
Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
| Keywords searched | ||
| “sickle cell disease”/“sickle cell anemia” | AND | “hemoglobinopathy”, “vaso-occlusive crisis”, “pathophysiology”, “clinical manifestations”, “bacterial infections”, “viral infections”, “parasitic infections”, “sepsis”, “pneumococcal infections”, “streptococcus infections/complications”, “pneumonia”, “osteomyelitis”, “meningitis”, “bacteremia” |
| “sickle cell” | AND | “COVID-19, prevention, therapeutics, and management” |
3. Results and Discussion
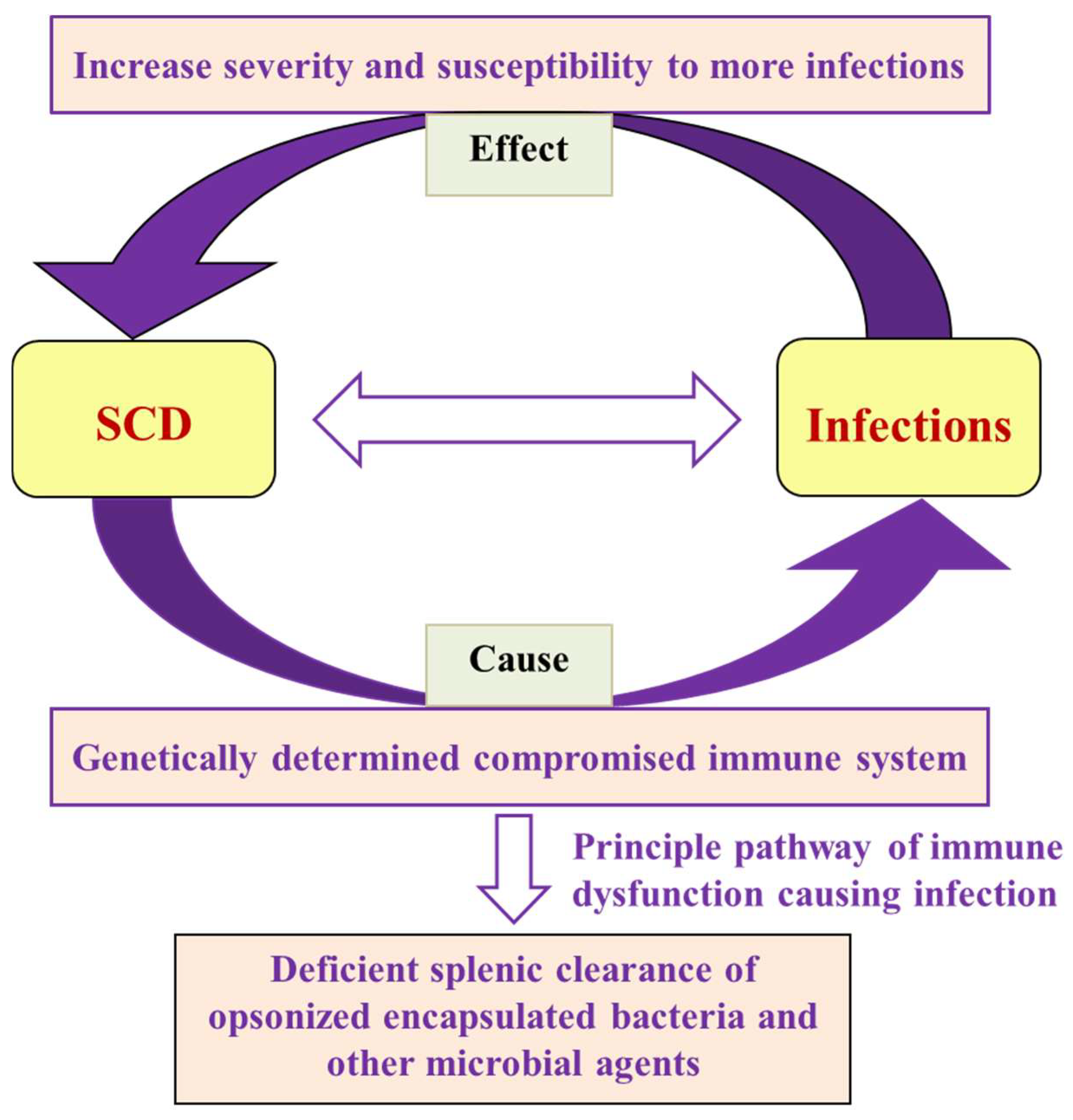
3.1. Immune Dysfunction and Susceptibility to Infection
3.2. Splenic Dysfunction
3.3. Opsonization
3.4. Lymphocytes
3.5. Nutritional Deficiencies
3.6. Hereditary Influences
3.7. Mechanical Factors
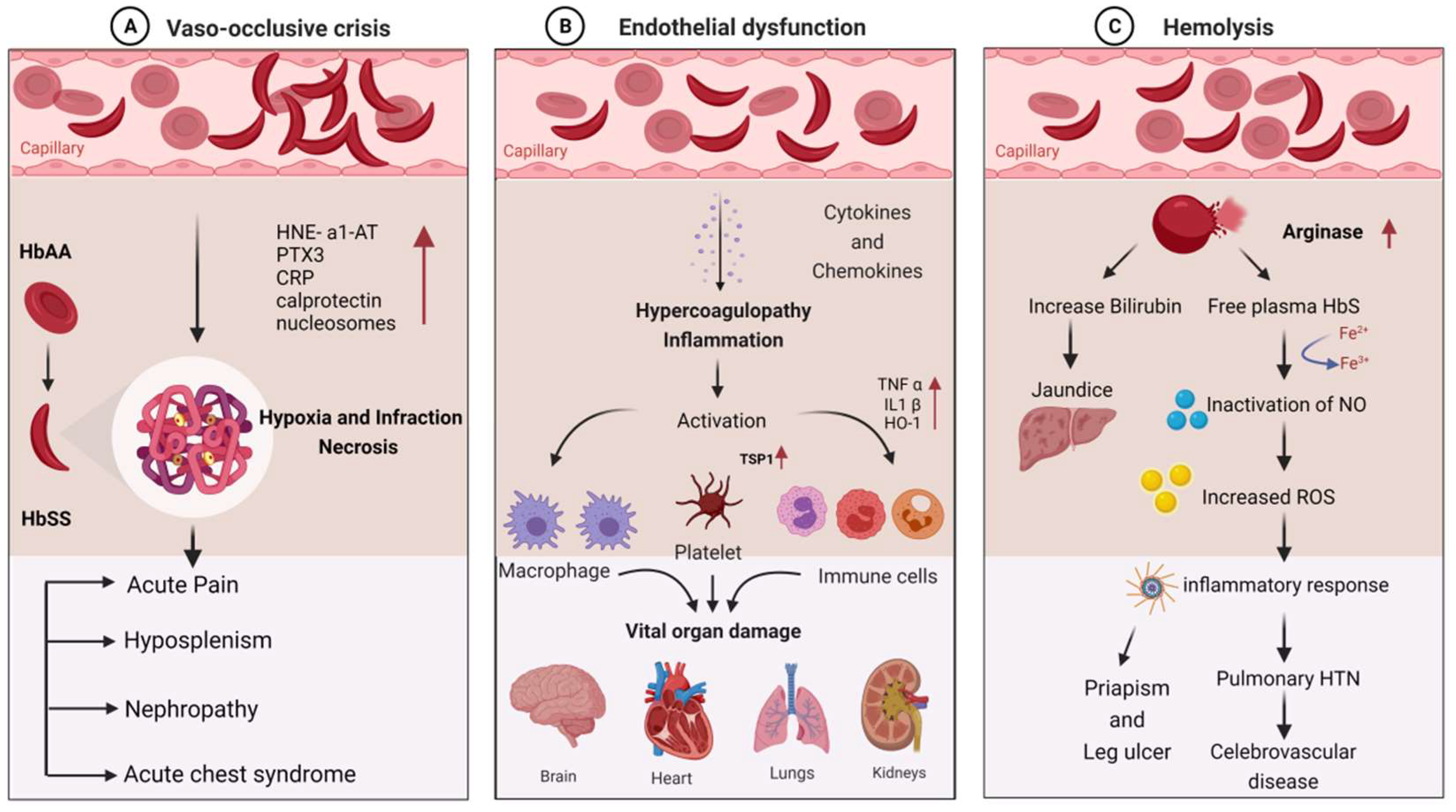
3.8. Molecular Mechanism of Clinical Manifestations in SCD
3.9. Infectious Complications Associated with SCD
3.10. Bacterial Infections
3.10.1. Bacteremia, Sepsis, and Pneumonia
3.10.2. Acute Chest Syndrome (ACS)
3.10.3. Meningitis
3.10.4. Osteomyelitis
3.10.5. Mycobacteria
3.10.6. Urinary Tract Infection (UTI)
3.10.7. Gastrointestinal Infections
3.11. Viral Infection
3.11.1. Respiratory Infections (RI)
3.11.2. Anemia Associated with Viral Infections
3.11.3. Hepatitis B and C Infections
3.11.4. HIV Infections
3.11.5. Dengue Virus
3.11.6. Coronavirus Disease (SARS-CoV-2)
3.12. Parasitic Infections
3.12.1. Malaria
3.12.2. Other Parasitic Infections
3.13. Treatment of Infections in SCD
4. Future Perspective and Roadmap for Research and Implementation
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kaul, D.K.; Finnegan, E.; Barabino, G.A. Sickle Red Cell–Endothelium Interactions. Microcirculation 2009, 16, 97–111. [Google Scholar] [CrossRef]
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Lakkakula, B.V.; Sahoo, R.; Verma, H.; Lakkakula, S. Pain Management Issues as Part of the Comprehensive Care of Patients with Sickle Cell Disease. Pain Manag. Nurs. 2018, 19, 558–572. [Google Scholar] [CrossRef]
- Patra, P.K.; Lakkakula, B.V.K.S.; Verma, H.K.; Choubey, M.; Patra, S.; Khodiar, P.K. Assessment of renal function in Indian patients with sickle cell disease. Saudi J. Kidney Dis. Transplant. 2017, 28, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, L.V.K.S.; Shukla, P.; Verma, H.; Patel, S.; Patra, P. Ocular manifestations of sickle cell disease and genetic susceptibility for refractive errors. Taiwan J. Ophthalmol. 2017, 7, 89–93. [Google Scholar] [CrossRef]
- Verma, H.K.; Lakkakula, S.; Lakkakula, B.V. Retrospection of the effect of hydroxyurea treatment in patients with sickle cell disease. Acta Haematol. Pol. 2018, 49, 1–8. [Google Scholar] [CrossRef]
- Jain, D.; Odame, I. Sickle cell disease: Progress made & challenges ahead. Indian J. Med Res. 2020, 151, 505–508. [Google Scholar] [CrossRef]
- Jha, A.N.; Mishra, H.; Verma, H.K.; Pandey, I. Compound Heterozygosity of beta-Thalassemia and the Sickle Cell Hemoglobin in Various Populations of Chhattisgarh Statel, India. Hemoglobin 2018, 2, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Bhanushali, A.A.; Patra, P.; Nair, D.; Verma, H.; Das, B. Genetic variant in the BCL11A (rs1427407), but not HBS1-MYB (rs6934903) loci associate with fetal hemoglobin levels in Indian sickle cell disease patients. Blood Cells Mol. Dis. 2015, 54, 4–8. [Google Scholar] [CrossRef]
- Al-Salem, A.H. Splenic Complications of Sickle Cell Anemia and the Role of Splenectomy. ISRN Hematol. 2011, 2011, 864257. [Google Scholar] [CrossRef]
- Monaco, C.P.; Fonseca, P.B.B.; Braga, J.A.P. Infectious complications after surgical splenectomy in children with sickle cell disease. Rev. Paul. Pediatr. (Engl. Ed.) 2015, 33, 150–153. [Google Scholar] [CrossRef]
- Hsu, L.; E Nnodu, O.; Brown, B.J.; Tluway, F.; King, S.; Dogara, L.G.; Patil, C.; Shevkoplyas, S.S.; Lettre, G.; Cooper, R.S.; et al. White Paper: Pathways to Progress in Newborn Screening for Sickle Cell Disease in Sub-Saharan Africa. J. Trop. Dis. 2018, 6, 260. [Google Scholar] [CrossRef] [PubMed]
- Bohnsack, J.F.; Brown, E.J. The role of the spleen in resistance to infection. Annu. Rev. Med. 1986, 37, 49–59. [Google Scholar] [CrossRef]
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718. [Google Scholar] [CrossRef]
- West, T.B.; West, D.W.; Ohene-Frempong, K. The presentation, frequency, and outcome of bacteremia among children with sickle cell disease and fever. Pediatr. Emerg. Care 1994, 10, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Halasa, N.B.; Shankar, S.M.; Talbot, T.R.; Arbogast, P.G.; Mitchel, E.F.; Wang, W.C.; Schaffner, W.; Craig, A.S.; Griffin, M.R. Incidence of Invasive Pneumococcal Disease among Individuals with Sickle Cell Disease before and after the Introduction of the Pneumococcal Conjugate Vaccine. Clin. Infect. Dis. 2007, 44, 1428–1433. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E. Salmonella infection in sickle cell disease: A clear and present danger. J. Pediatr. 1997, 130, 350–351. [Google Scholar]
- Dunkelberger, J.R.; Song, W.-C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Spirer, Z.; Weisman, Y.; Zakuth, V.; Fridkin, M.; Bogair, N. Decreased serum tuftsin concentrations in sickle cell disease. Arch. Dis. Child. 1980, 55, 566–567. [Google Scholar] [CrossRef]
- Deceulaer, K.; Wilson, W.A.; Morgan, A.G.; Serjeant, G.R. Plasma haemoglobin and complement activation in sickle cell disease. J. Clin. Lab. Immunol. 1981, 6, 57–60. [Google Scholar]
- Wilson, W.A.; Thomas, E.J. Activation of the alternative pathway of human complement by haemoglobin. Clin. Exp. Immunol. 1979, 36, 140–144. [Google Scholar]
- Cameron, P.U.; Jones, P.; Gorniak, M.; Dunster, K.; Paul, E.; Lewin, S.; Woolley, I.; Spelman, D. Splenectomy Associated Changes in IgM Memory B Cells in an Adult Spleen Registry Cohort. PLoS ONE 2011, 6, e23164. [Google Scholar] [CrossRef]
- Weller, S. Human blood IgM “memory” B cells are circulating splenic marginal zone B cells harboring a prediversified immunoglobulin repertoire. Blood 2004, 104, 3647–3654. [Google Scholar] [CrossRef] [PubMed]
- Ballester, O.F.; Abdallah, J.M.; Prasad, A.S. Impaired IgM antibody responses to an influenza virus vaccine in adults with sickle cell anemia. Am. J. Hematol. 1985, 20, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Koffi, K.G.; Sawadogo, D.; Meite, M.; Nanho, D.C.; Tanoh, E.S.; Attia, A.K.; Sanogo, I.; Sangare, A. Reduced levels of T-cell subsets CD4+ and CD8+ in homozygous sickle cell anaemia patients with splenic defects. Hematol. J. 2003, 4, 363–365. [Google Scholar] [CrossRef]
- Romagnani, S. T-cell subsets (Th1 versus Th2). Annals of allergy, asthma & immunology: Official publication of the American College of Allergy. Asthma Immunol. 2000, 85, 9–18. [Google Scholar]
- Hibbert, J.M.; Creary, M.S.; E Gee, B.; Buchanan, I.D.; Quarshie, A.; Hsu, L.L. Erythropoiesis and Myocardial Energy Requirements Contribute to the Hypermetabolism of Childhood Sickle Cell Anemia. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 680–687. [Google Scholar] [CrossRef]
- Prasad, A.S.; Schoomaker, E.B.; Ortega, J.; Brewer, G.J.; Oberleas, D.; Oelshlegel, F.J. Zinc Deficiency in Sickle Cell Disease. Clin. Chem. 1975, 21, 582–587. [Google Scholar] [CrossRef]
- Prasad, A.S. Zinc deficiency and effects of zinc supplementation on sickle cell anemia subjects. Prog. Clin. Biol. Res. 1981, 55, 99–122. [Google Scholar]
- Fraker, P.J.; King, L.E.; Laakko, T.; Vollmer, T.L. The Dynamic Link between the Integrity of the Immune System and Zinc Status. J. Nutr. 2000, 130, 1399S–1406S. [Google Scholar] [CrossRef]
- De Franceschi, L.; Bachir, D.; Galacteros, F.; Tchernia, G.; Cynober, Y.; Neuberg, D.; Beuzard, Y.; Brugnara, C. Oral magnesium pidolate: Effects of long-term administration in patients with sickle cell disease. Br. J. Haematol. 2000, 108, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Schall, J.I.; Zemel, B.S.; Kawchak, D.A.; Ohene-Frempong, K.; Stallings, V.A. Vitamin A status, hospitalizations, and other outcomes in young children with sickle cell disease. J. Pediatr. 2004, 145, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 9442, 1343–1360. [Google Scholar] [CrossRef]
- Tamouza, R.; Neonato, M.-G.; Busson, M.; Marzais, F.; Girot, R.; Labie, D.; Elion, J.; Charron, D. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum. Immunol. 2002, 63, 194–199. [Google Scholar] [CrossRef]
- Neonato, M.-G.; Lu, C.Y.; Guilloud-Bataille, M.; Lapouméroulie, C.; Nabeel-Jassim, H.; Dabit, D.; Girot, R.; Krishnamoorthy, R.; Feingold, J.; Besmond, C.; et al. Genetic polymorphism of the mannose-binding protein gene in children with sickle cell disease: Identification of three new variant alleles and relationship to infections. Eur. J. Hum. Genet. 1999, 7, 679–686. [Google Scholar] [CrossRef][Green Version]
- Norris, C.F.; Surrey, S.; Bunin, G.R.; Schwartz, E.; Buchanan, G.R.; McKenzie, S.E. Relationship between Fc receptor IIA polymorphism and infection in children with sickle cell disease. J. Pediatr. 1996, 128, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Powars, D.R.; Chan, L.; Schroeder, W.A. Beta S-gene-cluster haplotypes in sickle cell anemia: Clinical implications. Am. J. Pediatr. Hematol. Oncol. 1990, 12, 367–374. [Google Scholar] [CrossRef]
- Adewoye, A.H.; Nolan, V.G.; Ma, Q.; Baldwin, C.; Wyszynski, D.F.; Farrell, J.J.; Farrer, L.A.; Steinberg, M.H. Association of polymorphisms of IGF1R and genes in the transforming growth factor-beta/bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin. Infect. Dis.Off. Publ. Infect. Dis. Soc. Am. 2006, 43, 593–598. [Google Scholar] [CrossRef]
- West, D.C.; Romano, P.S.; Azari, R.; Rudominer, A.; Holman, M.; Sandhu, S. Impact of Environmental Tobacco Smoke on Children With Sickle Cell Disease. Arch. Pediatr. Adolesc. Med. 2003, 157, 1197–1201. [Google Scholar] [CrossRef]
- Cohen, R.T.; DeBaun, M.R.; Blinder, M.A.; Strunk, R.C.; Field, J.J. Smoking is associated with an increased risk of acute chest syndrome and pain among adults with sickle cell disease. Blood 2010, 115, 3852–3854. [Google Scholar] [CrossRef]
- Vichinsky, E.P. Current issues with blood transfusions in sickle cell disease. Semin. Hematol. 2001, 38 (Suppl. 1), 14–22. [Google Scholar] [CrossRef]
- Zarrouk, V.; Habibi, A.; Zahar, J.P.; Roudot-Thoraval, F.; Bachir, D.; Brun-Buisson, C.; Legrand, P.; Godeau, B.; Galacteros, F.; Lesprit, P. Bloodstream infection in adults with sickle cell disease: Association with venous catheters, Staphylococcus aureus, and bone-joint infections. Medicine 2006, 85, 43–48. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Cisneros, G.S.; Thein, S.L. Recent Advances in the Treatment of Sickle Cell Disease. Front. Physiol. 2020, 11, 435. [Google Scholar] [CrossRef]
- Krishnan, S.; Setty, Y.; Betal, S.G.; Vijender, V.; Rao, K.; Dampier, C.; Stuart, M. Increased levels of the inflammatory biomarker C-reactive protein at baseline are associated with childhood sickle cell vasocclusive crises. Br. J. Haematol. 2010, 148, 797–804. [Google Scholar] [CrossRef]
- Bargoma, E.M.; Mitsuyoshi, J.K.; Larkin, S.K.; Styles, L.A.; Kuypers, F.A.; Test, S.T. Serum C-reactive protein parallels secretory phospholipase A2 in sickle cell disease patients with vasoocclusive crisis or acute chest syndrome. Blood 2005, 105, 3384–3385. [Google Scholar] [CrossRef]
- Jaillon, S.; Peri, G.; Delneste, Y.; Frémaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804. [Google Scholar] [CrossRef]
- Kato, G.J.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Luken, B.M.; Nur, E.; van Tuijn, C.F.J.; Sins, J.W.; Brandjes, D.P.M.; Zeerleder, S.S.; Biemond, B.J. Inflammatory and endothelial markers during vaso-occlusive crisis and acute chest syndrome in sickle cell disease. Am. J. Hematol. 2017, 92, E634–E636. [Google Scholar] [CrossRef] [PubMed]
- Zeerleder, S.; Stephan, F.; Emonts, M.; de Kleijn, E.D.; Esmon, C.T.; Varadi, K.; Hack, C.E.; Hazelzet, J.A. Circulating nucleosomes and severity of illness in children suffering from meningococcal sepsis treated with protein C. Crit. Care Med. 2012, 40, 3224–3229. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, M.; Nur, E.; Biemond, B.J.; van Mierlo, G.J.; Solati, S.; Brandjes, D.P.; Otten, H.M.; Schnog, J.J.; Zeerleder, S. Nucleosomes and neutrophil activation in sickle cell disease painful crisis. Haematologica 2013, 98, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Camus, S.M.; De Moraes, J.A.; Bonnin, P.; Abbyad, P.; Le Jeune, S.; Lionnet, F.; Loufrani, L.; Grimaud, L.; Lambry, J.-C.; Charue, D.; et al. Circulating cell membrane microparticles transfer heme to endothelial cells and trigger vasoocclusions in sickle cell disease. Blood 2015, 125, 3805–3814. [Google Scholar] [CrossRef] [PubMed]
- Reeder, B.J.; Andersen, C.B.F.; Stødkilde, K.; Sæderup, K.L.; Kuhlee, A.; Raunser, S.; Graversen, J.H.; Moestrup, S.K.; Strader, M.B.; Alayash, A.I.; et al. The Redox Activity of Hemoglobins: From Physiologic Functions to Pathologic Mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Ofori-Acquah, S.F. Erythroid DAMPs drive inflammation in SCD. Blood 2014, 123, 3689–3690. [Google Scholar] [CrossRef]
- Mendonça, R.; Silveira, A.A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016, 65, 665–678. [Google Scholar] [CrossRef]
- Mpollo, M.-S.E.M.; Brandt, E.B.; Shanmukhappa, S.K.; Arumugam, P.I.; Tiwari, S.; Loberg, A.; Pillis, D.; Rizvi, T.; Lindsey, M.; Jonck, B.; et al. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. J. Clin. Investig. 2015, 126, 571–584. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Eckle, T. Ischemia and reperfusion—from mechanism to translation. Nat. Med. 2011, 17, 1391–1401. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef]
- Onwubalili, J.K. Sickle cell disease and infection. J. Infect. 1983, 7, 2–20. [Google Scholar] [CrossRef]
- Adeyokunnu, A.; Hendrickse, G. Salmonella osteomyelitis in childhood. Arch. Dis. Child. 1980, 55, 175–184. [Google Scholar] [CrossRef]
- El-Hazmi, M. Infections in Sickle Cell Disease. Ann. Saudi Med. 1986, 6, 33–40. [Google Scholar] [CrossRef]
- Powars, D.; Overturf, G.; Turner, E. Is There an Increased Risk of Haemophilus influenzae Septicemia in Children with Sickle Cell Anemia? Pediatrics 1983, 71, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Ochocinski, D.; Dalal, M.; Black, L.V.; Carr, S.; Lew, J.; Sullivan, K.; Kissoon, N. Life-Threatening Infectious Complications in Sickle Cell Disease: A Concise Narrative Review. Front. Pediatr. 2020, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.G. The Role of Infection in the Pathogenesis of Vaso-Occlusive Crisis in Patients with Sickle Cell Disease. Mediterr. J. Hematol. Infect. Dis. 2011, 3, e2011028. [Google Scholar] [CrossRef]
- Gonzales, J.; Chakraborty, T.; Romero, M.; Abu Mraheil, M.; Kutlar, A.; Pace, B.; Lucas, R. Streptococcus pneumoniae and Its Virulence Factors H2O2 and Pneumolysin Are Potent Mediators of the Acute Chest Syndrome in Sickle Cell Disease. Toxins 2021, 13, 157. [Google Scholar] [CrossRef]
- Brown, B.; Dada-adegbola, H.; Trippe, C.; Olopade, O. Prevalence and Etiology of Bacteremia in Febrile Children with Sickle Cell Disease at a Nigeria Tertiary Hospital. Mediterr. J. Hematol. Infect. Dis. 2017, 9, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, K.J.J.; Hambleton, I.R.; Wilson, R.M.; Alexander, H.; E Serjeant, B.; Serjeant, G.R. Significance of fever in Jamaican patients with homozygous sickle cell disease. Arch. Dis. Child. 2001, 84, 156–159. [Google Scholar] [CrossRef]
- Al Achkar, M.; Rogers, J.S.; Muszynski, M.J. Pantoea species sepsis associated with sickle cell crisis in a pregnant woman with a history of pica. Am. J. Case Rep. 2012, 13, 26–28. [Google Scholar] [CrossRef]
- Alkindi, S.; Matwani, S.; Al-Maawali, A.; Al-Maskari, B.; Pathare, A. Complications of PORT-A-CATH® in patients with sickle cell disease. J. Infect. Public Heal. 2012, 5, 57–62. [Google Scholar] [CrossRef]
- Cannas, G.; Merazga, S.; Virot, E. Sickle Cell Disease and Infections in High- and Low-Income Countries. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019042. [Google Scholar] [CrossRef]
- Quinn, C.T. Sickle cell disease in childhood: From newborn screening through transition to adult medical care. Pediatr. Clin. North. Am. 2013, 60, 1363–1381. [Google Scholar] [CrossRef]
- Battersby, A.J.; Knox-Macaulay, H.H.; Carrol, E.D. Susceptibility to invasive bacterial infections in children with sickle cell disease. Pediatr. Blood Cancer 2010, 55, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Rincón-López, E.M.; Gómez, M.L.N.; Matos, T.H.; Saavedra-Lozano, J.; de la Red, Y.A.; Rupérez, B.H.; de Julián, E.C. RETRO-DREP Study Group Low-risk factors for severe bacterial infection and acute chest syndrome in children with sickle cell disease. Pediatr. Blood Cancer 2019, 66, e27667. [Google Scholar] [CrossRef] [PubMed]
- BiscevicTokic, J.; Tokic, N.; Musanovic, A. Pneumonia as the Most Common Lower Respiratory Tract Infection. Med. Arch. 2013, 67, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Suarez, V.; Michel, F.; Toscano, C.M.; Bierrenbach, A.L.; Gonzales, M.; Alencar, A.P.; Matus, C.R.; Andrus, J.K.; de Oliveira, L.H. Impact of pneumococcal conjugate vaccine in children morbidity and mortality in Peru: Time series analyses. Vaccine 2016, 34, 4738–4743. [Google Scholar] [CrossRef]
- Yousif, T.I.; Elnazir, B. Approach to a child with recurrent pneumonia. Sudan. J. Paediatr. 2015, 15, 71–77. [Google Scholar]
- Yee, M.E.; Bakshi, N.; Graciaa, S.H.; Lane, P.A.; Jerris, R.C.; Wang, Y.F.; Yildirim, I. Incidence of invasive Haemophilus influenzae infections in children with sickle cell disease. Pediatr. Blood Cancer 2019, 66, e27642. [Google Scholar] [CrossRef]
- Jain, S.; Bakshi, N.; Krishnamurti, L. Acute Chest Syndrome in Children with Sickle Cell Disease. Pediatr. Allergy Immunol. Pulmonol. 2017, 30, 191–201. [Google Scholar] [CrossRef]
- Howard, J.; Hart, N.; Roberts-Harewood, M.; Cummins, M.; Awogbade, M.; Davis, B.; BCSH Committee. Guideline on the management of acute chest syndrome in sickle cell disease. Br. J. Haematol. 2015, 169, 492–505. [Google Scholar] [CrossRef]
- Clay, E.L.J.; Burrell, T.; Belhorn, T.; Redding-Lallinger, R. Immunogenicity of pneumococcal vaccination in a patient with sickle hemoglobinopathy: A case report. Clin. Case Rep. 2015, 3, 618–621. [Google Scholar] [CrossRef]
- Wahl, B.; Brien, K.L.O.; Greenbaum, A.; Majumder, A.; Liu, L.; Chu, Y.; Lukšić, I.; Nair, H.; McAllister, D.A.; Campbell, H.; et al. Articles Burden of Streptococcus pneumoniae and Haemophilus influenzae type b disease in children in the era of conjugate vaccines: Global, regional, and national estimates for. Lancet Glob. Health 2018, 6, e744–e757. [Google Scholar] [CrossRef]
- Mani, C.S. Acute Pneumonia and Its Complications. Princ. Pract. Pediatr. Infect. Dis. 2018, 238–249.e4. [Google Scholar] [CrossRef]
- Chenou, F.; Azevedo, J.; Leal, H.F.; Gonçalves, M.d.S.; Reis, J.N. Bacterial meningitis in patients with sickle cell anemia in Salvador, Bahia, Brazil: A report on ten cases. Hematol. Transfus. Cell Ther. 2020, 42, 139–144. [Google Scholar] [CrossRef]
- Kiriazopulos, D.; Pedroni, P.; Occhi, G.; Sassi, G.; Corna, A.; Cieri, F.; Cipolletta, E.; Orobello, M.; Colombo, M. Pneumococcal Meningitis in a Child With Sickle Cell Anemia: A Case Report. Int. J. Clin. Pediatr. 2015, 4, 168–170. [Google Scholar] [CrossRef][Green Version]
- Nottidge, V.A. Pneumococcal Meningitis in Sickle Cell Disease in Childhood. Arch. Pediatr. Adolesc. Med. 1983, 137, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Coyle, P. Overview of Acute and Chronic Meningitis. Neurol. Clin. 1999, 17, 691–710. [Google Scholar] [CrossRef] [PubMed]
- Rankine-Mullings, A.E.; Owusu-Ofori, S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. Cochrane Database Syst. Rev. 2017, 10, CD003427. [Google Scholar] [CrossRef]
- Thompson, A.A. Primary Prophylaxis in Sickle Cell Disease: Is It Feasible? Is It Effective? Hematology 2011, 2011, 434–439. [Google Scholar] [CrossRef]
- Junior, G.B.D.S.; Daher, E.D.F.; Da Rocha, F.A.C. Osteoarticular involvement in sickle cell disease. Rev. Bras. Hematol. E Hemoter. 2012, 34, 156–164. [Google Scholar] [CrossRef]
- Mary, P. Sickle cell disease as a cause of osteoarthritis. Arch. Pediatr. 2008, 15, 639–641. [Google Scholar] [CrossRef]
- Anand, A.J.; Glatt, A.E. Salmonella osteomyelitis and arthritis in sickle cell disease. Semin. Arthritis Rheum. 1994, 24, 211–221. [Google Scholar] [CrossRef] [PubMed]
- AlFawaz, T.; Alzumar, O.; AlShahrani, D.; Alshehri, M. Severity of Salmonella infection among sickle cell diseases pediatric patients: Description of the infection pattern. Int. J. Pediatr. Adolesc. Med. 2019, 6, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Fontalis, A.; Hughes, K.; Nguyen, M.P.; Williamson, M.; Yeo, A.; Lui, D.; Gelfer, Y. The challenge of differentiating vaso-occlusive crises from osteomyelitis in children with sickle cell disease and bone pain: A 15-year retrospective review. J. Child. Orthop. 2019, 13, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Thanni, L.O.A. Bacterial osteomyelitis in major sickling haemoglobinopathies: Geographic difference in pathogen prevalence. Afr. Health Sci. 2006, 6, 236–239. [Google Scholar]
- Almeida, A.; Roberts, I. Bone involvement in sickle cell disease. Br. J. Haematol. 2005, 129, 482–490. [Google Scholar] [CrossRef]
- Calhoun, J.H.; Manring, M.M.; Shirtliff, M. Osteomyelitis of the long bones. Semin. Plast. Surg. 2009, 23, 59–72. [Google Scholar] [CrossRef]
- Burnett, M.W.; Bass, J.W.; Cook, B.A. Etiology of Osteomyelitis Complicating Sickle Cell Disease. Pediatrics 1998, 101, 296–297. [Google Scholar] [CrossRef]
- Pszolla, N.; Sarkar, M.R.; Strecker, W.; Kern, P.; Kinzl, L.; Meyers, W.M.; Portaels, F. Buruli Ulcer: A Systemic Disease. Clin. Infect. Dis. 2003, 37, e78–e82. [Google Scholar] [CrossRef]
- Coates, T.D.; Wood, J.C. How we manage iron overload in sickle cell patients. Br. J. Haematol. 2017, 177, 703–716. [Google Scholar] [CrossRef]
- Raghupathy, R.; Manwani, D.; Little, J.A. Iron Overload in Sickle Cell Disease. Adv. Hematol. 2010, 2010, 272940. [Google Scholar] [CrossRef]
- Shemisa, K.; Jafferjee, N.; Thomas, D.; Jacobs, G.; Meyerson, H.J. Mycobacterium avium Complex Infection in a Patient with Sickle Cell Disease and Severe Iron Overload. Case Rep. Infect. Dis. 2014, 2014, 405323. [Google Scholar] [CrossRef] [PubMed]
- Thorell, E.A.; Sharma, M.; Jackson, M.A.; Selvarangan, R.; Woods, G.M. Disseminated Nontuberculous Mycobacterial Infections in Sickle Cell Anemia Patients. J. Pediatr. Hematol. 2006, 28, 678–681. [Google Scholar] [CrossRef]
- Edrees, N.; Howard, T.H. Unusual Presentation for Unusual Infection: Disseminated Mycobacterium Avium-Intracellulare complex (MAC) in Patient with Sickle Cell Anemia Mimicking Blood Transfusion Reaction. Blood 2014, 124, 4980. [Google Scholar] [CrossRef]
- Esnakula, A.K.; Mummidi, S.K.; Oneal, P.A.; Naab, T.J. Sepsis caused by Mycobacterium terrae complex in a patient with sickle cell disease. BMJ Case Rep. 2013, 2013, bcr2013009159. [Google Scholar] [CrossRef]
- Ashraf, M.S.; Swinker, M.; Augustino, K.L.; Nobles, D.; Knupp, C.; Liles, D.; Christie, J.; Ramsey, K.M. Outbreak of Mycobacterium mucogenicum Bloodstream Infections among Patients with Sickle Cell Disease in an Outpatient Setting. Infect. Control Hosp. Epidemiol. 2012, 33, 1132–1136. [Google Scholar] [CrossRef]
- Edun, B.; Shah, A.; Durkin, M.; Whitmire, M.; Williams, S.P.; Albrecht, H.; Al-Hasan, M.; Weissman, S. Non-tuberculous mycobacterial bloodstream infections in patients with indwelling vascular catheters—The role of sickle cell anaemia. Infect. Dis. 2017, 49, 341–346. [Google Scholar] [CrossRef]
- Droz, N.; De Lauzanne, A.; Holvoet, L.; Missud, F.; Benkerrou, M.; Brousse, V.; Odièvre, M.-H.; Faye, A.; Koehl, B. Tuberculosis in children with sickle cell anaemia: A retrospective study in French tertiary care centres. Eur. J. Pediatr. 2017, 176, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Yanda, A.N.A.; Nansseu, J.R.N.; Awa, H.D.M.; Tatah, S.A.; Seungue, J.; Eposse, C.; Koki, P.O.N. Burden and spectrum of bacterial infections among sickle cell disease children living in Cameroon. BMC Infect. Dis. 2017, 17, 211. [Google Scholar] [CrossRef]
- Musonda, T.; Zulu, M.; Samutela, M.; Kalonda, A.; Mantina, H.; Okuku, P.; Sinkala, M.; Nkhoma, P. Leucocytosis and Asymptomatic Urinary Tract Infections in Sickle Cell Patients at a Tertiary Hospital in Zambia. Anemia 2020, 2020, 3792728. [Google Scholar] [CrossRef] [PubMed]
- Chukwu, B.F.; Okafor, H.U.; Ikefuna, A.N. Asymptomatic bacteriuria in children with sickle cell anemia at The University of Nigeria teaching hospital, Enugu, South East, Nigeria. Ital. J. Pediatr. 2011, 37, 45. [Google Scholar] [CrossRef] [PubMed]
- Donkor, E.S.; Osei, J.A.; Anim-Baidoo, I.; Darkwah, S. Risk of Asymptomatic Bacteriuria among People with Sickle Cell Disease in Accra, Ghana. Diseases 2017, 5, 4. [Google Scholar] [CrossRef]
- Cumming, V.; Ali, S.; Forrester, T.; Roye-Green, K.; Reid, M. Asymptomatic bacteriuria in sickle cell disease: A cross-sectional study. BMC Infect. Dis. 2006, 6, 46. [Google Scholar] [CrossRef]
- Mava, Y.; Bello, M.; Ambe, J.; Zailani, S. Antimicrobial sensitivity pattern of organisms causing urinary tract infection in children with sickle cell anemia in Maiduguri, Nigeria. Niger. J. Clin. Pr. 2012, 15, 420–423. [Google Scholar] [CrossRef]
- Neves, P.D.M.d.M.; Reichert, B.V.; Bridi, R.A.; Yu, L.; Dias, C.B.; Pinheiro, R.B.B.; Testagrossa, L.d.A.; Cavalcante, L.B.; Malheiros, D.M.A.C.; Jorge, L.B.; et al. Atypical presentation of acute post-infectious glomerulonephritis in patients with sickle cell disease: Report of two cases. BMC Nephrol. 2020, 21, 56. [Google Scholar] [CrossRef]
- Dutta, D.; Methe, B.; Amar, S.; Morris, A.; Lim, S.H. Intestinal injury and gut permeability in sickle cell disease. J. Transl. Med. 2019, 17, 183. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Fast, L.; Morris, A. Sickle cell vaso-occlusive crisis: It’s a gut feeling. J. Transl. Med. 2016, 14, 334. [Google Scholar] [CrossRef] [PubMed]
- Green, B.T.; Branch, M.S. Ischemic colitis in a young adult during sickle cell crisis: Case report and review. Gastrointest. Endosc. 2003, 57, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H.; Methé, B.A.; Knoll, B.M.; Morris, A.; Obaro, S.K. Invasive non-typhoidal Salmonella in sickle cell disease in Africa: Is increased gut permeability the missing link? J. Transl. Med. 2018, 16, 239. [Google Scholar] [CrossRef]
- Sigaúque, B.; Roca, A.M.; Mandomando, I.D.; Morais, L.D.; Quintó, L.; Sacarlal, J.; Macete, E.; Nhamposa, T.; Machevo, S.; Aide, P.; et al. Community-Acquired Bacteremia Among Children Admitted to a Rural Hospital in Mozambique. Pediatr. Infect. Dis. J. 2009, 28, 108–113. [Google Scholar] [CrossRef]
- Obaro, S.K.; Hassan-hanga, F.; Olateju, E.K.; Umoru, D.; Lawson, L.; Olanipekun, G.; Ibrahim, S.; Munir, H.; Ihesiolor, G.; Maduekwe, A.; et al. Salmonella Bacteremia Among Children in Central and Northwest Nigeria, 2008–2015. Clin. Infect. Dis. 2015, 61 (Suppl. 4), 325–331. [Google Scholar] [CrossRef]
- MacLean, J.E.; Atenafu, E.; Kirby-Allen, M.; MacLusky, I.B.; Stephens, D.; Grasemann, H.; Subbarao, P. Longitudinal decline in lung volume in a population of children with sickle cell disease. Am. J. Respir. Crit. Care Med. 2009, 178, 1055–1059. [Google Scholar] [CrossRef] [PubMed]
- Lunt, A.; McGhee, E.; Sylvester, K.; Rafferty, G.; Dick, M.; Rees, D.; Height, S.; Thein, S.L.; Greenough, A. Longitudinal assessment of lung function in children with sickle cell disease. Pediatr. Pulmonol. 2015, 51, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Claudio, A.M.; Foltanski, L.; Delay, T.; Britell, A.; Duckett, A.; Weeda, E.R.; Bohm, N. Antibiotic Use and Respiratory Pathogens in Adults With Sickle Cell Disease and Acute Chest Syndrome. Ann. Pharmacother. 2019, 53, 991–996. [Google Scholar] [CrossRef]
- Alkindi, S.; Al-Yahyai, T.; Raniga, S.; Boulassel, M.R.; Pathare, A. Respiratory Viral Infections in Sickle Cell Anemia: Special Emphasis on H1N1 Co-infection. Oman Med. J. 2020, 35, e197. [Google Scholar] [CrossRef]
- Bundy, D.G.; Strouse, J.J.; Casella, J.F.; Miller, M.R. Burden of influenza-related hospitalizations among children with sickle cell disease. Pediatrics 2010, 125, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Gomez, E.; Morris, C.R. Asthma Management in Sickle Cell Disease. BioMed Res. Int. 2013, 2013, 604140. [Google Scholar] [CrossRef]
- Powars, D.; A Weidman, J.; Odom-Maryon, T.; Niland, J.C.; Johnson, C. Sickle cell chronic lung disease: Prior morbidity and the risk of pulmonary failure. Medicine 1988, 67, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Slavov, S.N.; Kashima, S.; Pinto, A.C.S.; Covas, D.T. Human parvovirus B19: General considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions. FEMS Immunol. Med. Microbiol. 2011, 62, 247–262. [Google Scholar] [CrossRef]
- Novelli, E.M.; Gladwin, M.T. Crises in Sickle Cell Disease. Chest 2016, 149, 1082–1093. [Google Scholar] [CrossRef]
- Bakarey, A.S.; Akinboade, I.O.; Aken’ova, Y.A. Transmission transmissible hepatitis B virus markers of infection among sickle cell disease patients receiving care at a tertiary health facility in Ibadan, southwest Nigeria. J. Immunoass. Immunochem. 2018, 39, 416–427. [Google Scholar] [CrossRef]
- Sonderup, M.W.; Afihene, M.; Ally, R.; Apica, B.; Awuku, Y.; Cunha, L.; Dusheiko, G.; Gogela, N.; Lohouès-Kouacou, M.-J.; Lam, P.; et al. Hepatitis C in sub-Saharan Africa: The current status and recommendations for achieving elimination by 2030. Lancet Gastroenterol. Hepatol. 2017, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Mulumba, L.L.; Wilson, L. Sickle cell disease among children in Africa: An integrative literature review and global recommendations. Int. J. Afr. Nurs. Sci. 2015, 3, 56–64. [Google Scholar] [CrossRef]
- Baseke, J.; Musenero, M.; Mayanja-Kizza, H. Prevalence of hepatitis B and C and relationship to liver damage in HIV infected patients attending Joint Clinical Research Centre Clinic (JCRC), Kampala, Uganda. Afr. Health Sci. 2015, 15, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Swaim, M.W.; Agarwal, S.; Rosse, W.F. Successful treatment of hepatitis C in sickle-cell disease. Ann. Intern. Med. 2000, 133, 750–751. [Google Scholar] [CrossRef]
- Babatola, A.O.; Olatunya, O.S.; Faboya, A.O.; Ojo, T.O.; Kayode, S.T.; Komolafe, A.K.; Oyelami, O.A.; Ajayi, O.A. Hepatitis B and C Infections Among Pediatric Patients with Sickle Cell Disease at a Tertiary Hospital in Nigeria. Arch. Pediatr. Infect. Dis. 2020, 8, e101632. [Google Scholar] [CrossRef]
- Mora, N.; Adams, W.H.; Kliethermes, S.; Dugas, L.; Balasubramanian, N.; Sandhu, J.; Nde, H.; Small, C.; Jose, J.; Scaglione, S.; et al. A Synthesis of Hepatitis C prevalence estimates in Sub-Saharan Africa: 2000–2013. BMC Infect. Dis. 2016, 16, 283. [Google Scholar] [CrossRef]
- Owusu, E.D.A.; Visser, B.J.; Nagel, I.M.; Mens, P.F.; Grobusch, M.P. The Interaction Between Sickle Cell Disease and HIV Infection: A Systematic Review. Clin. Infect. Dis. 2014, 60, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Robinson, T.M.; Lanzkron, S.M. Standard Definitions of Pneumococcal Immunity May Not Accurately Predict Protection in Adults with Sickle Cell Disease. Blood 2019, 134, 1014. [Google Scholar] [CrossRef]
- Belisário, A.R.; Blatyta, P.F.; Vivanco, D.; Oliveira, C.D.L.; Carneiro-Proietti, A.B.; Sabino, E.C.; de Almeida-Neto, C.; Loureiro, P.; Mateos, S.d.O.G.; Flor-Park, M.V.; et al. Association of HIV infection with clinical and laboratory characteristics of sickle cell disease. BMC Infect. Dis. 2020, 20, 638. [Google Scholar] [CrossRef]
- Nouraie, M.; Nekhai, S.; Gordeuk, V.R. Sickle cell disease is associated with decreased HIV but higher HBV and HCV comorbidities in U.S. hospital discharge records: A cross-sectional study. Sex. Transm. Infect. 2012, 88, 528–533. [Google Scholar] [CrossRef]
- Odera, E.B.; Kwobah, C.; Stone, G.; Some, F.; Vreeman, R.C. Sickle cell disease and HIV: A case highlighting management challenges for children in a resource-limited setting. J. Int. Assoc. Provid. AIDS Care 2014, 13, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Neto, J.P.M.; Lyra, I.M.; Reis, M.G.; Goncalves, M.S. The association of infection and clinical severity in sickle cell anaemia patients. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 121–126. [Google Scholar] [CrossRef]
- Kelly, S.; Jacobs, E.S.; Stone, M.; Keating, S.M.; Lee, T.-H.; Chafets, D.; Heitman, J.; Dimapasoc, M.; Operskalski, E.; Hagar, W.; et al. Influence of sickle cell disease on susceptibility to HIV infection. PLoS ONE 2020, 15, e0218880. [Google Scholar] [CrossRef]
- Halstead, S.; Wilder-Smith, A. Severe dengue in travellers: Pathogenesis, risk and clinical management. J. Travel Med. 2019, 26, taz062. [Google Scholar] [CrossRef]
- Martina, B.E.E.; Koraka, P.; Osterhaus, A.D.M.E. Dengue Virus Pathogenesis: An Integrated View. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Spiropoulou, C.F.; Srikiatkhachorn, A. The role of endothelial activation in dengue hemorrhagic fever and hantavirus pulmonary syndrome. Virulence 2013, 4, 525–536. [Google Scholar] [CrossRef]
- Rankine-mullings, A.; Reid, M.E.; Moo, M.; Richards-dawson, M.; Knight, J.M. A Retrospective Analysis of the Signi fi cance of Haemoglobin SS and SC in Disease Outcome in Patients With Sickle Cell Disease and Dengue Fever. EBioMedicine 2015, 2, 937–941. [Google Scholar] [CrossRef]
- Jentes, E.S.; Lash, R.R.; Johansson, M.A.; Sharp, T.M.; Henry, R.; Brady, O.J.; Sotir, M.J.; Hay, S.I.; Margolis, H.S.; Brunette, G.W. Evidence-based risk assessment and communication: A new global dengue-risk map for travellers and clinicians. J. Travel Med. 2016, 23, taw062. [Google Scholar] [CrossRef]
- Moesker, F.M.; Muskiet, F.D.; Koeijers, J.J.; Fraaij, P.L.A.; Gerstenbluth, I.; van Gorp, E.C.M.; Osterhaus, A.D.M.E. Fatal Dengue in Patients with Sickle Cell Disease or Sickle Cell Anemia in Curaçao: Two Case Reports. PLoS Negl. Trop. Dis. 2013, 7, e2203. [Google Scholar] [CrossRef] [PubMed]
- Elenga, N.; Celicourt, D.; Muanza, B.; Elana, G.; Hocquelet, S.; Tarer, V.; Maillard, F.; Sibille, G.; Doumdo, L.D.; Petras, M.; et al. Dengue in hospitalized children with sickle cell disease: A retrospective cohort study in the French departments of America. J. Infect. Public Health 2019, 13, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Elia, G.M.; Angel, A.; Regacini, R.; Nais, R.P.; dos Santos, A.R.A.; Vieira, P.P.M.G.; Braga, J.A.P. Acute chest syndrome and COVID-19 in sickle cell disease pediatric patients. Hematol. Transfus. Cell Ther. 2020, 43, 104–108. [Google Scholar] [CrossRef]
- Garg, S.; Kim, L.; Whitaker, M.; O’Halloran, A.; Cummings, C.; Holstein, R.; Prill, M.; Chai, S.J.; Kirley, P.D.; Alden, N.B.; et al. Hospitalization Rates and Characteristics of Patients Hospitalized with Laboratory-Confirmed Coronavirus Disease 2019—COVID-NET, 14 States, March 1–30, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 458–464. [Google Scholar] [CrossRef]
- Telfer, P.; De La Fuente, J.; Sohal, M.; Brown, R.; Eleftheriou, P.; Roy, N.; Piel, F.B.; Chakravorty, S.; Gardner, K.; Velangi, M.; et al. Real-time national survey of COVID-19 in hemoglobinopathy and rare inherited anemia patients. Haematologica 2020, 105, 2651–2654. [Google Scholar] [CrossRef]
- Minniti, C.P.; Zaidi, A.U.; Nouraie, M.; Manwani, D.; Crouch, G.D.; Crouch, A.S.; Callaghan, M.U.; Carpenter, S.; Jacobs, C.; Han, J.; et al. Clinical predictors of poor outcomes in patients with sickle cell disease and COVID-19 infection. Blood Adv. 2021, 5, 207–215. [Google Scholar] [CrossRef]
- Panepintoa, J.A.; Brandow, A.; Mucalo, L.; Yusuf, F.; Singh, A.; Taylor, B.; Woods, K.; Payne, A.B.; Peacock, G.; Schieve, L.A. Coronavirus Disease among Persons with Sickle Cell Disease, United States, March 20–May 21, 2020. Emerg. Infect. Dis. 2020, 26, 2473. [Google Scholar] [CrossRef]
- Chakravorty, S.; Padmore-Payne, G.; Ike, F.; Tshibangu, V.; Graham, C.; Rees, D.; Stuart-Smith, S. COVID-19 in patients with sickle cell disease—A case series from a UK Tertiary Hospital. Haematologica 2020, 105, 2691–2693. [Google Scholar] [CrossRef] [PubMed]
- Arlet, J.-B.; de Luna, G.; Khimoud, D.; Odièvre, M.-H.; de Montalembert, M.; Joseph, L.; Chantalat-Auger, C.; Flamarion, E.; Bartolucci, P.; Lionnet, F.; et al. Prognosis of patients with sickle cell disease and COVID-19: A French experience. Lancet Haematol. 2020, 7, e632–e634. [Google Scholar] [CrossRef] [PubMed]
- Menapace, L.A.; Thein, S.L. COVID-19 and sickle cell disease. Haematologica 2020, 105, 2501–2504. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Perisetti, A.; Kathirvelu, B.; Gajendran, M.; Ghanta, S.; Onukogu, I.; Lao, T.; Anwer, F. Low morbidity and mortality with COVID-19 in sickle cell disease: A single center experience. Ejhaem 2020, 1, 608–614. [Google Scholar] [CrossRef]
- AbdulRahman, A.; AlAli, S.; Yaghi, O.; Shabaan, M.; Otoom, S.; Atkin, S.L.; AlQahtani, M. COVID-19 and sickle cell disease in Bahrain. Int. J. Infect. Dis. 2020, 101, 14–16. [Google Scholar] [CrossRef]
- Hussain, F.A.; Njoku, F.U.; Saraf, S.L.; Molokie, R.E.; Gordeuk, V.R.; Han, J. COVID-19 infection in patients with sickle cell disease. Br. J. Haematol. 2020, 189, 851–852. [Google Scholar] [CrossRef]
- Balanchivadze, N.; Kudirka, A.A.; Askar, S.; Almadhoun, K.; Kuriakose, P.; Fadel, R.; Dabak, V. Impact of COVID-19 Infection on 24 Patients with Sickle Cell Disease. One Center Urban Experience, Detroit, MI, USA. Hemoglobin 2020, 44, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.C.; Murphy, M.L.; Hendricks, H.; Dulek, D.E.; Volanakis, E.J.; Borinstein, S.C. COVID-19 pneumonia in a pediatric sickle cell patient requiring red blood cell exchange. Clin. Case Rep. 2021, 9, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Sahu, K.K.; George, L.; Jones, N.; Mangla, A. COVID-19 in patients with sickle cell disease: A single center experience from Ohio, United States. J. Med. Virol. 2021, 93, 2591–2594. [Google Scholar] [CrossRef] [PubMed]
- Lakkakula, B.V.K.S.; Pattnaik, S. The HBG2 rs7482144 (C > T) Polymorphism is Linked to HbF Levels but not to the Severity of Sickle Cell Anemia. J. Pediatr. Genet. 2021, 12, 129–134. [Google Scholar] [CrossRef]
- Yazdany, J.; Kim, A.H.J. Use of Hydroxychloroquine and Chloroquine During the COVID-19 Pandemic: What Every Clinician Should Know. Ann. Intern. Med. 2020, 172, 754–755. [Google Scholar] [CrossRef]
- Luzzatto, L. Sickle Cell Anaemia and Malaria. Mediterr. J. Hematol. Infect. Dis. 2012, 4, e2012065. [Google Scholar] [CrossRef]
- Mwaiswelo, R.O.; Mawala, W.; Iversen, P.O.; de Montalembert, M.; Luzzatto, L.; Makani, J. Sickle cell disease and malaria: Decreased exposure and asplenia can modulate the risk from Plasmodium falciparum. Malar. J. 2020, 19, 165. [Google Scholar] [CrossRef]
- Achkar, M.A.; Rogers, J.S.; Muszynski, M.J. Resistance to Plasmodium falciparum in sickle cell trait erythrocytes is driven by oxygen-dependent growth inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, 7350–7355. [Google Scholar]
- Dada-Adegbola, H.O.; Brown, B.J.; Labaeka, A.A. Prevalence of malaria and performance of a rapid diagnostic test for malaria in febrile children with sickle cell disease. Pediatr. Hematol. Oncol. J. 2018, 3, 42–45. [Google Scholar] [CrossRef]
- Goheen, M.; Wegmüller, R.; Bah, A.; Darboe, B.; Danso, E.; Affara, M.; Gardner, D.; Patel, J.; Prentice, A.; Cerami, C. Anemia Offers Stronger Protection Than Sickle Cell Trait Against the Erythrocytic Stage of Falciparum Malaria and This Protection Is Reversed by Iron Supplementation. EBioMedicine 2016, 14, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cholera, R.; Brittain, N.J.; Gillrie, M.R.; Lopera-Mesa, T.M.; Diakité, S.A.S.; Arie, T.; Krause, M.A.; Guindo, A.; Tubman, A.; Fujioka, H.; et al. Impaired cytoadherence of Plasmodium falciparum -infected erythrocytes containing sickle hemoglobin. Proc. Natl. Acad. Sci. USA 2008, 105, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Eleonore, N.L.E.; Cumber, S.N.; Charlotte, E.E.; Lucas, E.E.; Edgar, M.M.L.; Nkfusai, C.N.; Geh, M.M.; Ngenge, B.M.; Bede, F.; Fomukong, N.H.; et al. Malaria in patients with sickle cell anaemia: Burden, risk factors and outcome at the Laquintinie hospital, Cameroon. BMC Infect. Dis. 2020, 20, 40. [Google Scholar] [CrossRef]
- White, N.J. Anaemia and malaria. Malar. J. 2018, 17, 371. [Google Scholar] [CrossRef]
- Atiku, S.M.; Louise, N.; Kasozi, D.M. Severe oxidative stress in sickle cell disease patients with uncomplicated Plasmodium falciparum malaria in Kampala, Uganda. BMC Infect. Dis. 2019, 19, 600. [Google Scholar] [CrossRef]
- Ahmed, S.; Uraka, J. Impact of intestinal parasites on haematological parameters of sickle-cell anaemia patients in Nigeria. East. Mediterr. Heal. J. 2011, 17, 710–713. [Google Scholar] [CrossRef]
- Mahdi, N.K.; Ali, N. Intestinal parasites, including Cryptosporidium species, in Iraqi patients with sickle-cell anaemia. East. Mediterr. Heal. J. 2002, 8, 345–349. [Google Scholar] [CrossRef]
- Ahmed, S.G.; Ibrahim, U. A compendium of pathophysiologic basis of etiologic risk factors for painful vaso-occlusive crisis in sickle cell disease. Niger. J. Basic Clin. Sci. 2017, 14, 57. [Google Scholar] [CrossRef]
- Hernigou, P.; Daltro, G.; Flouzat-Lachaniette, C.-H.; Roussignol, X.; Poignard, A. Septic Arthritis in Adults with Sickle Cell Disease Often is Associated with Osteomyelitis or Osteonecrosis. Clin. Orthop. Relat. Res. 2010, 468, 1676–1681. [Google Scholar] [CrossRef]
- Guabiraba, R.; Ryffel, B. Dengue virus infection: Current concepts in immune mechanisms and lessons from murine models. Immunology 2014, 141, 143–156. [Google Scholar] [CrossRef]
- Motran, C.C.; Silvane, L.; Chiapello, L.S.; Theumer, M.G.; Ambrosio, L.F.; Volpini, X.; Celias, D.P.; Cervi, L. Helminth Infections: Recognition and Modulation of the Immune Response by Innate Immune Cells. Front. Immunol. 2018, 9, 664. [Google Scholar] [CrossRef] [PubMed]
- Seed, J.R. Protozoa: Pathogenesis and Defenses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; The University of Texas Medical Branch: Galveston, TX, USA, 1996. [Google Scholar]
- Alsayegh, F.; Mousa, S.A. Challenges in the Management of Sickle Cell Disease During SARS-CoV-2 Pandemic. Clin. Appl. Thromb. Hemost. 2020, 26, 1076029620955240. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| System/Infection | Mechanism | Microorganisms Involved | Clinical Manifestation | Prophylaxis | Treatment | Reference |
|---|---|---|---|---|---|---|
| Bacteremia/sepsis | Lack of IgG and IgM antibody response, impairments in splenic complement and opsonophagocytic functions | S. pneumoniae, N. meningitides, H. influenza, E. coli, K. pneumonia, S. aureus, Salmonella sp. | Septic shock with multi-organ failure | Diphtheria/tetanus/pertussis/HIB/polio/13-valent pneumococcal vaccine; penicillin V; Salmonella typhi vaccine | S. pneumonia and other Bacteroides: third-generation cephalosporins; S. aureus: oxacillin, nafcillin, or cefazolin; vancomycin, clindamycin | [67,68,69,70] |
| Tuberculosis (Mycobacterium tuberculosis) | Cold agglutinations of anti-I specificity, hyperhaemolysis episodes | Mycobacterium tuberculosis | Vaso-occlusive pain episodes, hemolysis, acute chest syndrome; pulmonary dysfunction, anemia, bone and joint infections | Bacillus Calmette–Guerin (BCG) vaccine | Rifampin, isoniazid, pyrazinamide, and ethambutol for the first 2 months, and isoniazid and rifampin for the remaining 4 months | [61,103,104,105] |
| Meningitis | HLA polymorphism | H. influenzae, H. meningitis, E. coli, S. pneumonia | Renal dysfunction, stroke, thrombosis, silent cerebral infarction, cognitive abnormalities | Diphtheria/tetanus/pertussis/HIB/pneumococcal vaccine/meningococcal vaccine; S. pneumoniae 23-valent vaccine; meningococcal vaccine; penicillin V prophylaxis | Third-generation cephalosporins; beta-lactam antibiotics; amphotericin B; intravenous acyclovir | [84,87,88] |
| Human immunodeficiency infection | Low CD4/CD8 ratio | HIV-1 and HIV-2 | Acute chest syndrome, pneumonia, sepsis, hemorrhagic stroke, abnormal transcranial Doppler, and pulmonary hypertension | Using protection while having sex; blood should be screened for HIV before transfusion; using new and sterile needle for injection, and not using same needle for different persons | HIV expert consultation is recommended | [124] |
| Osteomyelitis | Abnormal opsonizing and complement function; HLA polymorphism | Staphylococcus, Pneumococcus, and actinomycetes; Typhi and non-typhi Salmonella, Gram-negative enteric bacteria, S. aureus | Avascular necrosis, leg ulceration (skin), osteonecrosis, bone inflammation, bowel ischemia, and vaso-occlusive crisis | Diphtheria/tetanus/pertussis/HIB; S. pneumoniae 23-valent vaccine; penicillin V prophylaxis | Ceftriaxone, cefotaxime; S. aureus: oxacillin, nafcillin, orcefazolin (MSSA); vancomycin, clindamycin | [89,93,95,179] |
| Urinary tract infection | Renal lesions, medullary ischemia, and perturb | Gram-negative pathogens, Staphylococcus species E. coli | Impaired kidney function, scarification, severe septicemia, acute post-infection | Prevention against hypoxia, acidosis, hypothermia, infection, and hypovolemia, which give rise to vaso-occlusive crisis | Third-generation cephalosporin (ceftriaxone, cefotaxime) | [111,112,113,114] |
| Gastrointestinal | Accumulation of microbes and their products activate neutrophils, resulting in VOC | Gram-negative bacteria, including Typhi and non-typhi Salmonella, Enterococci, and anaerobic bacteria | Common biliary duct obstruction, cholestasis, hepatic vaso-occlusive crisis, hepatic sequestration, hepatic fibrosis, and bowel infarcts | Prevention against hypoxia, acidosis, hypothermia, infection, and hypovolemia, which give rise to vaso-occlusive crisis; for gallstone formation: hydroxyurea, ursodiol, ursodoxylic acid; Salmonella typhi vaccine for typhoid fever | Piperacillin–tazobactam or a carbapenem; third-generation cephalosporin, piperacillin, or trimethoprim-sulfamethoxazole | [123,124,125] |
| Respiratory tract infection | Endothelial dysfunction caused by hemolysis and release of free hemoglobin, which depletes endothelial nitric oxide, resulting in increased vasoconstriction, ischemia, and free radicals | Influenza, S. pneumoniae, Mycoplasma pneumoniae, S. aureus, rhinoviruses, human metapneumo, and para-influenza | Acute chest syndrome, pneumonia, chronic lung disease, pulmonary hypertension | Annual influenza vaccine; diphtheria/tetanus/pertussis/HIB/ 13-valent pneumococcal vaccine; S. pneumoniae 23-valent vaccine; penicillin V prophylaxis, erythromycin if penicillin allergy. | Influenza: oseltamivir, inhaled zanamivir; S. pneumoniae, H. influenza type B: third-generation cephalosporins; S. aureus: oxacillin, nafcillin, or cefazolin; vancomcyin, clindamycin; Mycoplasma pneumoniae: macrolides, quinolones | [123] |
| Dengue | Imbalanced and dysregulated cell-mediated immunity | Arbovirus (Flaviviridae family; genus Flavivirus) | Headaches, fever, abdominal pain, bleeding, myalgias, capillary fragility, vaso-occlusive pain, splenic sequestration, leg ulcers, heart block, plasma leakage, secondary pulmonary and brain edema, hemorrhage, and multiorgan failure | Avoid mosquito exposure by eliminating local mosquito breeding sites by eliminating standing water repositories; clogged rain gutters must be cleared; mosquito repellents: (a) wear long-sleeved clothing; (b) sleep with a mosquito net; and (c) use mosquito repellents; in Dengue-endemic areas, avoid outdoor activities during daylight hours; Dengvaxia vaccine has been approved by the FDA for children aged 9 to 45. | High fluid intake; soft diet; nonsteroidal anti-inflammatory drugs (ibuprofen); blood product transfusion (platelets); steroids, anti-viral therapy (chloroquine, balapiravir, celgosivir) | [145,146,147,180] |
| Malaria | Hypoxia, acidosis, and sickling; decreased deoxyhemoglobin solubility ultimately leads to VOC | P. falciparum, P. vivax, P. ovale, P. malariae, and P. knowlesi | Vaso-occlusive pain episodes, splenic sequestration, severe anemia necessitating blood transfusions causing folate-deficiency anemia, hypoglycemia, acidosis, thrombocytopenia, and multi-organ failure | Avoid mosquito exposure; eliminate local mosquito breeding sites by eliminating standing water repositories; clogged rain gutters must be cleared; mosquito repellents: (a) wear long-sleeved clothing; (b) sleep with a mosquito net; and (c) use mosquito repellents; (d) avoid going outside at dawn and dusk. | Intravenous quinidine until the parasite density < 1% and able to tolerate oral therapy; oral therapy: based on the infecting species, possible drug resistance, and severity of disease | [172,175] |
| Parasitic infections | Stimulation of the growth of antibody-producing B cells rather than stimulation of the proliferation of specific antiparasite B-cells; proliferation of suppressor T-cells and macrophages, which inhibit the immune system by excretion of regulatory cytokines; production by the parasite of specific immune-suppressor substances; damages host tissues, causing the release of stimuli that activate various cells, including innate immune cells such as macrophages, dendritic cells, eosinophils, basophils, and mast cells | Protozoa (other than malaria): E. histolytica, E. coli and G. lamblia Helminths: Ascaris lumbricoides, Ancylostoma duodenale, Trichuris trichiura, Strongyloides stercoralis Schistosomiasis (S. mansoni, S. haematobium, S. japonicum), Toxocara canis, filariasis (Onchocerca volvulus) | Chronic Giardia infection with secondary chronic intestinal malabsorption and failure to thrive; toxic megacolon, fulminant colitis, ulcerations on the colonic mucosa, secondary perforation; hepatic, pleural, lung, and pericardium abscesses (E. histolytica); vaso-occlusive pain episodes, chronic iron deficiency, and chronic eosinophilia; malnutrition, delayed growth, and cognitive deficit; acute intestinal obstruction accompanied by peritonitis and intestinal perforation; appendicitis; common bile duct obstruction accompanied by secondary biliary colic, cholangitis, or pancreatitis; hepatosplenomegaly, bloody diarrhea, portal hypertension, ascites, esophageal varices, and hematemesis are all symptoms of hepatosplenomegaly; visual impairment/blindness (filariasis, T. canis) | -E. histolytica: (a) Asymptomatic cyst excretion: paromomycin or diiodohydroxyquinoline/iodoquinol; (b) invasive colitis or extraintestinal disease: metronidazole or tinidazole, followed by diiodohydroxyquinoline/iodoquinol or paromomycin; (c) percutaneous or surgical aspiration of large liver abscesses; (d) piperacillin–tazobactam or meropenem if peritonitis; -Giardiasis: metronidazole, nitazoxanide, or tinidazole; hand hygiene after defecation; sanitary disposal of fecal material; treatment of drinking water (boiling, chemical disinfection with iodine or chlorine, use of filters); avoid using recreational water venues (e.g., swimming pools, water parks) until asymptomatic and treatment is complete; chemotherapy prophylaxis: albendazole, mebendazole; community-wide mass ivermectin treatment | Albendazole, mebendazole, pyrantel pamoate, ivermectin, doxycycline albendazole, mebendazole, pyrantel pamoate, ivermectin, doxycycline | [176,177,178,181,182] |
| COVID-19 | Progressive endothelial activation with the risk of micro- and macrothrombi, and disseminated intravascular coagulation (DIC) | SARS-CoV-2 | Acute chest syndrome, severe pneumonia, hemolysis, vaso-occlusive pain episodes, stroke, reduced oxygen saturation, fever, headache, sore throat, mild to severe cough, weakness, fatigue, difficulty breathing | Frequent hand sanitization; avoid touching face; wear mask; maintain at least 1 m distance from other persons to avoid infection | Hydroxyurea, L-glutamine, voxelotor, and crizanlizumab with vitamins D, C, and zinc; azithromycin; ivermectin; tocilizumab dexamethasone; convalescent plasma infusion; remdesivir with intravenous dexamethasone | [153,163,164,183] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sahu, T.; Pande, B.; Verma, H.K.; Bhaskar, L.V.K.S.; Sinha, M.; Sinha, R.; Rao, P.V. Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalass. Rep. 2023, 13, 206-229. https://doi.org/10.3390/thalassrep13030019
Sahu T, Pande B, Verma HK, Bhaskar LVKS, Sinha M, Sinha R, Rao PV. Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalassemia Reports. 2023; 13(3):206-229. https://doi.org/10.3390/thalassrep13030019
Chicago/Turabian StyleSahu, Tarun, Babita Pande, Henu Kumar Verma, L V K S Bhaskar, Meenakshi Sinha, Ramanjan Sinha, and Pasupuleti Visweswara Rao. 2023. "Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective" Thalassemia Reports 13, no. 3: 206-229. https://doi.org/10.3390/thalassrep13030019
APA StyleSahu, T., Pande, B., Verma, H. K., Bhaskar, L. V. K. S., Sinha, M., Sinha, R., & Rao, P. V. (2023). Infection and Potential Challenge of Childhood Mortality in Sickle Cell Disease: A Comprehensive Review of the Literature from a Global Perspective. Thalassemia Reports, 13(3), 206-229. https://doi.org/10.3390/thalassrep13030019