Abstract
Clostridioides difficile is a major nosocomial pathogen and has a considerable burden on healthcare systems. Our objective was to determine the transmission patterns of C. difficile in a non-epidemic setting using whole-genome multi-locus sequence typing (wgMLST) and core-genome single-nucleotide polymorphism (cgSNP) analyses. A retrospective study was conducted in a 650-bed university hospital between January 2016 and February 2017. In total, 191 strains isolated from 169 symptomatic C. difficile infection (CDI) patients were analyzed by WGS. Sequences were compared using wgMLST and cgSNP analyses. Genetic data and ward movements were then combined to identify the transmission rate and the type of transmission. The transmission rate varied from 55/169 (19.5%) (wgMLST) to 33/169 (32.5%) (cgSNP). Most transmission was considered cryptic, irrespective of the genetic analysis (38/55 [69.1%] by wgMLST to 25/33 [75.8%] by cgSNP). No transmission within the same ward was observed. In a non-epidemic setting, most C. difficile transmission occurs from sources other than symptomatic CDI patients.
1. Introduction
Clostridioides difficile is a major cause of healthcare-associated diarrhea and is frequently involved in community-acquired diarrhea [1]. The epidemiology of C. difficile infections (CDI) is now well-known, but there are still uncertainties concerning the transmission of C. difficile within the hospital in a non-epidemic setting. Typing methods are essential for the identification of transmission routes and monitoring of the emergence and spread of C. difficile clones worldwide. High-resolution capillary gel-based electrophoresis PCR-ribotyping is the reference typing method in Europe [2]. However, this method lacks the discriminatory power to investigate outbreaks due to common PCR ribotype (RT) strains. Indeed, more than 50% of the strains circulating in France belong to only ten major RTs [3]. More discriminant typing methods (e.g., multi-locus variable number tandem repeat analysis (MLVA)) have been used to document patient-to-patient transmission of strains or investigate outbreaks [4]. The availability of new next-generation sequencing (NGS) techniques enables laboratories to sequence entire genomes at a low cost. Whole-genome sequencing (WGS) provides the most comprehensive overview of a bacterial strain and therefore has greater discriminatory power than conventional molecular typing methods [5]. Strain typing based on WGS is being increasingly used in bacterial epidemiology, both in public health and in infection-control settings, as it allows more accurate monitoring of both transmission mechanisms and the emergence of new clones [6,7,8,9,10,11,12,13,14,15].
Here, we report the first study in France using wgMLST (whole-genome multi-locus sequence typing) and cgSNPs (core-genome single-nucleotide polymorphisms) to determine the transmission rate of C. difficile strains within a single hospital in a non-epidemic setting.
2. Materials and Methods
2.1. Population
This was a single-center retrospective study conducted in a 650-bed, university-affiliated acute-care hospital with 28,250 admissions per year. We included all patients hospitalized between January 2016 and February 2017 presenting with a symptomatic CDI. The following information was collected from the electronic medical files for each patient: age, sex, length of hospital stay, date of CDI diagnosis (positive stool specimen), and initial hospital ward and movements.
2.2. CDI Diagnosis
A CDI case was defined as a patient with clinical diarrhea (three or more loose or watery stools over at least 24 h), without an alternative explanation, and a stool sample positive for C. difficile. CDI testing was performed upon the physician’s request or systematically in cases of healthcare-associated diarrhea. CDI testing was based on a two-step algorithm using glutamate dehydrogenase (GDH) and free-toxin detection by enzyme immunoassay (EIA) (C.Diff Quik Chek Complete®, Abbott Diagnostics, Waltham, MA, USA) as a screening method, followed by a reflex PCR assay targeting toxin genes (GenXpert® C. difficile, Cepheid, Sunnyvale, CA, USA) in cases of toxin-negative GDH-positive results. In addition, for all CDI cases, stool samples were cultured on ChromID plates (bioMérieux, Marcy l’Etoile, France), as described by Couturier et al. [16], and C. difficile isolates were characterized by molecular typing.
A healthcare-associated CDI was defined as a CDI episode occurring 48 h after admission to the hospital or diagnosed within 48 h of admission for patients who had been hospitalized in the previous four weeks [17]. A recurrent CDI was defined by a new CDI episode within six months following the first episode.
2.3. Transmission Analysis
Transmission analysis was performed in two successive steps. The first step consisted of determining the genetic relationship between each C. difficile isolate based on PCR ribotyping and WGS results, including wgMLST and cgSNP analysis. The second step consisted of identifying the epidemiological link between isolates that belonged to a clonal complex using WGS-based methods.
2.4. Molecular Typing and WGS-Based Analysis
2.4.1. PCR-Ribotyping
C. difficile isolates were characterized by high-resolution capillary gel-based electrophoresis PCR-ribotyping [2] on an ABI 3500 sequencer (Applied Biosystems, Waltham, MA, USA), using primers described by Bidet et al. [18], according to the European standardized protocol of the European Centre for Disease Prevention and Control (ECDC) [19]. The RTs were determined using the freely available Webribo database (https://webribo.ages.at/ accessed on 20 June 2019). When the RT was not found in the Webribo database, the strain was identified using the prefix FR-XXX.
2.4.2. Whole Genome Sequencing (WGS)
We performed WGS on 191 isolates. C. difficile isolates were subcultured on Columbia agar (bioMérieux) for 48 h in an anaerobic atmosphere. DNA was extracted using a DNeasy UltraClean Microbial kit (Qiagen, Hilden, Germany). Then, the DNA library was prepared using a Nextera XT DNA library kit (Illumina, San Diego, CA, USA). Finally, we generated 2 × 150 base paired-end reads using a NextSeq500 High Output v2.0 300 cycles kit on a NextSeq 500 instrument (Illumina). Once the quality of the reads was evaluated, de novo assembly was performed using the SPAdes algorithm [20]. Both wgMLST and cgSNP analyses were performed for all isolates using BioNumerics 8.0 (Applied Maths, Sint-Martens-Latem, Belgium) [4]. The data have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB54703 (https://www.ebi.ac.uk/ena/browser/view/PRJEB54703 accessed on 20 June 2019).
For wgMLST, alleles were identified by combining a k-mer frequency approach using sequence reads (assembly-free) and a BLAST approach using contigs (assembly-based). Allele calling was performed from a pan-genome locus scheme of C. difficile developed by Applied Maths. This wgMLST scheme contains 8745 coding loci, representing a pan-genome of C. difficile identified from 259 previously published genomes. Once all alleles were assigned to each genome, a minimum spanning tree (MST) was constructed. Loci with no allele calls and those with <80% identity were ignored in the pairwise comparison during the tree construction. The genetic relationship between two isolates was assessed by calculating the number of different alleles for wgMLST. Isolates with an allelic difference ≤20 were defined as belonging to the same clonal complex, those with an allelic difference between 21 and 200 as genetically related, and those with an allele difference >200 as unrelated [4].
For cgSNP analysis, one reference sequence was chosen among each set of isolate sequences of the same RT. Among each, the isolate with the highest quality assembly (lowest number of contigs and highest N50 value) was defined as the reference core genome sequence for mapping using the Bowtie 2 algorithm [21]. The resulting SNPs were filtered using the strict SNP filtering template, as described by Gateau et al. [4]. Isolates with ≤10 SNP differences were defined as belonging to the same clonal complex, those with between 11 and 100 SNP differences as genetically related, and those with >100 SNP differences as unrelated. In addition, an overall cgSNP analysis including all isolates was performed using the C. difficile CD630 strain as the reference genome (GenBank: AM180355.1) and a MST containing all strains was generated.
2.5. Epidemiological Link Determination
2.5.1. Hypotheses
Hypotheses were formulated for the infectious period (duration between the first positive sample from the “donor” and contact with the “recipient”), the incubation period (duration between contact with the “donor” and the first positive sample from the “recipient”), and the persistence of spores in the environment. The maximum times for each of these periods were those used by Kong et al. [11]: eight weeks for the infectious period, 12 weeks for the incubation period, and 26 weeks for the persistence of spores in the environment. Furthermore, we considered that a patient could be contagious for up to a week before the diagnosis of the CDI [13].
2.5.2. Interpretation of the Epidemiological Link
We analyzed the epidemiological links solely for isolates belonging to the same clonal complex by wgMLST and/or cgSNP analysis. The criteria for determining the most likely transmission routes were those used by Kong et al. [11]. Each case of CDI was classified according to the route of transmission: (i) transmission in the same ward (the “donor” and “recipient” shared time on the same ward), (ii) environmental transmission, i.e., transmission through the environment contaminated with persistent spores (patients were hospitalized in the same ward at different times, provided the “donor” was diagnosed before leaving the ward and the “recipient” was diagnosed after admission to the same ward), (iii) intrahospital transmission (infected patients were hospitalized at the same time in the same hospital but in different wards), and (iv) indeterminate or cryptic transmission (patients with no obvious spatio-temporal epidemiological link, but with clonal C. difficile isolates).
3. Results
3.1. Population
In total, 169 patients were included in this study and 191 isolates (including 22 from recurrent infections) of C. difficile were characterized. The mean age at the time of the CDI was 63 years (range: 16–96 years). More than half of the patients were female (n = 93, 55.0% of cases) and had a healthcare-related CDI (n = 89, 52.7% of cases).
3.2. PCR-Ribotyping
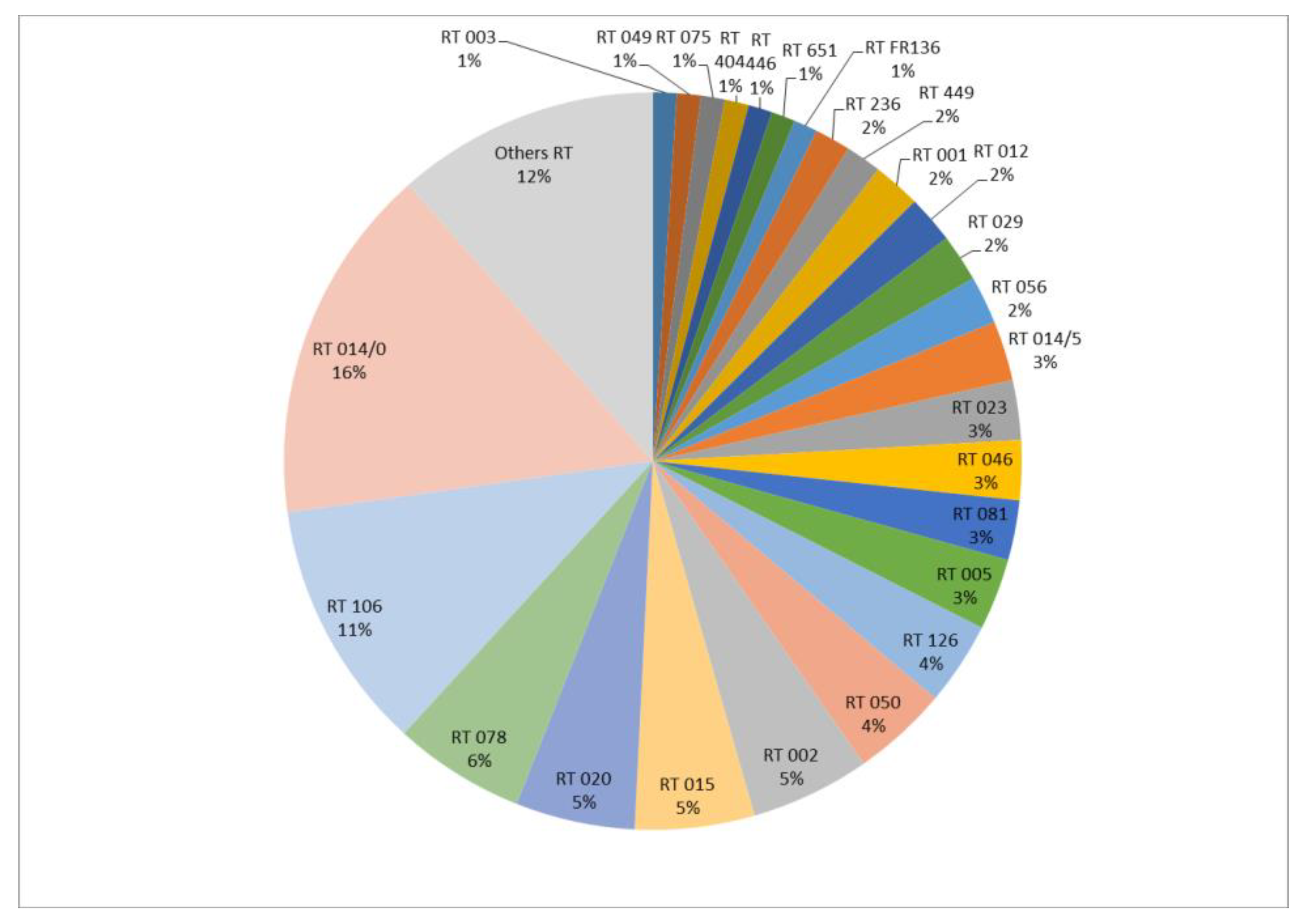
The 191 isolates were divided into 48 RTs. The most frequent RTs were RT 014/0 and RT 106, representing 15.7% (n = 30) and 11% (n = 21) of the isolates, respectively (Figure 1).
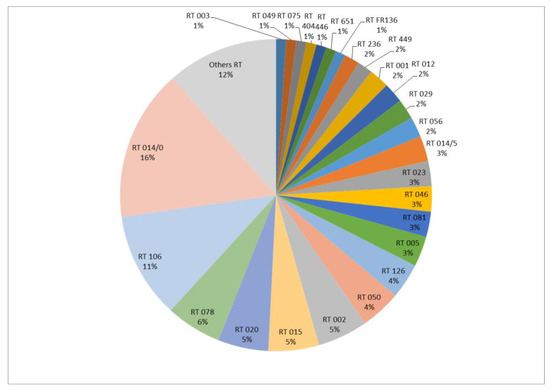
Figure 1.
Distribution of PCR-ribotypes (%) (n = 191 isolates).
Among the 191 isolates, 22 (11.5%) were isolated from recurrent CDIs. Based on the PCR-ribotyping results, 77% of recurrences (n = 17/22) were considered to be relapses (same RT as the initial episode), with a mean time interval of 52 days (min = 8, max = 140), and 23% (n = 5/22) were re-infections (different RTs), with a mean time interval of 74 days (min = 2, max = 182).
3.3. Analysis of the Clonal Link between Strains
Strains belonged to the same clonal complex in 55/169 (32.5%) by wgMLST, and 33/169 (19.5%) by cgSNP analysis (Table 1). The results of wgMLST and cgSNP analysis were concordant for 75.7% of the strains (n = 128/169) (Table 1). No major discordance (i.e., strains belonging to a clonal complex by one method that would not be genetically related by the other method) was observed.

Table 1.
Comparison of the genetic links between wgMLST and cgSNP analyses (n = 169 patients).
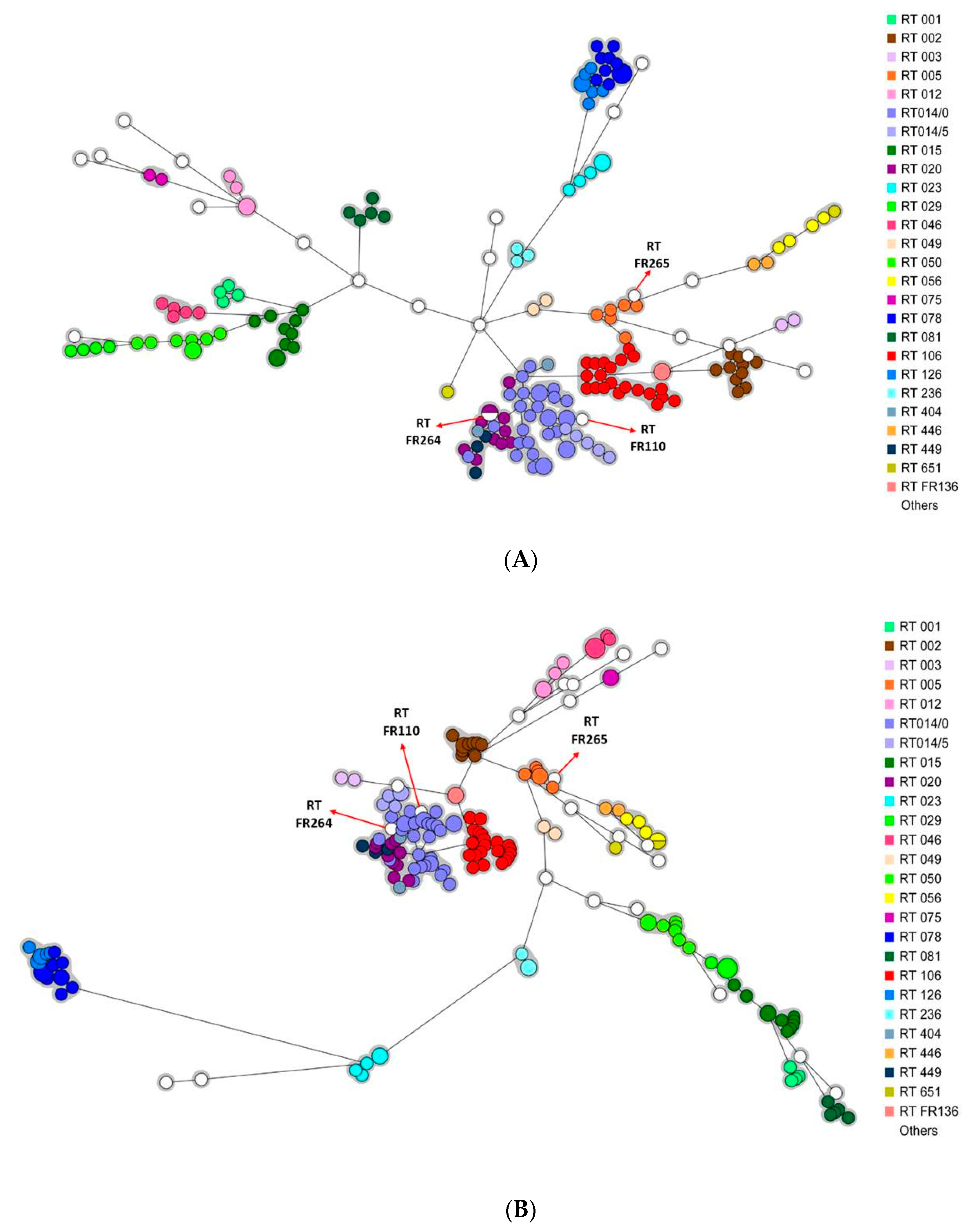
Two minimum spanning trees (MSTs) containing all the strains were generated after wgMLST and cgSNP analysis. Irrespective of the analysis (wgMLST or cgSNP), the strains were mostly grouped according to their RT (Figure 2A,B).
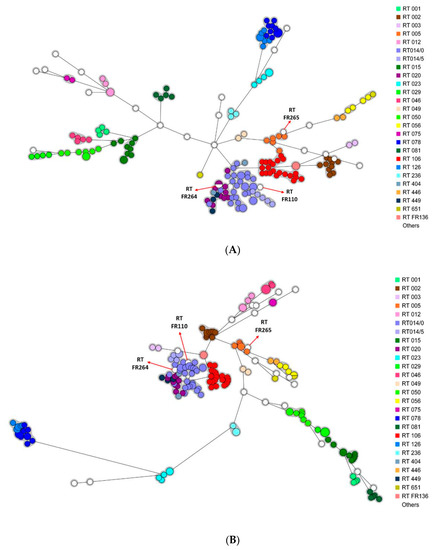
Figure 2.
Minimum spanning trees including all isolates (n = 191). Each color represents a PCR-ribotype (26 different colors). The white color corresponds to PCR-ribotypes including only one isolate (n = 22). A circle represents an isolate, the size of the circle being proportional to the number of identical isolates. (A) wgMLST analysis. The grey area represents a clonal complex (≤20 different alleles). (B) cgSNP analysis. The grey area represents a clonal complex (≤10 different SNPs).
Please note that three strains from RT FR110, FR264, and FR265 were genetically related to strains from another RT (RT 014/0, RT 020, and RT 005, respectively), with 0 to 26 allelic differences by wgMLST and 1 to 43 SNP differences by cgSNP analysis. Comparison of the PCR-ribotyping electrophoresis profiles of each pair of related strains showed only slight variations by one or two peaks.
3.4. Analysis of Transmission Routes
Clonal strains (55 by wgMLST and 33 by cgSNP) were analyzed with respect to ward movement to identify the different transmission routes according to the criteria previously defined (Table 2). Most transmission was considered to be cryptic, irrespective of the genetic analysis (69.1% (n = 38/55) by wgMLST and 75.8% (n = 25/33) by cgSNP analysis). No transmission within the same ward was observed.
Table 2.
Epidemiological links among CDIs due to isolates belonging to clonal complexes.
3.5. Analysis of Strains from Recurrences by wgMLST and cgSNP Analysis
We used wgMLST and cgSNP analysis to compare strains responsible for CDI relapses and re-infections as initially defined by PCR-ribotyping analysis. The results showed 16/17 (94%) strains corresponding to relapses belong to the same clonal complex and 4/5 (80%) strains corresponding to re-infections were genetically unrelated by both wgMLST and cgSNP analysis.
We noticed two discordant results between the PCR-ribotyping and WGS methods. Based on the PCR-ribotyping results, one patient had two relapses due to strain RT 014/0. However, the isolates from the relapses belonged to the same clonal complex but were different from the isolate of the initial episode by 23 alleles by wgMLST and 25 SNPs by cgSNP analysis. Conversely, two isolates from the same patient were considered to be a re-infection based on PCR-ribotyping (RT020 and FR264), although sharing a very similar PCR-ribotype profile. These two isolates were clonal by wgMLST and cgSNP analysis (0 allelic and 1 SNP difference, respectively). More detailed analysis of the profiles obtained by PCR-ribotyping of these two strains showed the only difference between RT 020 and FR264 to correspond to an additional peak for RT FR264 (circled in red in Figure S1). This peak was well individualized and too intense to be an artefact.
4. Discussion
In a non-epidemic setting, we found that the transmission rate of C. difficile varies from 19.5% to 32.5%, depending on the type of WGS analysis. Irrespective of the method used for genetic analysis, most (69.1% to 75.8%) transmission was considered to be cryptic, i.e., from an unknown reservoir. Several authors have used WGS approaches to estimate the transmission of C. difficile within a hospital or at a regional level. In a large study conducted in the United Kingdom, Eyre et al. [13] reported a transmission rate among symptomatic patients of 35%. They found that among the patients whose strains presented a clonal link, 36% showed no evidence of previous contact with another case (cryptic transmission). In another study conducted in Canada, Kong et al. found a transmission rate among symptomatic patients of 46% [11].
The variability observed in the transmission rates can be explained by the epidemiological context of the different studies. For example, the higher transmission rate observed by Kong et al. (46%) can be explained by the epidemic setting in which the study was carried out. Indeed, during the study period, Canada was facing a major outbreak of CDI caused by the RT 027 (NAP1/BI/ST1) strain and most patient-to-patient transmission was due to clone RT 027. This clone has a higher capacity of transmission and dissemination, mainly due to its greater virulence and higher capacity to sporulate [22].
Another source of variability relies on the interpretation of WGS data [15] and the criteria used to define clonal complexes. In our study, the thresholds used to define a clonal link between two strains were 20 alleles for wgMLST and 10 SNPs for cgSNP analysis [4]. However, Eyre et al. used different thresholds for their wgSNV analysis; a genetic relationship was defined as ≤2 SNVs and distinct subtypes were defined by more than 10 different SNVs [13].
This is the first study to compare the minimum spanning trees generated by cgSNP analysis and wgMLST. Our results showed phylogenetic trees to provide relatively consistent information and the strains to be grouped according to their RT (Figure 2A,B) [4].
We observed a low rate of intra- and inter-ward transmission and environmental transmission, suggesting that infection control measures implemented for symptomatic CDI cases are effective in reducing transmission. Most transmission was considered to be “cryptic” (69.1% by wgMLST and 75.8% by cgSNP analysis) and to come from other sources than symptomatic CDI patients. The hypothetical other sources include food transmission or transmission from CDI patients from the community or asymptomatic carriers of C. difficile. Foodborne transmission has been suspected but never clearly demonstrated. Transmission through community contacts has been shown by Eyre et al. [13] using WGS analysis. The contribution of asymptomatic colonized patients in transmission has been documented in several molecular epidemiology studies. Curry et al. [23] used multilocus variable number tandem repeat analysis and showed that 29% of CDI cases could be linked to colonized patients.
This study had several limitations. First, it was conducted in a single acute-care hospital. Therefore, the results cannot be extrapolated to other healthcare settings. Second, only symptomatic patients with a CDI were included. The role of asymptomatic carriers was not considered, although they have shown to be responsible for the transmission of toxigenic strains. Finally, we picked and analyzed only one colony of C. difficile per episode of CDI. However, we cannot exclude that several patients were coinfected by several different strains.
5. Conclusions
In conclusion, the transmission rate of C. difficile varied from 19.5% to 32.5% in a non-epidemic setting, depending on the WGS analysis. No transmission within the same ward was observed. Most transmission was considered to be cryptic and occurred from sources other than symptomatic CDI patients.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microbiolres13030037/s1. Figure S1: Diagrams representing the electrophoregrams of the strains from PCR-ribotypes FR 264 (bottom) and 020 (top). Each peak represents a fragment of different size. The intensity of the fluorescent signal corresponds to the y-axis and the size of DNA fragments (in base pairs) to the x-axis. The additional peak observed for RT FR264 is circled in red.
Author Contributions
Conceptualization: F.B.; methodology, F.B. and V.C.; software, R.S.Z.; validation, F.B., K.L.N. and V.C.; formal analysis, V.C., J.C., C.G., K.L.N. and A.Y.; investigation, J.C. and V.C.; data curation, R.S.Z.; writing—original drafting preparation, V.C.; writing review and editing, F.B.; visualization, F.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This study did not require ethical approval.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data for this study have been deposited in the European Nucleotide Archive (ENA) at EMBL-EBI under accession number PRJEB54703 (https://www.ebi.ac.uk/ena/browser/view/PRJEB54703 accessed on 20 June 2019).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Primers 2016, 2, 16020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawley, W.N.; Knetsch, C.W.; MacCannell, D.R.; Harmanus, C.; Du, T.; Mulvey, M.R.; Paulick, A.; Anderson, L.; Kujiper, E.J.; Wilcox, M.H. Development and validation of an internationally-standardized, high-resolution capillary gel-based electrophoresis PCR-ribotyping protocol for Clostridium difficile. PLoS ONE 2015, 10, e0118150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.A.; Ashwin, H.; Longshaw, C.M.; Burns, D.A.; Davis, G.L.; Wilcox, M.H.; EUCLID Study Group. Diversity of Clostridium difficile PCR ribotypes in Europe: Results from the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID), 2012 and 2013. Eurosurveillance 2016, 21, 30294. [Google Scholar] [CrossRef] [PubMed]
- Gateau, C.; Deboscker, S.; Couturier, J.; Vogel, T.; Schmitt, E.; Muller, J.; Ménard, C.; Turcan, B.; Syed Zaidi, R.; Youssouf, A.; et al. Local outbreak of Clostridioides difficile PCR-Ribotype 018 investigated by multi locus variable number tandem repeat analysis, whole genome multi locus sequence typing and core genome single nucleotide polymorphism typing. Anaerobe 2019, 60, 102087. [Google Scholar] [CrossRef]
- Parkhill, J.; Wren, B.W. Bacterial epidemiology and biology—Lessons from genome sequencing. Genome Biol. 2011, 12, 230. [Google Scholar] [CrossRef] [Green Version]
- Köser, C.U.; Holden, M.T.; Ellington, M.J.; Cartwright, E.J.; Brown, N.M.; Ogilvy-Stuart, A.L.; Hsu, L.Y.; Chewapreecha, C.; Croucher, N.J.; Harris, S.R.; et al. Rapid whole-genome sequencing for investigation of a neonatal MRSA outbreak. N. Engl. J. Med. 2012, 366, 2267–2275. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Fawley, W.N.; Best, E.L.; Griffiths, D.; Stoesser, N.E.; Crook, D.W.; Peto, T.E.A.; Walker, A.S.; Wilcox, M.H. Comparison of multilocus variable-number tandem-repeat analysis and whole-genome sequencing for investigation of Clostridium difficile transmission. J. Clin. Microbiol. 2013, 51, 4141–4149. [Google Scholar] [CrossRef] [Green Version]
- Didelot, X.; Eyre, D.W.; Cule, M.; Ip, C.L.; Ansari, M.A.; Griffiths, D.; Vaughan, A.; O’Connor, L.; Golubchik, T.; Batty, E.M.; et al. Microevolutionary analysis of Clostridium difficile genomes to investigate transmission. Genome Biol. 2012, 13, R118. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Walker, A.S.; Freeman, J.; Baines, S.D.; Fawley, W.N.; Chilton, C.H.; Griffiths, D.; Vaughan, A.; Crook, D.W.; Peto, T.E.A.; et al. Short-term genome stability of serial Clostridium difficile ribotype 027 isolates in an experimental gut model and recurrent human disease. PLoS ONE 2013, 8, e63540. [Google Scholar] [CrossRef]
- Groß, U.; Brzuszkiewicz, E.; Gunka, K.; Starke, J.; Riedel, T.; Bunk, B.; Spröer, C.; Wetzel, D.; Poehlein, A.; Chibani, C.; et al. Comparative genome and phenotypic analysis of three Clostridioides difficile strains isolated from a single patient provide insight into multiple infection of C. difficile. BMC Genom. 2018, 19, 1. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.Y.; Eyre, D.W.; Corbeil, J.; Raymond, F.; Walker, A.S.; Wilcox, M.H.; Crook, D.W.; Michaud, S.; Toye, B.; Frost, E.; et al. Clostridium difficile: Investigating transmission patterns between infected and colonized patients using whole genome sequencing. Clin. Infect. Dis. 2019, 68, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D.W.; Griffiths, D.; Vaughan, A.; Golubchik, T.; Acharya, M.; O’Connor, L.; Crook, D.W.; Walker, A.S.; Peto, T.E.A. Asymptomatic Clostridium difficile colonisation and onward transmission. PLoS ONE 2013, 8, e78445. [Google Scholar] [CrossRef]
- Eyre, D.W.; Cule, M.L.; Wilson, D.J.; Griffiths, D.; Vaughan, A.; O’Connor, L.; Ip, C.L.C.; Golubchick, T.; Batty, E.M.; Finney, J.M.; et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N. Engl. J. Med. 2013, 369, 1195–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, D.R.; Riley, T.V. Genomic delineation of zoonotic origins of Clostridium difficile. Front. Public Health 2019, 7, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janezic, S.; Rupnik, M. Development and implementation of whole genome sequencing-based typing schemes for Clostridioides difficile. Front. Public Health 2019, 7, 309. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Franconeri, L.; Janoir, C.; Ferraris, L.; Syed-Zaidi, R.; Youssouf, A.; Gateau, C.; Hoys, S.; Aires, J.; Barbut, F. Characterization of non-toxigenic Clostridioides difficile strains isolated from preterm neonates and in vivo study of their protective effect. J. Clin. Med. 2020, 9, 3650. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.C.; Coignard, B.; Dubberke, E.; Song, X.; Horan, T.; Kutty, P.K.; The Ad Hoc Clostridium difficile Surveillance Working Group. Recommendations for surveillance of Clostridium difficile-associated disease. Infect. Control. Hosp. Epidemiol. 2007, 28, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Bidet, P.; Barbut, F.; Lalande, V.; Burghoffer, B.; Petit, J.-C. Development of a new PCR-ribotyping method for Clostridium difficile based on ribosomal RNA gene sequencing. FEMS Microbiol. Lett. 1999, 175, 261–266. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Laboratory Procedures for Diagnosis and Typing of Human Clostridium difficile Infection. LU: Publications Office. 2018. Available online: https://data.europa.eu/doi/10.2900/04291 (accessed on 2 April 2021).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prijibelski, A.V.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Merrigan, M.; Venugopal, A.; Mallozzi, M.; Roxas, B.; Viswanathan, V.K.; Johnson, S.; Gerding, D.N.; Vedantam, G. Human hypervirulent Clostridium difficile strains exhibit increased sporulation as well as robust toxin production. J. Bacteriol. 2010, 192, 4904–4911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, S.R.; Muto, C.A.; Schlackman, J.L.; Pasculle, A.W.; Shutt, K.A.; Marsh, J.W.; Harrison, L.H. Use of multilocus variable number of tandem repeats analysis genotyping to determine the role of asymptomatic carriers in Clostridium difficile transmission. Clin. Infect. Dis. 2013, 57, 1094–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).