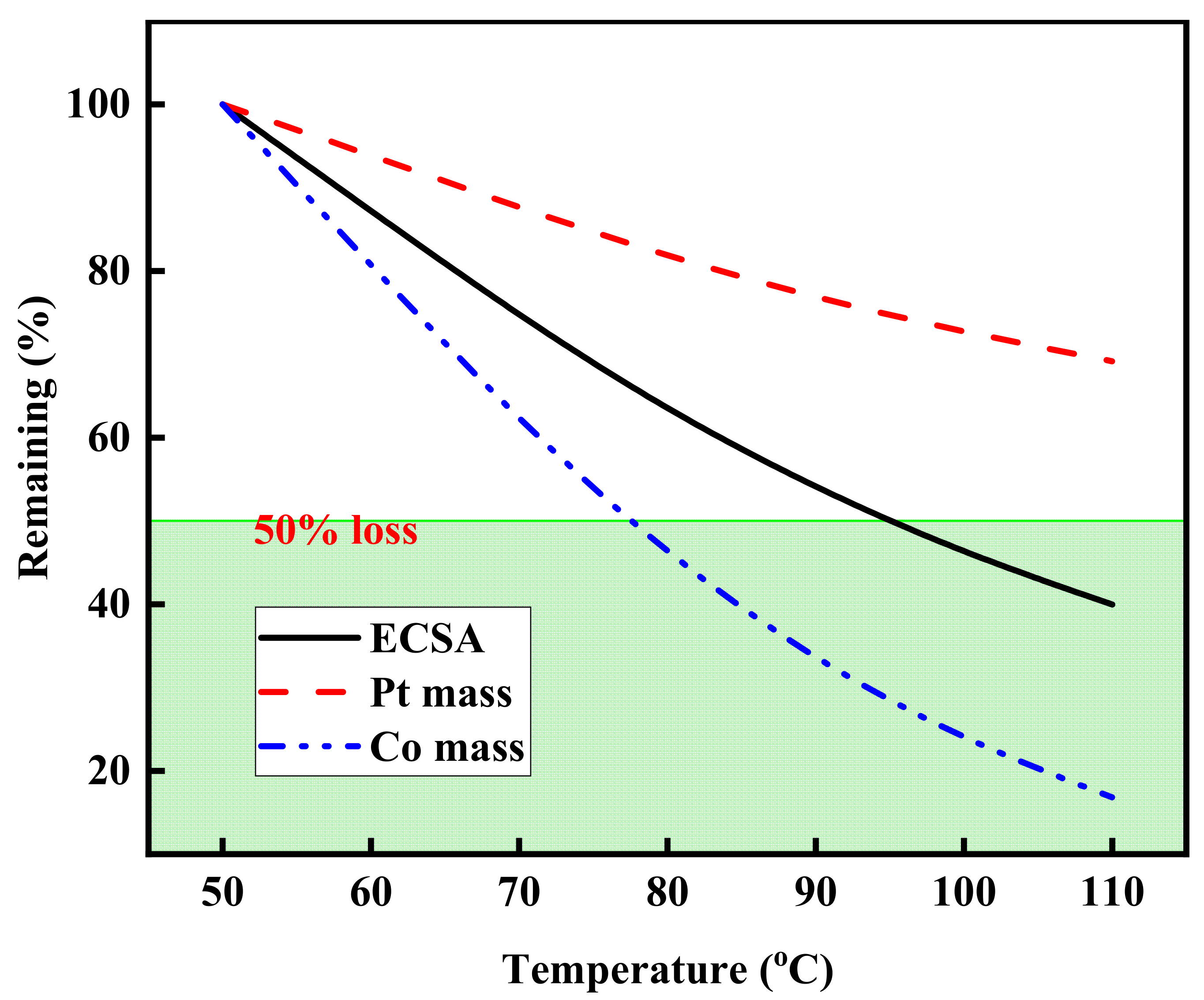
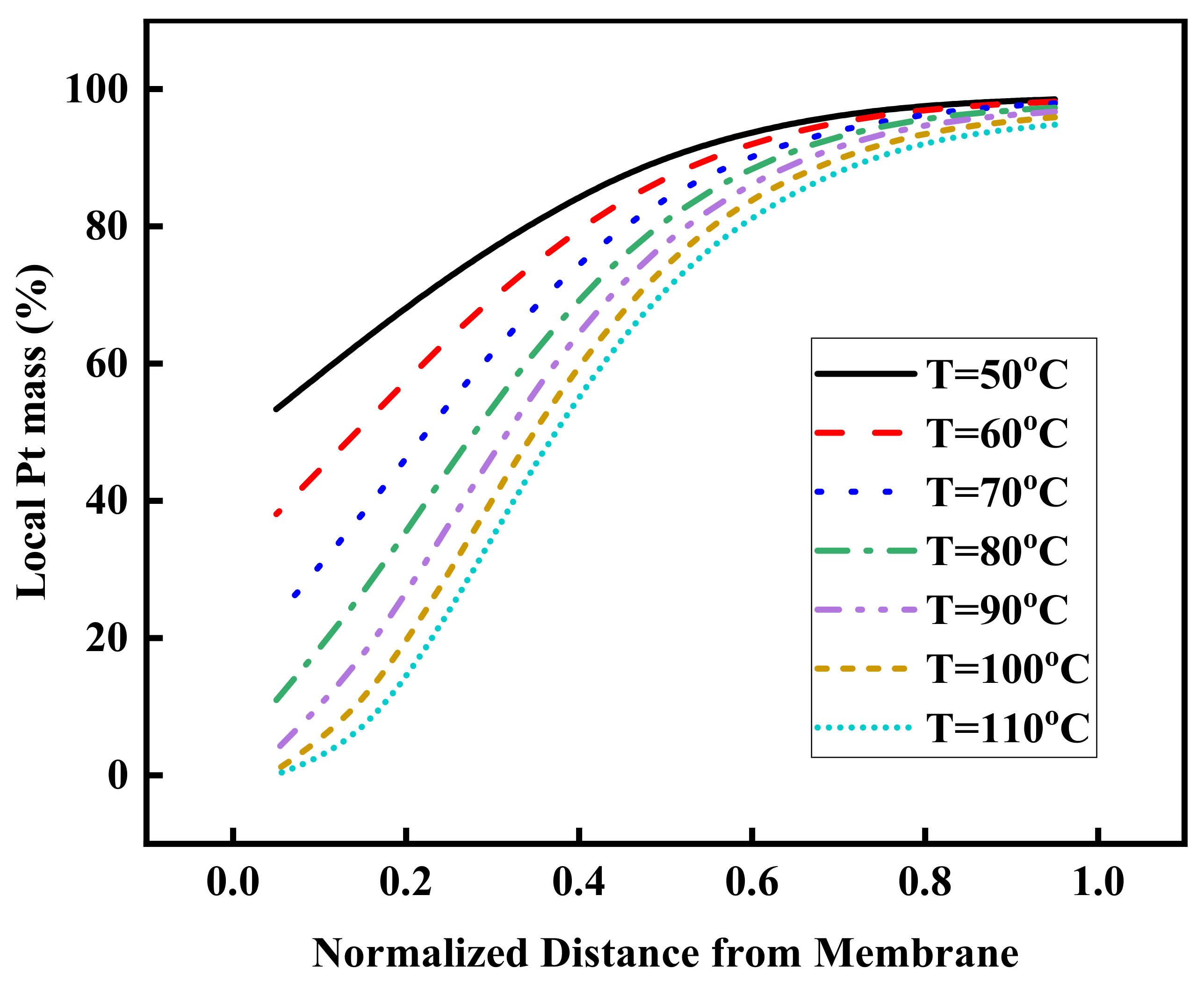
3.4. Effect of Operating Temperature
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sandbeck, D.J.S.; Secher, N.M.; Speck, F.D.; Sorensen, J.E.; Kibsgaard, J.; Chorkendorff, I.; Cherevko, S. Particle Size Effect on Platinum Dissolution: Considerations for Accelerated Stability Testing of Fuel Cell Catalysts. ACS Catal. 2020, 10, 6281–6290. [Google Scholar] [CrossRef]
- Paul, M.T.Y.; Gates, B.D. Mesoporous Platinum Prepared by Electrodeposition for Ultralow Loading Proton Exchange Membrane Fuel Cells. Sci. Rep. 2019, 9, 4161. [Google Scholar] [CrossRef] [PubMed]
- Proietti, E.; Jaouen, F.; Lefevre, M.; Larouche, N.; Tian, J.; Herranz, J.; Dodelet, J.P. Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2011, 2, 416. [Google Scholar] [CrossRef]
- Lee, W.J.; Bera, S.; Kim, C.M.; Koh, E.K.; Hong, W.P.; Oh, S.J.; Cho, E.; Kwon, S.H. Synthesis of highly dispersed Pt nanoparticles into carbon supports by fluidized bed reactor atomic layer deposition to boost PEMFC performance. Npg. Asia Mater. 2020, 12, 40. [Google Scholar] [CrossRef]
- Sun, Y.Y.; Polani, S.; Luo, F.; Ott, S.; Strasser, P.; Dionigi, F. Advancements in cathode catalyst and cathode layer design for proton exchange membrane fuel cells. Nat. Commun. 2021, 12, 5984. [Google Scholar] [CrossRef]
- Cullen, D.A.; Neyerlin, K.C.; Ahluwalia, R.K.; Mukundan, R.; More, K.L.; Borup, R.L.; Weber, A.Z.; Myers, D.J.; Kusoglu, A. New roads and challenges for fuel cells in heavy-duty transportation. Nat. Energy 2021, 6, 462–474. [Google Scholar] [CrossRef]
- Sun, C.; Zhang, H. Review of the Development of First-Generation Redox Flow Batteries: Iron-Chromium System. ChemSusChem 2022, 15, 1–15. [Google Scholar] [CrossRef]
- Jiao, K.; Xuan, J.; Du, Q.; Bao, Z.M.; Xie, B.A.; Wang, B.W.; Zhao, Y.; Fan, L.H.; Wang, H.Z.; Hou, Z.J.; et al. Designing the next generation of proton-exchange membrane fuel cells. Nature 2021, 595, 361–369. [Google Scholar] [CrossRef]
- Sun, C.; Negro, E.; Nale, A.; Pagot, G.; Vezzù, K.; Zawodzinski, T.A.; Meda, L.; Gambaro, C.; Di Noto, V. An efficient barrier toward vanadium crossover in redox flow batteries: The bilayer [Nafion/(WO3)x] hybrid inorganic-organic membrane. Electrochim. Acta 2021, 378, 138133. [Google Scholar] [CrossRef]
- Eslamibidgoli, M.J.; Huang, J.; Kadyk, T.; Malek, A.; Eikerling, M. How theory and simulation can drive fuel cell electrocatalysis. Nano Energy 2016, 29, 334–361. [Google Scholar] [CrossRef]
- Huang, J.; Eikerling, M. Modeling the oxygen reduction reaction at platinum-based catalysts: A brief review of recent developments. Curr. Opin. Electrochem. 2019, 13, 157–165. [Google Scholar] [CrossRef]
- Wang, X.X.; Swihart, M.T.; Wu, G. Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation. Nat. Catal. 2019, 2, 578–589. [Google Scholar] [CrossRef]
- Dai, S.; Hou, Y.S.; Onoue, M.; Zhang, S.Y.; Gao, W.P.; Yan, X.X.; Graham, G.W.; Wu, R.Q.; Pan, X.Q. Revealing Surface Elemental Composition and Dynamic Processes Involved in Facet-Dependent Oxidation of Pt3Co Nanoparticles via in Situ Transmission Electron Microscopy. Nano Lett. 2017, 17, 4683–4688. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.J.; la O’, G.J.; Shao-Horn, Y.; Morgan, D.; Makharia, R.; Kocha, S.; Gasteiger, H.A. Instability of Pt/C electrocatalysts in proton exchange membrane fuel cells—A mechanistic investigation. J. Electrochem. Soc. 2005, 152, A2256–A2271. [Google Scholar] [CrossRef]
- Carlton, C.E.; Chen, S.; Ferreira, P.J.; Allard, L.F.; Shao-Horn, Y. Sub-Nanometer-Resolution Elemental Mapping of “Pt3Co” Nanoparticle Catalyst Degradation in Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2012, 3, 161–166. [Google Scholar] [CrossRef]
- Chen, S.; Gasteiger, H.A.; Hayakawa, K.; Tada, T.; Shao-Horn, Y. Platinum-Alloy Cathode Catalyst Degradation in Proton Exchange Membrane Fuel Cells: Nanometer-Scale Compositional and Morphological Changes. J. Electrochem. Soc. 2010, 157, A82–A97. [Google Scholar] [CrossRef]
- Xin, H.L.; Mundy, J.A.; Liu, Z.Y.; Cabezas, R.; Hovden, R.; Kourkoutis, L.F.; Zhang, J.L.; Subramanian, N.P.; Makharia, R.; Wagner, F.T.; et al. Atomic-Resolution Spectroscopic Imaging of Ensembles of Nanocatalyst Particles Across the Life of a Fuel Cell. Nano Lett. 2012, 12, 490–497. [Google Scholar] [CrossRef]
- Yu, Y.; Xin, H.L.; Hovden, R.; Wang, D.; Rus, E.D.; Mundy, J.A.; Muller, D.A.; Abruna, H.D. Three-dimensional tracking and visualization of hundreds of Pt-Co fuel cell nanocatalysts during electrochemical aging. Nano Lett. 2012, 12, 4417–4423. [Google Scholar] [CrossRef]
- Hasché, F.; Oezaslan, M.; Strasser, P. Activity, Stability, and Degradation Mechanisms of Dealloyed PtCu3 and PtCo3 Nanoparticle Fuel Cell Catalysts. ChemCatChem 2011, 3, 1805–1813. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Ashton, S.J.; Meier, J.C.; Wiberg, G.K.H.; Hanzlik, M.; Arenz, M. Non-destructive transmission electron microscopy study of catalyst degradation under electrochemical treatment. J. Power Sources 2008, 185, 734–739. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Meier, J.C.; Ashton, S.J.; Wiberg, G.K.H.; Kraus, F.; Hanzlik, M.; Arenz, M. Fuel cell catalyst degradation on the nanoscale. Electrochem. Commun. 2008, 10, 1144–1147. [Google Scholar] [CrossRef]
- Yasuda, K.; Taniguchi, A.; Akita, T.; Ioroi, T.; Siroma, Z. Platinum dissolution and deposition in the polymer electrolyte membrane of a PEM fuel cell as studied by potential cycling. Phys. Chem. Chem. Phys. 2006, 8, 746–752. [Google Scholar] [CrossRef]
- Zhang, H.; Haas, H.; Hu, J.; Kundu, S.; Davis, M.; Chuy, C. The Impact of Potential Cycling on PEMFC Durability. J. Electrochem. Soc. 2013, 160, F840–F847. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Kinetic model of platinum dissolution in PEMFCs. J. Electrochem. Soc. 2003, 150, A1523–A1527. [Google Scholar] [CrossRef]
- Darling, R.M.; Meyers, J.P. Mathematical model of platinum movement in PEM fuel cells. J. Electrochem. Soc. 2005, 152, A242–A247. [Google Scholar] [CrossRef]
- Franco, A.A.; Tembely, M. Transient multiscale modeling of aging mechanisms in a PEFC cathode. J. Electrochem. Soc. 2007, 154, B712–B723. [Google Scholar] [CrossRef]
- Franco, A.A.; Schott, P.; Jallut, C.; Maschke, B. A dynamic mechanistic model of an electrochemical interface. J. Electrochem. Soc. 2006, 153, A1053–A1061. [Google Scholar] [CrossRef]
- Bi, W.; Fuller, T.F. Modeling of PEM fuel cell Pt/C catalyst degradation. J. Power Sources 2008, 178, 188–196. [Google Scholar] [CrossRef]
- Holby, E.F.; Sheng, W.; Shao-Horn, Y.; Morgan, D. Pt nanoparticle stability in PEM fuel cells: Influence of particle size distribution and crossover hydrogen. Energy Environ. Sci. 2009, 2, 865–871. [Google Scholar] [CrossRef]
- Holby, E.F.; Morgan, D. Application of Pt Nanoparticle Dissolution and Oxidation Modeling to Understanding Degradation in PEM Fuel Cells. J. Electrochem. Soc. 2012, 159, B578–B591. [Google Scholar] [CrossRef]
- Li, Y.; Moriyama, K.; Gu, W.; Arisetty, S.; Wang, C.Y. A One-Dimensional Pt Degradation Model for Polymer Electrolyte Fuel Cells. J. Electrochem. Soc. 2015, 162, F834–F842. [Google Scholar] [CrossRef]
- Zheng, Z.F.; Yang, F.; Lin, C.; Zhu, F.J.; Shen, S.Y.; Wei, G.H.; Zhang, J.L. Design of gradient cathode catalyst layer (CCL) structure for mitigating Pt degradation in proton exchange membrane fuel cells (PEMFCs) using mathematical method. J. Power Sources 2020, 451, 227729. [Google Scholar] [CrossRef]
- Zheng, Z.; Luo, L.; Zhu, F.; Cheng, X.; Yang, F.; Shen, S.; Wei, G.; Zhang, J. Degradation of core-shell Pt3Co catalysts in proton exchange membrane fuel cells (PEMFCs) studied by mathematical modeling. Electrochim. Acta 2019, 323, 134751. [Google Scholar] [CrossRef]
- Wang, X.P.; Kumar, R.; Myers, D.J. Effect of voltage on platinum dissolution relevance to polymer electrolyte fuel cells. Electrochem. Solid State Lett. 2006, 9, A225–A227. [Google Scholar] [CrossRef]
- Dukic, T.; Moriau, L.J.; Pavko, L.; Kostelec, M.; Prokop, M.; Ruiz-Zepeda, F.; Sala, M.; Drazic, G.; Gatalo, M.; Hodnik, N. Understanding the Crucial Significance of the Temperature and Potential Window on the Stability of Carbon Supported Pt-Alloy Nanoparticles as Oxygen Reduction Reaction Electrocatalysts. ACS Catal. 2022, 12, 101–115. [Google Scholar] [CrossRef]
- Yoshida, T.; Kojima, K. Toyota MIRAI fuel cell vehicle and progress toward a future hydrogen society. Electrochem. Soc. Interface 2015, 24, 45–49. [Google Scholar] [CrossRef]
- Lochner, T.; Kluge, R.M.; Fichtner, J.; El-Sayed, H.A.; Garlyyev, B.; Bandarenka, A.S. Temperature Effects in Polymer Electrolyte Membrane Fuel Cells. ChemElectroChem 2020, 7, 3545–3568. [Google Scholar] [CrossRef]
- Ahluwalia, R.K.; Arisetty, S.; Peng, J.-K.; Subbaraman, R.; Wang, X.; Kariuki, N.; Myers, D.J.; Mukundan, R.; Borup, R.; Polevaya, O. Dynamics of Particle Growth and Electrochemical Surface Area Loss due to Platinum Dissolution. J. Electrochem. Soc. 2014, 161, F291–F304. [Google Scholar] [CrossRef]
- Dubau, L.; Maillard, F. Unveiling the crucial role of temperature on the stability of oxygen reduction reaction electrocatalysts. Electrochem. Commun. 2016, 63, 65–69. [Google Scholar] [CrossRef]
- Gatalo, M.; Jovanovic, P.; Petek, U.; Sala, M.; Selih, V.S.; Ruiz-Zepeda, F.; Bele, M.; Hodnik, N.; Gaberscek, M. Comparison of Pt-Cu/C with Benchmark Pt-Co/C: Metal Dissolution and Their Surface Interactions. ACS Appl. Energy Mater. 2019, 2, 3131–3141. [Google Scholar] [CrossRef] [Green Version]
- Moriau, L.J.; Hrnjic, A.; Pavlisic, A.; Kamsek, A.R.; Petek, U.; Ruiz-Zepeda, F.; Sala, M.; Pavko, L.; Selih, V.S.; Bele, M.; et al. Resolving the nanoparticles’ structure-property relationships at the atomic level: A study of Pt-based electrocatalysts. IScience 2021, 24, 102102. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Tetteh, E.B.; Lee, H.Y.; Kang, T.H.; Yu, J.S. Tailor-Made Pt Catalysts with Improved Oxygen Reduction Reaction Stability/Durability. ACS Catal. 2019, 9, 8622–8645. [Google Scholar] [CrossRef]
- Wang, C.; Spendelow, J.S. Recent developments in Pt–Co catalysts for proton-exchange membrane fuel cells. Curr. Opin. Electrochem. 2021, 28, 100715. [Google Scholar] [CrossRef]
- Colón-Mercado, H.R.; Popov, B.N. Stability of platinum based alloy cathode catalysts in PEM fuel cells. J. Power Sources 2006, 155, 253–263. [Google Scholar] [CrossRef]
- Han, A.; Fu, C.; Yan, X.; Chen, J.; Cheng, X.; Ke, C.; Hou, J.; Shen, S.; Zhang, J. Effect of cobalt ion contamination on proton conduction of ultrathin Nafion film. Int. J. Hydrogen Energy 2020, 45, 25276–25285. [Google Scholar] [CrossRef]
- Li, H.; Gazzarri, J.; Tsay, K.; Wu, S.H.; Wang, H.J.; Zhang, J.J.; Wessel, S.; Abouatallah, R.; Joos, N.; Schrooten, J. PEM fuel cell cathode contamination in the presence of cobalt ion (Co2+). Electrochim. Acta 2010, 55, 5823–5830. [Google Scholar] [CrossRef]
- Braaten, J.P.; Xu, X.M.; Cai, Y.; Kongkanand, A.; Litster, S. Contaminant Cation Effect on Oxygen Transport through the Ionomers of Polymer Electrolyte Membrane Fuel Cells. J. Electrochem. Soc. 2019, 166, F1337–F1343. [Google Scholar] [CrossRef]
- Papadias, D.D.; Ahluwalia, R.K.; Kariuki, N.; Myers, D.; More, K.L.; Cullen, D.A.; Sneed, B.T.; Neyerlin, K.C.; Mukundan, R.; Borup, R.L. Durability of Pt-Co Alloy Polymer Electrolyte Fuel Cell Cathode Catalysts under Accelerated Stress Tests. J. Electrochem. Soc. 2018, 165, F3166–F3177. [Google Scholar] [CrossRef]
- Wang, Y.; Seo, B.; Wang, B.; Zamel, N.; Jiao, K.; Adroher, X.C. Fundamentals, materials, and machine learning of polymer electrolyte membrane fuel cell technology. Energy AI 2020, 1, 100014. [Google Scholar] [CrossRef]
- Tang, L.; Han, B.; Persson, K.; Friesen, C.; He, T.; Sieradzki, K.; Ceder, G. Electrochemical Stability of Nanometer-Scale Pt Particles in Acidic Environments. J. Am. Chem. Soc. 2010, 132, 596–600. [Google Scholar] [CrossRef]
- Uchimura, M.; Sugawara, S.; Suzuki, Y.; Zhang, J.B.; Kocha, S.S. Electrocatalyst Durability under Simulated Automotive Drive Cycles. ECS Trans. 2008, 16, 225–234. [Google Scholar] [CrossRef]
- Yang, Y.J.; Bai, M.L.; Lv, J.Z.; Gao, L.S.; Li, Y.; Lv, X.C.; Li, Y.B.; Song, Y.C. One-Dimensional Modeling for Aging of Pt-Co Core-Shell Catalysts in Proton Exchange Membrane Fuel Cells. J. Electrochem. Soc. 2022, 169, 054526. [Google Scholar] [CrossRef]
- Oezaslan, M.; Hasché, F.; Strasser, P. Pt-Based Core–Shell Catalyst Architectures for Oxygen Fuel Cell Electrodes. J. Phys. Chem. Lett. 2013, 4, 3273–3291. [Google Scholar] [CrossRef]
- Jomori, S.; Nonoyama, N.; Yoshida, T. Analysis and modeling of PEMFC degradation: Effect on oxygen transport. J. Power Sources 2012, 215, 18–27. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Value | Units | Description |
|---|---|---|---|
| 1 × 104 | Hz | Forward Pt oxide formation rate [30] | |
| 2 × 10−2 | Hz | Backward Pt oxide formation rate [30] | |
| 2.2 × 10−9 | mol/cm2 | Pt surface site density [30] | |
| 1.2 × 104 | J/mol | Partial molar oxide formation activation enthalpy (0 coverage) [30] | |
| 0.5 | (-) | Butler–Volmer transfer coefficient for Pt oxide formation [30] | |
| n | 2 | (-) | Electrons transferred during Pt dissolution [30] |
| 2 | (-) | Electrons transferred during Pt oxide formation [30] | |
| 1.03 | V | Pt oxide formation bulk equilibrium voltage [30] | |
| 2.0 × 104 | J/mol | Pt oxide dependent kinetic barrier constant [30] | |
| 5.0 × 104 | J/mol | Pt oxide-oxide interaction energy [30] | |
| 1 × 104 | Hz | Dissolution attempt frequency [30] | |
| 8 × 105 | Hz | Backward dissolution rate factor [30] | |
| 0.5 | (-) | Butler–Volmer transfer coefficient for Pt dissolution [30] | |
| 1.118 | V | Pt dissolution bulk equilibrium voltage [30] | |
| ΩPt | 9.09 | cm3/mol | Molar volume of Pt [30] |
| ΩCo | 6.62 | cm3/mol | Molar volume of Co |
| 2.4 × 10−4 | J/cm2 | Pt [1 1 1] surface tension [30] | |
| 4.0 × 10−3 | mol/L | Reference Pt2+ concentration | |
| 4.1 × 104 | J/mol | Pt dissolution activation enthalpy under fully humidified condition | |
| 0.2 | (-) | Volume fraction of the ionomer increment in cathode electrode [14] | |
| L | 5.0 × 10−4 | cm | Thickness of CCL [16] |
| ni, j | 4.7 | (-) | Atomic ratio of Pt and Co in the catalyst (initial) [16] |
| DPt2+ | 2 × 10−8 | cm2/s | Diffusion coefficient of Pt2+ through ionomer |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Bai, M.; Su, L.; Lv, J.; Hu, C.; Gao, L.; Li, Y.; Li, Y.; Song, Y. One-Dimensional Numerical Simulation of Pt-Co Alloy Catalyst Aging for Proton Exchange Membrane Fuel Cells. Sustainability 2022, 14, 11462. https://doi.org/10.3390/su141811462
Yang Y, Bai M, Su L, Lv J, Hu C, Gao L, Li Y, Li Y, Song Y. One-Dimensional Numerical Simulation of Pt-Co Alloy Catalyst Aging for Proton Exchange Membrane Fuel Cells. Sustainability. 2022; 14(18):11462. https://doi.org/10.3390/su141811462
Chicago/Turabian StyleYang, Yunjie, Minli Bai, Laisuo Su, Jizu Lv, Chengzhi Hu, Linsong Gao, Yang Li, Yubai Li, and Yongchen Song. 2022. "One-Dimensional Numerical Simulation of Pt-Co Alloy Catalyst Aging for Proton Exchange Membrane Fuel Cells" Sustainability 14, no. 18: 11462. https://doi.org/10.3390/su141811462
APA StyleYang, Y., Bai, M., Su, L., Lv, J., Hu, C., Gao, L., Li, Y., Li, Y., & Song, Y. (2022). One-Dimensional Numerical Simulation of Pt-Co Alloy Catalyst Aging for Proton Exchange Membrane Fuel Cells. Sustainability, 14(18), 11462. https://doi.org/10.3390/su141811462