Complexity Stage Model of the Medical Device Development Based on Economic Evaluation—MedDee

 , , , , and
, , , , and 
Abstract
1. Introduction
2. Theoretical Background
2.1. MD Development Stages
- Step1: Initiation
- Step 2: Concept proposing
- Step 3: Design and development
- Step 4: Verification and validation
- Step 5: Production
- Step 6: Market device deployment
2.2. Economic Evaluation Methods
2.3. Legislative Environment
3. Methods
3.1. Analysis and Comparison of Available MD Development Phases and Investment Evaluation Methods
3.2. Evaluation of the Stage Investment Model
- (a)
- The preliminary assessment of the MD investment practices and procedures from seven interviewees who actively cooperate with researchers (5) and companies (2).
- (b)
- The detection of the team members involved in the evaluation of prospective investments.
4. Results
4.1. Phases and Processes of the Model of the Medical Device Development
4.1.1. Initiation
- Active implantable devices;
- Active non-implantable devices for imaging, monitoring and/or diagnosis;
- Active non-implantable therapeutic devices and general active non-implantable devices;
- Non-active implants and long-term surgically invasive devices;
- Non-active, non-implantable devices;
- Specifics of MDs;
- Technologies for MDs—auditing.
- -
- setting an appropriate time horizon for the economic evaluation;
- -
- identification of all types of resources needed for the use of technology and comparators;
- -
- calculation of the unit costs of each resource and the total costs of public health insurance and;
- -
- costs from a societal perspective;
- -
- indicate the costs that have implications for non-medical budgets (generally social costs).
4.1.2. Concept Proposal
4.1.3. Design and Development
- “Product design, design verification, and validation (determination of the tests to be carried out before and after the design freeze).
- Regulatory—verification of the protection of the technology, and possibly specification of accompanying documentation”.
- Purchase and validation strategy (technical solutions to fulfil customer requirements, specification of all key parameters’ specifications, design of production technologies, materials design, supplier decisions).
- Regulatory (post market clinical follow up (PMCF) plan, post-performance qualification, clinical evaluation plan).
- Risk management design failure mode and effects analysis (dFMEA), update advanced failure modes and effects analysis (aFMEA).
- Technical specifications, block diagram, eclectic diagram, detailed design hardware(HW), software (SW), mechanical design, prototype.
- Testing.
4.1.4. Verification and Validation
4.1.5. Production
- -
- determination of all direct and indirect costs for the rated technology and comparators, a summary of the costs related to public health insurance;
- -
- benefits for the rated technology and comparators;
- -
- the resulting ICER value in terms of the costs of the public health insurance payer and then also of the society-wide perspective, taking into account all direct and indirect costs;
- -
- direct costs of the patient and his/her family (out of pocket money, co-payments). Although these costs are included in the ICER calculation from a societal perspective, their amount may influence the decision on the cost-effectiveness of the technology or the amount of the patient’s supplement;
- -
- sensitivity analyzes.
4.1.6. Market Device Deployment
4.2. Economic Methods of Investment Efficiency Evaluation for Individual Stages of MD Development
4.3. Qualitative Investigation—Evaluation of the Proposed Meddee Model
4.4. Economic Evaluation of the MD Development—A Case Study
5. Discussion
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| MD according to the Risk | Classification Rules |
|---|---|
| Class I—Provided sterile and/or have a measuring function (low/medium risk) or reusable low risk Class I devices placed on the market in sterile condition, have a measuring function, or are reusable surgical instruments: Assessment of the technical documentation relating only to those specific features of the device, e.g., sterility, measurement, or reprocessing. | Rule 1—Non-invasive devices. Rule 2—Non-invasive devices intended for channelling or storing (now includes cells). Rule 4—Non-invasive devices in contact with injured skin or mucous membrane. Rule 5—Devices invasive in body orifices. Rule 6—Surgically invasive devices for transient use. Rule 10—Active devices for diagnosis and monitoring, emit ionising radiation. Rule 11—Software intended to provide information used to make decisions with diagnosis or therapeutic purposes (from class I to class III). Rule 13—All other active devices. |
| Class IIa (medium risk) Class IIa devices: Assessment of the technical documentation for at least one representative device for each category of devices. | Rule 2—Non-invasive devices intended for channelling or storing (now includes cells). Rule 3—Non-invasive devices that modify biological or chemical. composition of blood, body liquids, other liquids and cells. Rule 4—Non-invasive devices in contact with injured skin or mucous membrane. Rule 5—Devices invasive in body orifices. Rule 6—Surgically invasive devices for transient use. Rule 7—Surgically invasive devices for short-term use. Rule 8—Surgically invasive devices for long-term use and implantable (including any device administering medicinal products, surgical mesh or spinal disc). Rule 10—Active devices for diagnosis and monitoring, emit ionising radiation. Rule 11—Software intended to provide information used to make decisions with diagnosis or therapeutic purposes (from class I to class III). Rule 12—Active devices intended to administer and/or remove medicinal products, body liquids or other substances. Rule 17—Devices specifically intended for recording of diagnostic images generated by X-ray radiation. Rule 19—Devices incorporating or consisting of nanomaterial. Rule 20—Invasive devices with respect to body orifices to administer medicinal products by inhalation. Rule 21—Substances or of combinations of substances that are intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed. |
| Class IIb (medium/high risk) Class IIb implantable devices (except sutures, staples, dental filings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips, and connectors) and class IIb active devices intended to administer and/or remove a medicinal product: Assessment of the technical documentation for every device. All other class IIb devices: Assessment of the technical documentation for at least one representative device per generic device group | Rule 2—Non-invasive devices intended for channelling or storing (now includes cells). Rule 3—Non-invasive devices that modify biological or chemical composition of blood, body liquids, other liquids and cells. Rule 4—Non-invasive devices in contact with injured skin or mucous membrane. Rule 5—Devices invasive in body orifices. Rule 6—Surgically invasive devices for transient use. Rule 7—Surgically invasive devices for short-term use. Rule 8—Surgically invasive devices for long-term use and implantable (including any device administering medicinal products, surgical mesh or spinal disc). Rule 9—Active therapeutic devices intended to exchange or administer energy. Rule 10—Active devices for diagnosis and monitoring, emit ionising radiation. Rule 11—Software intended to provide information used to make decisions with diagnosis or therapeutic purposes (from class I to class III). Rule 12—Active devices intended to administer and/or remove medicinal products, body liquids, or other substances. Rule 15—Contraception or prevention of the transmission of sexually transmitted diseases. Rule 16—Specific disinfecting, cleaning, and rinsing devices. Rule 19—Devices incorporating or consisting of nanomaterial. Rule 20—Invasive devices with respect to body orifices to administer medicinal products by inhalation. Rule 21—Substances or of combinations of substances intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed. |
| Class III (high risk). Class III devices: Assessment of the technical documentation for every device | Rule 2—Non-invasive devices intended for channelling or storing (now includes cells). Rule 3—Non-invasive devices that modify biological or chemical composition of blood, body liquids, other liquids and cells. Rule 6—Surgically invasive devices for transient use. Rule 7—Surgically invasive devices for short-term use. Rule 8—Surgically invasive devices for long-term use and implantable (including any device administering medicinal products, surgical mesh or spinal disc). Rule 9—Active therapeutic devices intended to exchange or administer energy. Rule 11—Software intended to provide information used to make decisions with diagnosis or therapeutic purposes (from class I to class III). Rule 14—Devices incorporating a medicinal substance including human blood or plasma. Devices used for contraception or prevention of sexually transmitted diseases. Rule 15—Contraception or prevention of the transmission of sexually transmitted diseases. Rule 18—Devices utilising non-viable tissues or cells of human origin or tissues of animal or derivatives. Four new rules: Rule 19—Devices incorporating or consisting of nanomaterial. Rule 21—Substances or of combinations of substances intended to be introduced into the human body via a body orifice or applied to the skin and that are absorbed. Rule 22—Active therapeutic devices with an integrated or incorporated diagnostic function that significantly determines the patient management. |
References
- Lee, M.; Yoon, Y.; Ryu, G.H.; Bok, H.S.; Yoon, K.; Park, S.; Lee, K.-S. Innovative Distribution Priorities for the Medical Devices Industry in the Fourth Industrial Revolution. Int. Neurourol. J. 2018, 22, S83–S90. [Google Scholar] [CrossRef]
- Lee, M.; Park, S.; Lee, K.-S. What are the features of successful medical device start-ups? Evidence from KOREA. Sustainability 2019, 11, 1948. [Google Scholar] [CrossRef]
- Juanola-Feliu, E.; Colomer-Farrarons, J.; Miribel-Català, P.; Samitier, J.; Valls-Pasola, J. Market challenges facing academic research in commercializing nano-enabled implantable devices for in-vivo biomedical analysis. Technovation 2012, 32, 193–204. [Google Scholar] [CrossRef]
- Vernon, J.A.; Goldberg, R.; Golec, J. Economic Evaluation and Cost-Effectiveness Thresholds: Signals to Firms and Implications for R&D Investment and Innovation. Pharmacoeconomics 2009, 27, 797–806. [Google Scholar] [PubMed]
- Freiberg, F.; Scholz, P. Evaluation of Investment in Modern Manufacturing Equipment Using Discrete Event Simulation. Procedia Econ. Financ. 2015, 34, 217–224. [Google Scholar] [CrossRef]
- Kiliç, M.; Kaya, İ. The prioritisation of provinces for public grants allocation by a decision-making methodology based on type-2 fuzzy sets. Urban Stud. 2016, 53, 755–774. [Google Scholar] [CrossRef]
- Vallejo-Torres, L.; Steuten, L.; Parkinson, B.; Girling, A.J.; Buxton, M.J. Integrating Health Economics Into the Product Development Cycle: A Case Study of Absorbable Pins for Treating Hallux Valgus. Med. Decis. Mak. 2011, 31, 596–610. [Google Scholar] [CrossRef]
- Money, A.G.; Barnett, J.; Kuljis, J.; Craven, M.P.; Martin, J.L.; Young, T. The role of the user within the medical device design and development process: medical device manufacturers’ perspectives. BMC Med. Inf. Decis. Mak. 2011, 11, 15. [Google Scholar] [CrossRef]
- IJzerman, M.J.; Koffijberg, H.; Fenwick, E.; Krahn, M. Emerging Use of Early Health Technology Assessment in Medical Product Development: A Scoping Review of the Literature. Pharmacoeconomics 2017, 35, 727–740. [Google Scholar] [CrossRef]
- Johal, S.S.; Oliver, P.; Williams, H.C. Better decision making for evaluating new medical device projects: A real options approach. J. Med. Mark. 2008, 8, 101–112. [Google Scholar] [CrossRef]
- MacKeigan, L.D.; Bootman, J.L. A Review of Cost-Benefit and Cost-Effectiveness Analyses of Clinical Pharmacy Services. J. Pharm. Mark. Manag. 1988, 2, 63–84. [Google Scholar] [CrossRef] [PubMed]
- Danner, M.; Hummel, J.M.; Volz, F.; van Manen, J.G.; Wiegard, B.; Dintsios, C.-M.; Bastian, H.; Gerber, A.; IJzerman, M.J. Integrating patients’ views into health technology assessment: Analytic hierarchy process (AHP) as a method to elicit patient preferences. Int. J. Technol. Assess. Health Care 2011, 27, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Akoka, J.; Leune, B.; Koster, A. An expert system for feasibility assessment of product development. Expert Syst. Appl. 1994, 7, 291–303. [Google Scholar] [CrossRef]
- Winterhalter, S.; Zeschky, M.B.; Neumann, L.; Gassmann, O. Business Models for Frugal Innovation in Emerging Markets: The Case of the Medical Device and Laboratory Equipment Industry. Technovation 2017, 66–67, 3–13. [Google Scholar] [CrossRef]
- Ball, G.P.; Shah, R.; Donohue, K. The decision to recall: A behavioral investigation in the medical device industry. J. Oper. Manag. 2018, 62, 1–15. [Google Scholar] [CrossRef]
- Martin, J.L.; Clark, D.J.; Morgan, S.P.; Crowe, J.A.; Murphy, E. A user-centred approach to requirements elicitation in medical device development: a case study from an industry perspective. Appl. Ergon. 2012, 43, 184–190. [Google Scholar] [CrossRef]
- Panescu, D. Medical device development. In Proceedings of the 2009 Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Minneapolis, MN, USA, 2–6 September 2009; pp. 5591–5594. [Google Scholar]
- Pietzsch, J.B.; Shluzas, L.A.; Paté-Cornell, M.E.; Yock, P.G.; Linehan, J.H. Stage-Gate Process for the Development of Medical Devices. J. Med. Devices 2009, 3, 021004. [Google Scholar] [CrossRef]
- Tomaskova, H.; Maresova, P.; Penhaker, M.; Augustynek, M.; Klimova, B.; Fadeyi, O.; Kuca, K. The Business Process Model and Notation of Open Innovation: The Process of Developing Medical Instrument. Open Innov. Technol. Mark. Complex. 2019, 5, 101. [Google Scholar] [CrossRef]
- Medina, L.A.; Kremer, G.E.O.; Wysk, R.A. Supporting medical device development: a standard product design process model. J. Eng. Des. 2013, 24, 83–119. [Google Scholar] [CrossRef]
- Shah, S.G.S.; Robinson, I.; AlShawi, S. Developing medical device technologies from users’ perspectives: A theoretical framework for involving users in the development process. Int. J. Technol. Assess. Health Care 2009, 25, 514–521. [Google Scholar] [CrossRef]
- Songkajorn, Y.; Thawesaengskulthai, N. Medical Device Innovation Development Process. Int. J. Innov. Technol. Manag. 2014, 11, 1450027. [Google Scholar] [CrossRef]
- Korol, T. Early warning models against bankruptcy risk for Central European and Latin American enterprises. Econ. Model. 2013, 31, 22–30. [Google Scholar] [CrossRef]
- Sun, J.; Shang, Z.; Li, H. Imbalance-oriented SVM methods for financial distress prediction: a comparative study among the new SB-SVM-ensemble method and traditional methods. J. Oper. Res. Soc. 2014, 65, 1905–1919. [Google Scholar] [CrossRef]
- Ashby, R.L.; Gabe, R.; Ali, S.; Saramago, P.; Chuang, L.-H.; Adderley, U.; Bland, J.M.; Cullum, N.A.; Dumville, J.C.; Iglesias, C.P.; et al. VenUS IV (Venous leg Ulcer Study IV)—Compression hosiery compared with compression bandaging in the treatment of venous leg ulcers: a randomised controlled trial, mixed-treatment comparison and decision-analytic model. Health Technol. Assess. 2014, 18, 1–294. [Google Scholar] [CrossRef] [PubMed]
- Downing, A.; Morris, E.J.; Corrigan, N.; Sebag-Montefiore, D.; Finan, P.J.; Thomas, J.D.; Chapman, M.; Hamilton, R.; Campbell, H.; Cameron, D.; et al. High hospital research participation and improved colorectal cancer survival outcomes: a population-based study. Gut 2017, 66, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Heintz, U.; Schlichting, I. Blue light-induced LOV domain dimerization enhances the affinity of Aureochrome 1a for its target DNA sequence. eLife 2016, 5, e11860. [Google Scholar] [CrossRef]
- Castañeda-Orjuela, C.; García-Molina, M.; De la Hoz-Restrepo, F. Is There Something Else Beyond Cost-Effectiveness Analysis for Public Health Decision Making? Value Health Reg. Issues 2020, 23, 1–5. [Google Scholar] [CrossRef]
- Dozet, A.; Ivarsson, B.; Eklund, K.; Klefsgård, R.; Geijer, M. Radiography on wheels arrives to nursing homes - an economic assessment of a new health care technology in southern Sweden: Mobile radiography in nursing homes. J. Eval. Clin. Pract. 2016, 22, 994–1001. [Google Scholar] [CrossRef]
- Brockis, E.; Marsden, G.; Cole, A.; Devlin, N. A Review of NICE Methods Across Health Technology Assessment Programmes: Differences, Justifications and Implications. OHE 2016, 16, 3–37. [Google Scholar]
- Mathes, T.; Jacobs, E.; Morfeld, J.-C.; Pieper, D. Methods of international health technology assessment agencies for economic evaluations—A comparative analysis. BMC Health Serv. Res. 2013, 13, 371. [Google Scholar] [CrossRef]
- Hartz, S.; John, J. Contribution of economic evaluation to decision making in early phases of product development: A methodological and empirical review. Int. J. Technol. Assess. Health Care 2008, 24, 465–472. [Google Scholar] [CrossRef]
- McAteer, H.; Cosh, E.; Freeman, G.; Pandit, A.; Wood, P.; Lilford, R. Cost-effectiveness analysis at the development phase of a potential health technology: examples based on tissue engineering of bladder and urethra. J. Tissue Eng. Regen. Med. 2007, 1, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Girling, A.; Young, T.; Brown, C.; Lilford, R. Early-Stage Valuation of Medical Devices: The Role of Developmental Uncertainty. Value Health 2010, 13, 585–591. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fotr, J. Podnikatelský Plán a Investiční Rozhodování; Grada: Prague, Czech Republic, 1999; ISBN 978-80-7169-812-8. [Google Scholar]
- Kotler, P. Marketing Management: Analysis, Planning, Implementation, and Control; Prentice-Hall: Upper Saddle River, NJ, USA, 1991; ISBN 978-0-13-552480-0. [Google Scholar]
- EU Medical Device Regulation. Available online: https://www.tuv-sud.com/industries/medical-devices-healthcare/market-approval-amp-certification/eu-market-access/eu-medical-device-regulation (accessed on 25 December 2018).
- Paul Brooks Medical Device Regulation: What’s the Impact on Notified Bodies? Available online: https://www.med-technews.com/features/medical-device-regulation-whats-the-impact-on-notified-bodi/ (accessed on 13 December 2018).
- Bernasconi, S. How MDR and IVDR Are Reshaping Europe’s Medtech Industry; Pulse of the Industry 2017; EY-Ernst Young: Durham, NC, USA, 2017; pp. 28–29. [Google Scholar]
- EUCOMED. Medical Technology Financial Impact of the Revision of the EU Medical Devices Directives on European SMEs and Industry; New Medtech Regulations; MedTechEurope: Granada, Spain, 2013; p. 3. [Google Scholar]
- Hede, S.; Nunes, M.J.L.; Ferreira, P.F.V.; Rocha, L.A. Incorporating sustainability in decision-making for medical device development. Technol. Soc. 2013, 35, 276–293. [Google Scholar] [CrossRef]
- Adams, W.C. Conducting Semi-Structured Interviews. In Handbook of Practical Program Evaluation; Newcomer, K.E., Hatry, H.P., Wholey, J.S., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 492–505. ISBN 978-1-119-17138-6. [Google Scholar]
- Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on Medical Devices, Amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and Repealing Council Directives 90/385/EEC and 93/42/EEC (Text with EEA Relevance.); European Commission: Brussels, Belgium, 2017; Volume 117.
- Shah, S.G.S.; Robinson, I. Benefits of and barriers to involving users in medical device technology development and evaluation. Int. J. Technol. Assess. Health Care 2007, 23, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Home—Eurostat. Available online: https://ec.europa.eu/eurostat (accessed on 25 December 2018).
- Soenksen, L.R.; Yazdi, Y. Stage-gate process for life sciences and medical innovation investment. Technovation 2017, 62–63, 14–21. [Google Scholar] [CrossRef]
- Smith, D.; Gravelle, H. The Practice of Discounting Economic Evaluation of Health Care Interventions; CHE Technical Paper Series 19; The University of York: York, UK, 2000. [Google Scholar]
- Wright, D. Discount Rate; York Health Economics Consortium: York, UK, 2016. [Google Scholar]


| Purpose | Indicator, Type of Analysis |
|---|---|
| Evaluation of benefits, typically only for technologies where the impact on human health and required by legislation is measured. | QALY is the combination of quantity and quality of life generated by an intervention; One QALY = 1 year of perfect health, numerically compare the benefit gained from a variety of interventions. |
| Comparison of two solutions in terms of costs for one year of QALY. | ICER (Calculates the cost to generate one additional year of perfect (one QALY) health if using intervention B in place of intervention A). |
| This is important with respect to the decision of whether to start the investment. Industry and investors seeking informed product development decisions based on health technology assessment; marketing managers seeking to differentiate products based on cost-effectiveness; sales managers seeking to articulate the value proposition of innovative products; R&D managers wishing to integrate health economic considerations into new product development systems | CEA—A cost-effectiveness analysis (means of assessing both the costs and health benefits (measured using a non-monetary indicator) of a new MD Health) [32,33], estimating product value [34]. |
| Economic indicators in CEA or CBA [35,36] | average annual costs discounted costs net present value and profitability index internal yield percentage average return (yield) payback time |
| Health Technology Assessment for MD | Cost benefit analysis (CBA)—Transformed to (measured in) money units (e.g., CZK, EUR, USD); Cost-effectiveness analysis (CEA)—Expressed as one figure in natural units (e.g., number of life-years saved, incidence of the disease); Cost-utility analysis (CUA)—Expressed in QALYs (i.e., quality-adjusted life-years); Cost-consequence analysis (CCA)—Listed in natural units. |
| MD according to the Risk | Classification Rules |
|---|---|
| Class I—“Provided all of low risk medical devices (class I) or sterile (class Is) and/or have a measuring function (low/medium risk class Im) or reusable low risk devices (Class Ir), which are placed on the market in sterile condition, have a measuring function or are reusable surgical instruments: Assessment of the technical documentation relating only to those specific features of the device, e.g., sterility, measurement or reprocessing.” | Rules: 1–2, 4–6, 10–11, 13 |
| Class IIa (medium risk) Class IIa devices: “Assessment of the technical documentation for at least one representative device for each category of devices”. | Rules: 2–8, 10–12, 17, 19–21 |
| Class IIb (medium/high risk) Class IIb “implantable devices (except sutures, staples, dental filings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors), and class IIb active devices intended to administer and/or remove a medicinal product: Assessment of the technical documentation for every device. All other class IIb devices: Assessment of the technical documentation for at least one representative device per generic device group”. | Rules: 2–12, 15–16, 19–21 |
| Class III (high risk) Class III devices: “Assessment of the technical documentation for every device”. | Rules: 2–3, 6–9, 14–15, 18–22 |
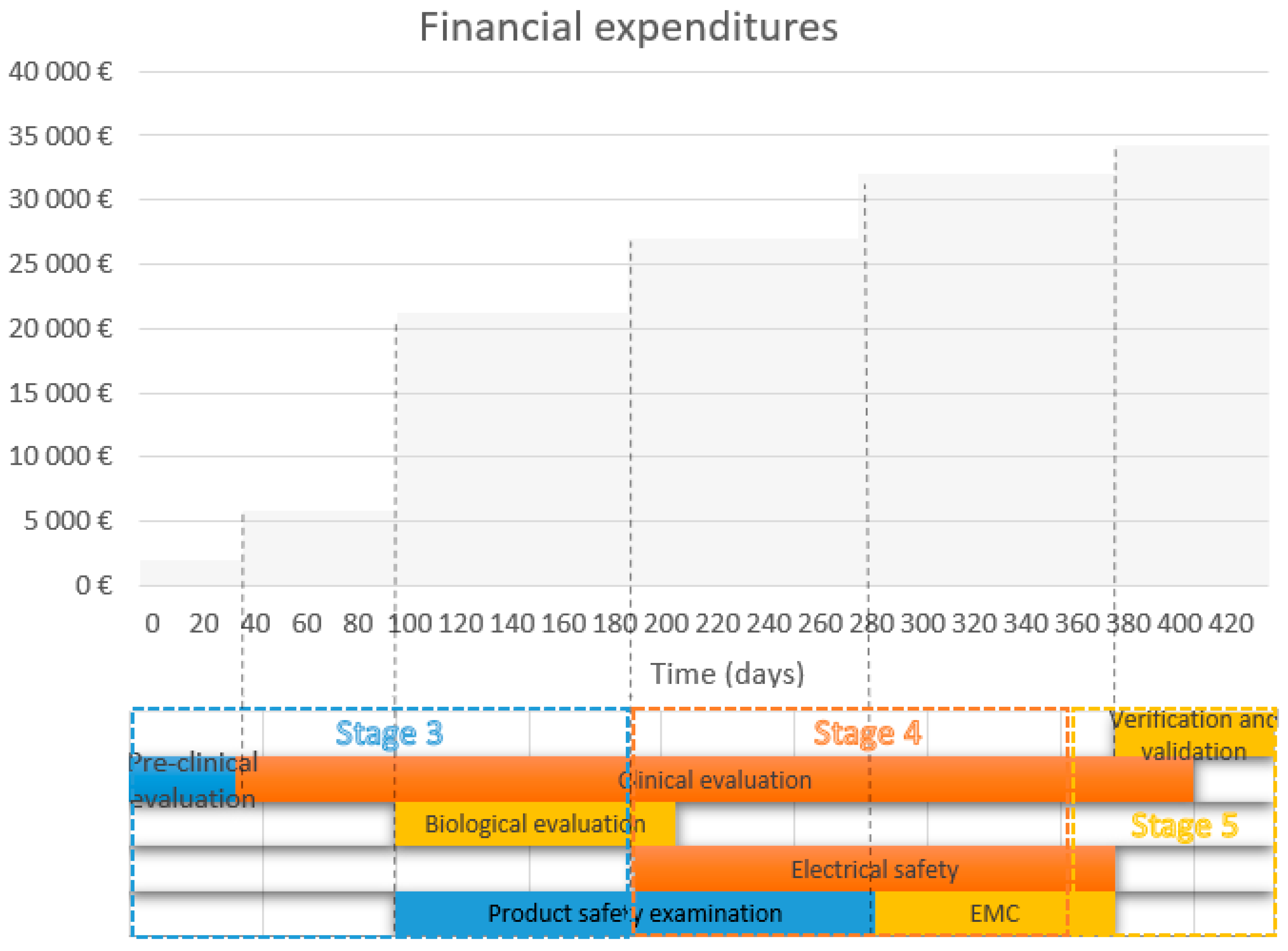
| TESTING 1 | TIME | PRICE |
|---|---|---|
| EMC | 90 days | EUR 5200 |
| Electrical safety | 180 days | EUR 6000 |
| Type test | 180 days | EUR 6000 |
| Biological evaluation | 90–120 days | EUR 10,000 |
| Preclinical evaluation | 40 days | EUR 2000 (worker’s salary per 2 months) |
| Clinical evaluation | Up to 360 days | CZK 50,000–10,000 (annual clinical trial, 100 patients, device IIa, CZK 8 mil., in the case of a clinical trial based on the use of its own clinical data, CZK 50,000 per worker’s salary for 2 months) |
| Verification and validation | 60 days | CZK 60,000 |
| Conformity Assessment | Time | Price | Complexity | Price |
|---|---|---|---|---|
| CZE Estimate | US estimate | |||
| Class I | 1 | EUR 4500–13500 | ||
| Class Ir, Is, Im | 360 days | EUR 6000 | 2 | EUR 13,500–27,000 |
| Class IIa | 3 | EUR 27,000–45,000 | ||
| Class IIb | 480 days | EUR 14,000–20,000 | 4 | EUR 45,000+ |
| Class III | 5 | EUR 45,000+ |
| Initiation | Concept Proposing | Design and Development | Verification and Validation | Production | Market Device Deployment | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Is my product MD? | Time schedule | Patent search | Patent industrial design | |||||||
| Selection of project’s team | Management of MD | |||||||||
| Specification of the product | Risk analysis according to ISO/GMP | |||||||||
| Specification of the production | Quality management system—GMP/ISO | |||||||||
| Specification of the Regulatory | VIGILANCE/CAPA system | |||||||||
| Post-market surveillance/Post-Market Clinical Follow-up | ||||||||||
| Validation/Verification according to QMS | ||||||||||
| Technical File | ||||||||||
| Risk analysis according to design of the product | ||||||||||
| Design | Testing | EMC | ||||||||
| Safety tests | ||||||||||
| Biological evaluation | ||||||||||
| Preclinical evaluation | Clinical evaluation | |||||||||
| Validation/Verification according to technical specification | ||||||||||
| DOCUMENTATION—User Manual/Usability/Clinical evaluation/Test report/Risk analysis/Labelling/Packaging/PMCF/PMS/CAPA | ||||||||||
| Economics methods | CEA/QALY/ICER update of CEA/CBA parameters; early stage HTA | HTA | ||||||||
| Phase | Economic Methods |
|---|---|
| Initiation | Comparison of the current situation and the new solution, specification of all benefits and costs, within the general CBA method, transfer of qualitative benefits to quantitative expression (substitute market methods, shadow prices, hedonic prices, labor market analysis. Depending on the type of medical device (if relevant), a more specific CEA method may be considered. A cost-effectiveness analysis, especially for higher risk classes, should include one QALY calculation, i.e., health if using new intervention/ new technology in place of the original technology. This is a period of preparatory work in which the project is being prepared and decides on its implementation or rejection. In terms of cash flows, this usually includes the costs of project documentation, administrative costs of project preparation, costs of processing economic studies, and costs of evaluating the effectiveness of the investment plan itself. The content of this phase is in any case the initial calculation of dynamic and static economic indicators as part of the CBA. In the case of instrumentation (i.e., risk class III), it is advisable to use the HTA concept, which includes the assessment of societal impacts, i.e., the CBA method. |
| Concept proposing | Update of benefits and costs, conversion to financial statements, use of economic indicators: average annual costs, discounted costs, net present value and profitability index, internal rate of return, average profitability (return), payback period. |
| Design and development | |
| Verification and validation | |
| Production | |
| Market device deployment | Final HTA/ CBA calculation with real cost information. Calculation of sensitivity analysis to identify cash flow scenarios where its approach may be as follows: characterization of factors affecting cash flows, change of each factor by a certain percentage and for each change separately calculation of new indicator value, calculation of change of resulting criterion indicator. |
| Direct costs | Non-medical costs | Time spent on the patient and his/her family—time spent on unpaid work (e.g., housework), performed by the patient or the family providing care. |
| Medical costs | Direct costs of the patient and his/her family—amounts paid by the patient (including surcharges) for drugs, medical devices, dental care, home care services. Transport costs, nursing costs. Premiums paid to private insurance companies and premiums paid by them. | |
| Costs of public health insurance. Drugs, medical devices and aids, diagnostic, therapeutic, rehabilitation, spa, screening and preventive health care covered by the general health insurance fund. Prevention Fund of Public Health Insurance Companies. | ||
| Direct costs of the health system financed from other public sources Costs of the state budget, budget of regions, towns and municipalities for the health system, which are thus paid outside the system of public health insurance. | ||
| Non-direct costs | Indirect costs of social security paid sickness benefits, pensions, care allowances. | |
| Productivity costs—loss of productivity due to the reduced work capacity, short or long term absence at work. Costs incurred by employers for recruiting and training a worker to replace a patient. |
| Thematic Area | Traditional Concept Approach/Current State | New Concept Approach—Meddee |
|---|---|---|
| Description of the process | There is a methodology for the development of medical devices in one of the companies. There is nothing like this in another company, the initial idea is an estimate of costs and development time, the expected return is calculated, no other methods are used. At present in the MPS, in the Czech Republic often dealt with randomly, within the procedure of the given device and its development, a generalized methodology often does not exist. | Setting and specification of stages of development of medical devices, determination of activities in each stage, and related types of costs is considered beneficial, it is necessary to specify in practice, it is a suitable input and starting point for setting up in the company. It is a possibility to specify the estimates, for real practice it would be appropriate to automate processes, list other individual tasks, go to a high level of detail. |
| Impact of the affected technology on the process | From the point of view of business practice, there is currently no customized methodology in place for the risk classes of a medical device or other technology breakdown, everything is modified in relation to a new idea, innovative solutions. From the theory point of view, approaches are available, specifying the phases of resource development, focusing on the activities within each of them, the focus lies, again with minimal linkage to the type of technology. | The model is beneficial from the point of view of the simultaneous transition to new legislation on new medical devices, taking into account risk classes, cost links related to increased registration fees. To some extent, MedDee tries to capture the nature of the solution, the type of technology. This aspect is essential for considering different business models. |
| Evaluation of the investment effects on the process | • In financing decisions, different business models are considered in relation to the target group and the method of financing, both being a key input for calculating the return. • In terms of economic methods, basic profitability indicators or break-even point calculations are included. It is found to be sufficient. | Evaluation of the investment includes consideration of all possible forms of sales, including the possibility of product modification in relation to the target group (medical facility or individualized solution for users). This area is marginally presented in the model and in the individual phases. The work on the business model itself is very complex, different in relation to the target country, legislative conditions of financing. It is one of the key places, it opens up an area that could be addressed in the future, there is also an interest from companies, to contribute, and then to use all the analyzed data. Setting up a more detailed description of the economic methods involved is applicable, close to business practice, to the requirements of approval authorities. |
| Potential cost savings and potential risks | The level of the current state of business practice is such that the area is dealt with in a relatively heterogeneous way, in most Czech companies on the basis of basic economic indicators of return or calculation of the break-even point. From the point of view of theory, it is not often solved within the framework of economic efficiency. The situation is different when dealing with a risk issue where aspects related to the risk class are dealt with intensively, in particular at the level of approval authorities. | The proposed methods make it possible to compare the new solution with the existing one. Along with the setting of new legislative conditions, it can be expected that pressure will be increased to systematically specify and evaluate savings and risks in relation to the new product. |
| Calculation of return | Different types of rentability indicators, more complex indicators are not usually used. | More complex evaluation methods (CBA/CEA, HTA) are included in the new model, but they are not addressed in terms of business practice. Given the ongoing legislative changes, it can be assumed that even more complex evaluations will become more and more part of business practice. |
| Phase | Type of Costs |
|---|---|
| Initiation | Wages |
| Concept proposal | Wages |
| Design and development | Wages Electrical safety (test costs in the accredited testing facility) EUR 6000–12,000 Biological evaluation EUR 4000–10,000 Clinical testing EUR 2000–400,000l Type examination EUR 6000 thousand |
| Verification and validation | Wages Conformity assessment EUR 14,000–20,000 thousand (Is, Im, Ir—EUR 6000, for IIa/IIb EUR 14,000) |
| Production | Wages Material Output control (electrical MD—production of own tester, for mechanical MD—preparation and production of own test system) |
| Market device deployment | Wage per hour Fee for patent filing (patent application in relation to the type of patent protection—European, national, worldwide) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marešová, P.; Peter, L.; Honegr, J.; Režný, L.; Penhaker, M.; Augustýnek, M.; Mohelská, H.; Klímová, B.; Kuča, K. Complexity Stage Model of the Medical Device Development Based on Economic Evaluation—MedDee. Sustainability 2020, 12, 1755. https://doi.org/10.3390/su12051755
Marešová P, Peter L, Honegr J, Režný L, Penhaker M, Augustýnek M, Mohelská H, Klímová B, Kuča K. Complexity Stage Model of the Medical Device Development Based on Economic Evaluation—MedDee. Sustainability. 2020; 12(5):1755. https://doi.org/10.3390/su12051755
Chicago/Turabian StyleMarešová, Petra, Lukáš Peter, Jan Honegr, Lukáš Režný, Marek Penhaker, Martin Augustýnek, Hana Mohelská, Blanka Klímová, and Kamil Kuča. 2020. "Complexity Stage Model of the Medical Device Development Based on Economic Evaluation—MedDee" Sustainability 12, no. 5: 1755. https://doi.org/10.3390/su12051755
APA StyleMarešová, P., Peter, L., Honegr, J., Režný, L., Penhaker, M., Augustýnek, M., Mohelská, H., Klímová, B., & Kuča, K. (2020). Complexity Stage Model of the Medical Device Development Based on Economic Evaluation—MedDee. Sustainability, 12(5), 1755. https://doi.org/10.3390/su12051755