
Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP)

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Subjects and Study Design
2.2. Data Collection
2.3. Statistical Analysis
3. Results
3.1. Clinical Presentation of Probands and Relatives
3.2. Electrocardiography and Arrhythmias
3.3. Cardiovascular Imaging
3.4. Histopathology
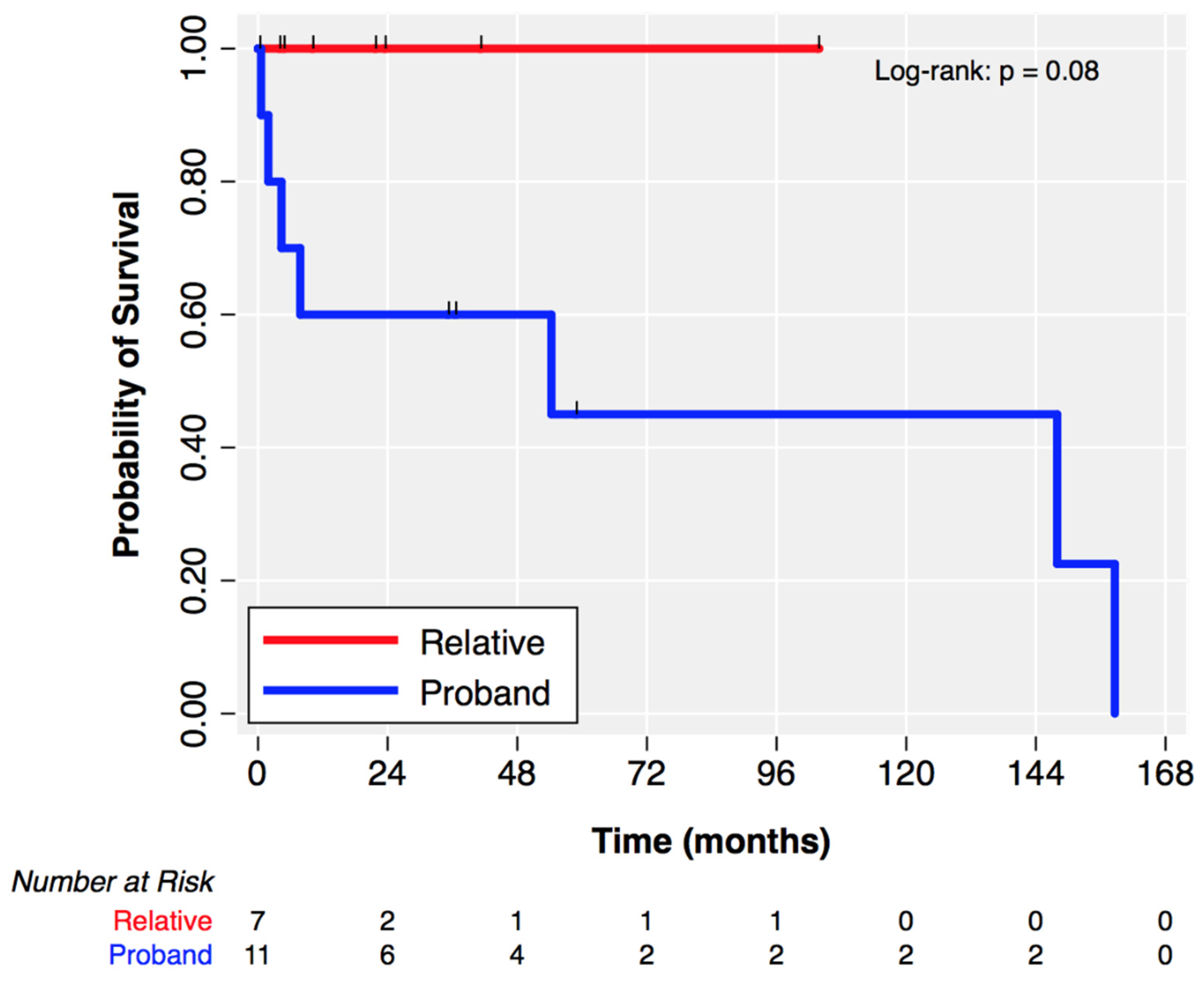
3.5. Outcomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McNally, E.M.; Mestroni, L. Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 2017, 121, 731–748. [Google Scholar] [CrossRef] [PubMed]
- Mestroni, L.; Maisch, B.; McKenna, W.J.; Schwartz, K.; Charron, P.; Rocco, C.; Tesson, F.; Richter, R.; Wilke, A.; Komajda, M.; et al. Guidelines for the study of familial dilated cardiomyopathies. Eur. Heart J. 1999, 20, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Spezzacatene, A.; Sinagra, G.; Merlo, M.; Barbati, G.; Graw, S.L.; Brun, F.; Slavov, D.; Di Lenarda, A.; Salcedo, E.E.; Towbin, J.A.; et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J. Am. Heart Assoc. 2015, 4, e002149. [Google Scholar] [CrossRef] [Green Version]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.; Ackerman, M.J.; Calkins, H.; Darrieux, F.; Daubert, J.P.; De Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.A.; Fuchs, E. Defining the interactions between intermediate filaments and desmosomes. J. Cell Biol. 1998, 141, 1229–1241. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Reuter, C.M.; Dries, A.M.; Parikh, V.N. Arrhythmogenic Cardiomyopathy: Mechanisms, Genetics, and Their Clinical Implications. Curr. Cardiovasc. Risk Rep. 2021, 15, 7. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Protonotarios, A.; Brodehl, A.; Asimaki, A.; Jager, J.; Quinn, E.; Stanasiuk, C.; Ratnavadivel, S.; Futema, M.; Akhtar, M.M.; Gossios, T.D.; et al. The Novel Desmin Variant p.Leu115Ile Is Associated with a Unique Form of Biventricular Arrhythmogenic Cardiomyopathy. Can. J. Cardiol. 2020, 37, 857–866. [Google Scholar] [CrossRef]
- Jiménez, F.J.B.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.; Álvarez, M.; López-Fernández, S.; et al. Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity, and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef]
- Castelletti, S.; Vischer, A.S.; Syrris, P.; Crotti, L.; Spazzolini, C.; Ghidoni, A.; Parati, G.; Jenkins, S.; Kotta, M.-C.; McKenna, W.J.; et al. Desmoplakin missense and non-missense mutations in arrhythmogenic right ventricular cardiomyopathy: Genotype-phenotype correlation. Int. J. Cardiol. 2017, 249, 268–273. [Google Scholar] [CrossRef] [Green Version]
- Bauce, B.; Basso, C.; Rampazzo, A.; Beffagna, G.; Daliento, L.; Frigo, G.; Malacrida, S.; Settimo, L.; Danieli, G.; Thiene, G.; et al. Clinical profile of four families with arrhythmogenic right ventricular cardiomyopathy caused by dominant desmoplakin mutations. Eur. Heart J. 2005, 26, 1666–1675. [Google Scholar] [CrossRef] [Green Version]
- Sen-Chowdhry, S.; Syrris, P.; Ward, D.; Asimaki, A.; Sevdalis, E.; McKenna, W.J. Clinical and Genetic Characterization of Families with Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Provides Novel Insights into Patterns of Disease Expression. Circulation 2007, 115, 1710–1720. [Google Scholar] [CrossRef] [Green Version]
- Quarta, G.; Muir, A.; Pantazis, A.; Syrris, P.; Gehmlich, K.; Garcia-Pavia, P.; Ward, D.; Sen-Chowdhry, S.; Elliott, P.; McKenna, W.J. Familial Evaluation in Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2011, 123, 2701–2709. [Google Scholar] [CrossRef]
- Bhonsale, A.; Groeneweg, J.A.; James, C.A.; Dooijes, D.; Tichnell, C.; Jongbloed, J.D.H.; Murray, B.; Te Riele, A.S.J.M.; Van Den Berg, M.P.; Bikker, H.; et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated mutation carriers. Eur. Heart J. 2015, 36, 847–855. [Google Scholar] [CrossRef]
- Zorzi, A.; Rigato, I.; Rampazzo, A.; Basso, C.; Bauce, B.; Corrado, D.; Pilichou, K.; Marra, M.P.; Migliore, F.; Mazzotti, E.; et al. Phenotypic expression is a prerequisite for malignant arrhythmic events and sudden cardiac death in arrhythmogenic right ventricular cardiomyopathy. EP Europace 2016, 18, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneweg, J.A.; Bhonsale, A.; James, C.A.; Te Riele, A.S.; Dooijes, D.; Tichnell, C.; Murray, B.; Wiesfeld, A.C.; Sawant, A.C.; Kassamali, B.; et al. Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 2015, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Haan, A.D.D.; Tan, B.Y.; Zikusoka, M.N.; Lladó, L.I.; Jain, R.; Daly, A.; Tichnell, C.; James, C.; Amat-Alarcon, N.; Abraham, T.; et al. Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Cardiovasc. Genet. 2009, 2, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. J. Card. Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Chitipiralla, S.; Brown, G.R.; Chen, C.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; Kaur, K.; Liu, C.; et al. ClinVar: Improvements to accessing data. Nucleic Acids Res. 2020, 48, D835–D844. [Google Scholar] [CrossRef]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy/Dysplasia. Circulation 2010, 121, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Fressart, V.; Duthoit, G.; Donal, E.; Probst, V.; Deharo, J.-C.; Chevalier, P.; Klug, D.; Dubourg, O.; Delacretaz, E.; Cosnay, P.; et al. Desmosomal gene analysis in arrhythmogenic right ventricular dysplasia/cardiomyopathy: Spectrum of mutations and clinical impact in practice. EP Europace 2010, 12, 861–868. [Google Scholar] [CrossRef]
- Sen-Chowdhry, S.; Syrris, P.; Prasad, S.K.; Hughes, S.E.; Merrifield, R.; Ward, D.; Pennell, D.; McKenna, W.J. Left-dominant arrhythmogenic cardiomyopathy: An under-recognized clinical entity. J. Am. Coll. Cardiol. 2008, 52, 2175–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, P.; O’Mahony, C.; Syrris, P.; Evans, A.; Sorensen, C.R.; Sheppard, M.; Carr-White, G.; Pantazis, A.; McKenna, W.J. Prevalence of desmosomal protein gene mutations in patients with dilated cardiomyopathy. Circ. Cardiovasc. Genet. 2010, 3, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ayala, J.M.; Gómez-Milanés, I.; Muñoz, J.J.S.; Ruiz-Espejo, F.; Ortíz, M.; González-Carrillo, J.; López-Cuenca, D.; Oliva-Sandoval, M.J.; Monserrat, L.; Valdés, M.; et al. Desmoplakin truncations and arrhythmogenic left ventricular cardiomyopathy: Characterizing a phenotype. EP Europace 2014, 16, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Pavia, P.; Syrris, P.; Salas, C.; Evans, A.; Mirelis, J.G.; Cobo-Marcos, M.; Vilches, C.; Bornstein, B.; Segovia, J.; Alonso-Pulpon, L.; et al. Desmosomal protein gene mutations in patients with idiopathic dilated cardiomyopathy undergoing cardiac transplantation: A clinicopathological study. Heart 2011, 97, 1744–1752. [Google Scholar] [CrossRef]
- Singh, S.M.; Casey, S.A.; Berg, A.A.; Abdelhadi, R.H.; Katsiyiannis, W.T.; Bennett, M.K.; Mackey-Bojack, S.; Duncanson, E.R.; Sengupta, J.D. Autosomal-dominant biventricular arrhythmogenic cardiomyopathy in a large family with a novel in-frame DSP nonsense mutation. Am. J. Med. Genet. Part A 2018, 176, 1622–1626. [Google Scholar] [CrossRef]
- Ng, R.; Manring, H.; Papoutsidakis, N.; Albertelli, T.; Tsai, N.; See, C.J.; Li, X.; Park, J.; Stevens, T.L.; Bobbili, P.J.; et al. Patient mutations linked to arrhythmogenic cardiomyopathy enhance calpain-mediated desmoplakin degradation. JCI Insight 2019, 4, e128643. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Murray, B.; Tichnell, C.; Gilotra, N.A.; Zimmerman, S.L.; Gasperetti, A.; Scheel, P.; Tandri, H.; Calkins, H.; James, C.A. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. EP Europace 2021, euab183. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Chen, X.; Chen, K.; Ren, J.; Zhang, N.; Rao, M.; Hu, Z.; Zhang, Y.; Gu, M.; et al. A novel genotype-based clinicopathology classification of arrhythmogenic cardiomyopathy provides novel insights into disease progression. Eur. Heart J. 2019, 40, 1690–1703. [Google Scholar] [CrossRef]
- Reza, N.; Hoffman-Andrews, L.; Chowns, J.L.; Marzolf, A.; Shields, B.E.; Owens, A.T. Left-Dominant Arrhythmogenic Cardiomyopathy, Palmoplantar Keratoderma, and Curly Hair Associated with a Rare Autosomal Dominant Truncating Variant in Desmoplakin. Circ. Genomic. Precis. Med. 2020, 13, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Ayala, J.M.; Pastor-Quirante, F.; Gonzalez-Carrillo, J.; Lopez-Cuenca, D.; Sanchez-Munoz, J.J.; Oliva-Sandoval, M.J.; Gimeno, J.R. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm 2015, 12, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Caforio, A.L.; Re, F.; Avella, A.; Marcolongo, R.; Baratta, P.; Seguso, M.; Gallo, N.; Plebani, M.; Izquierdo-Bajo, A.; Cheng, C.-Y.; et al. Evidence From Family Studies for Autoimmunity in Arrhythmogenic Right Ventricular Cardiomyopathy: Associations of Circulating Anti-Heart and Anti-Intercalated Disk Autoantibodies With Disease Severity and Family History. Circulation 2020, 141, 1238–1248. [Google Scholar] [CrossRef]
- Xu, T.; Yang, Z.; Vatta, M.; Rampazzo, A.; Beffagna, G.; Pillichou, K.; Scherer, S.E.; Saffitz, J.; Kravitz, J.; Zareba, W.; et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, T.; Kaneko, Y.; Irie, T.; Takahashi, R.; Kato, T.; Iijima, T.; Iso, T.; Kurabayashi, M. Compound and digenic heterozygosity in desmosome genes as a cause of arrhythmogenic right ventricular cardiomyopathy in Japanese patients. Circ. J. 2012, 76, 737–743. [Google Scholar] [CrossRef] [Green Version]
- Rigato, I.; Bauce, B.; Rampazzo, A.; Zorzi, A.; Pilichou, K.; Mazzotti, E.; Migliore, F.; Marra, M.P.; Lorenzon, A.; De Bortoli, M.; et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ. Cardiovasc. Genet. 2013, 6, 533–542. [Google Scholar] [CrossRef] [Green Version]
- König, E.; Volpato, C.B.; Motta, B.M.; Blankenburg, H.; Picard, A.; Pramstaller, P.; Casella, M.; Rauhe, W.; Pompilio, G.; Meraviglia, V.; et al. Exploring digenic inheritance in arrhythmogenic cardiomyopathy. BMC Med. Genet. 2017, 18, 145. [Google Scholar] [CrossRef] [Green Version]
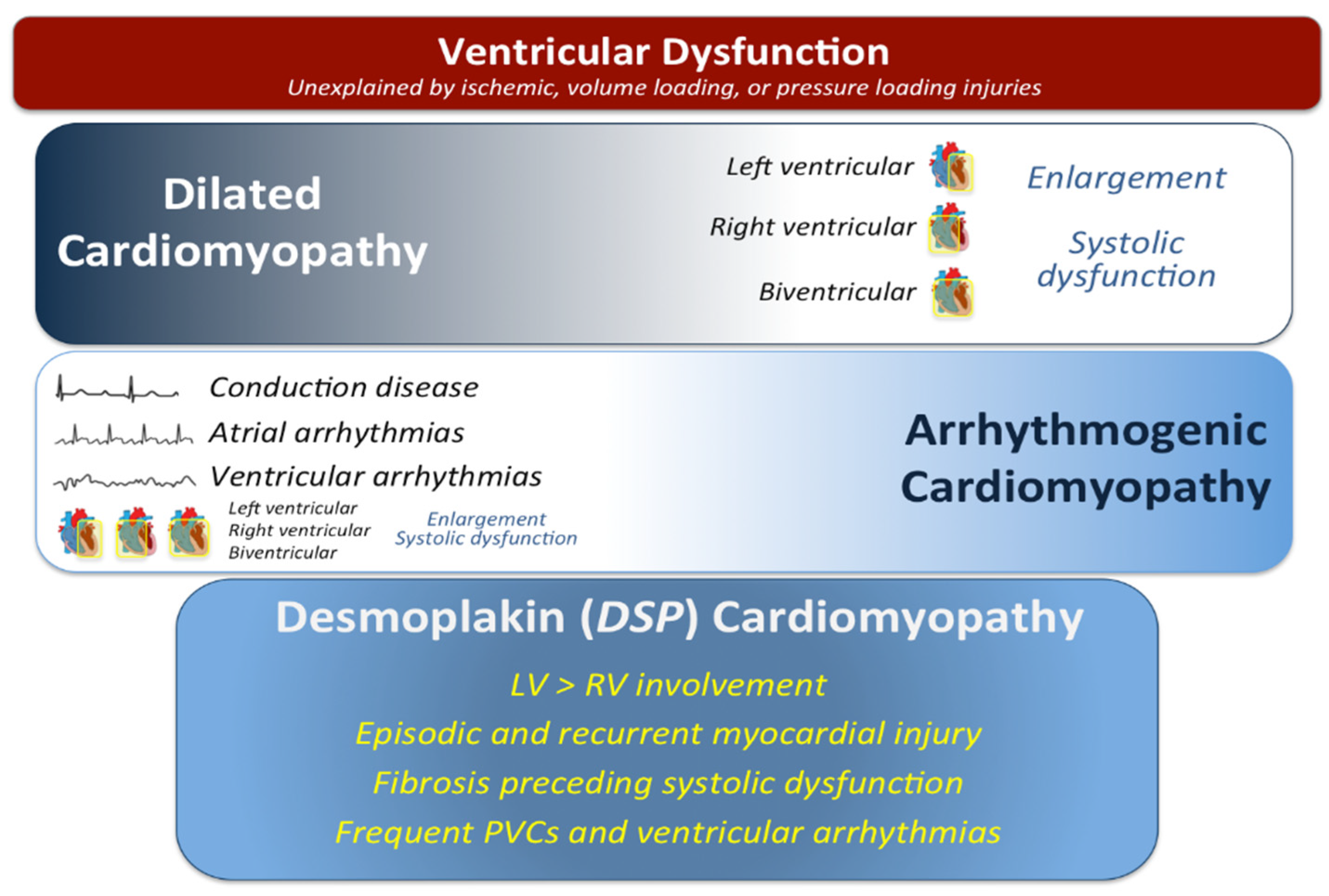


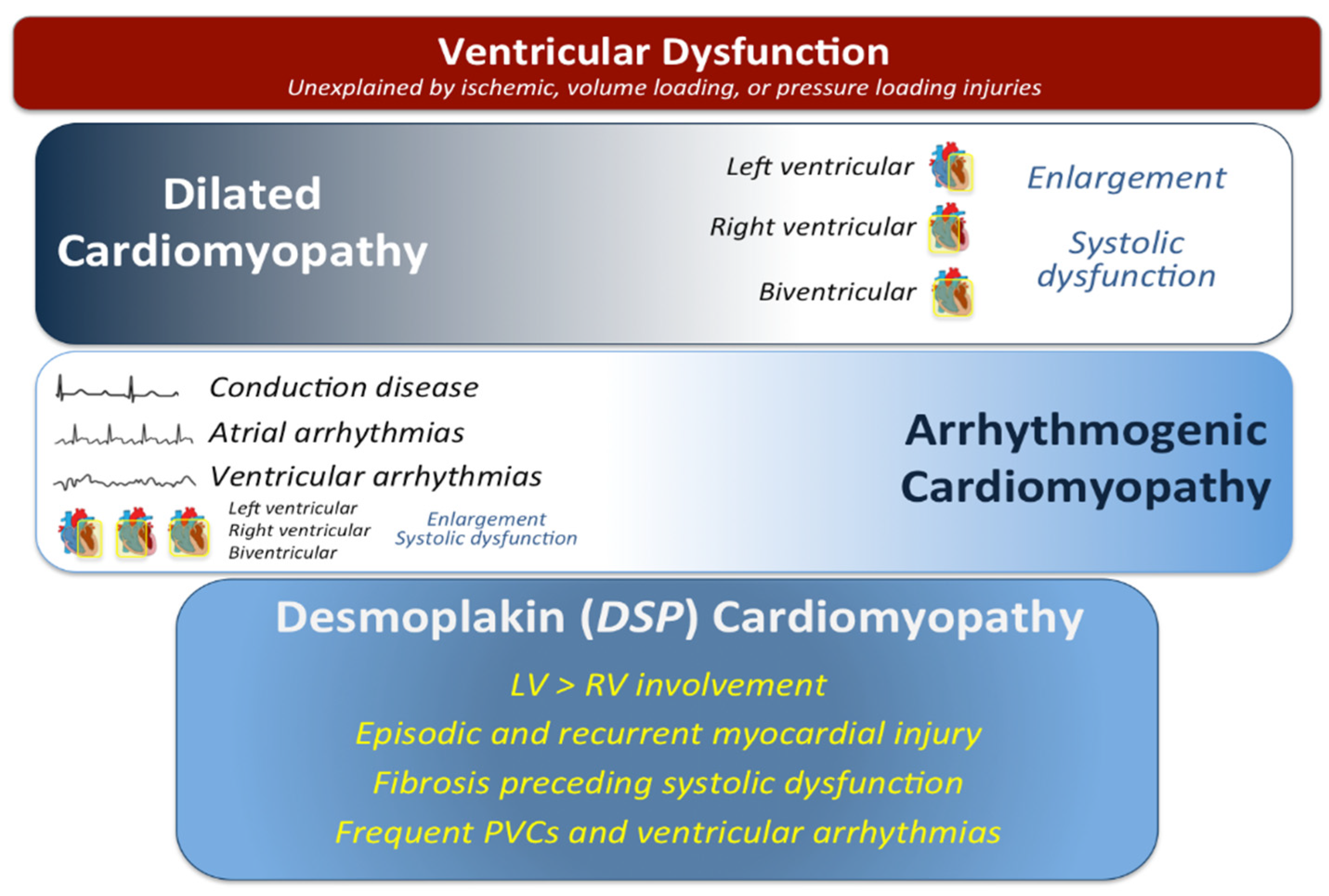{kind=link}
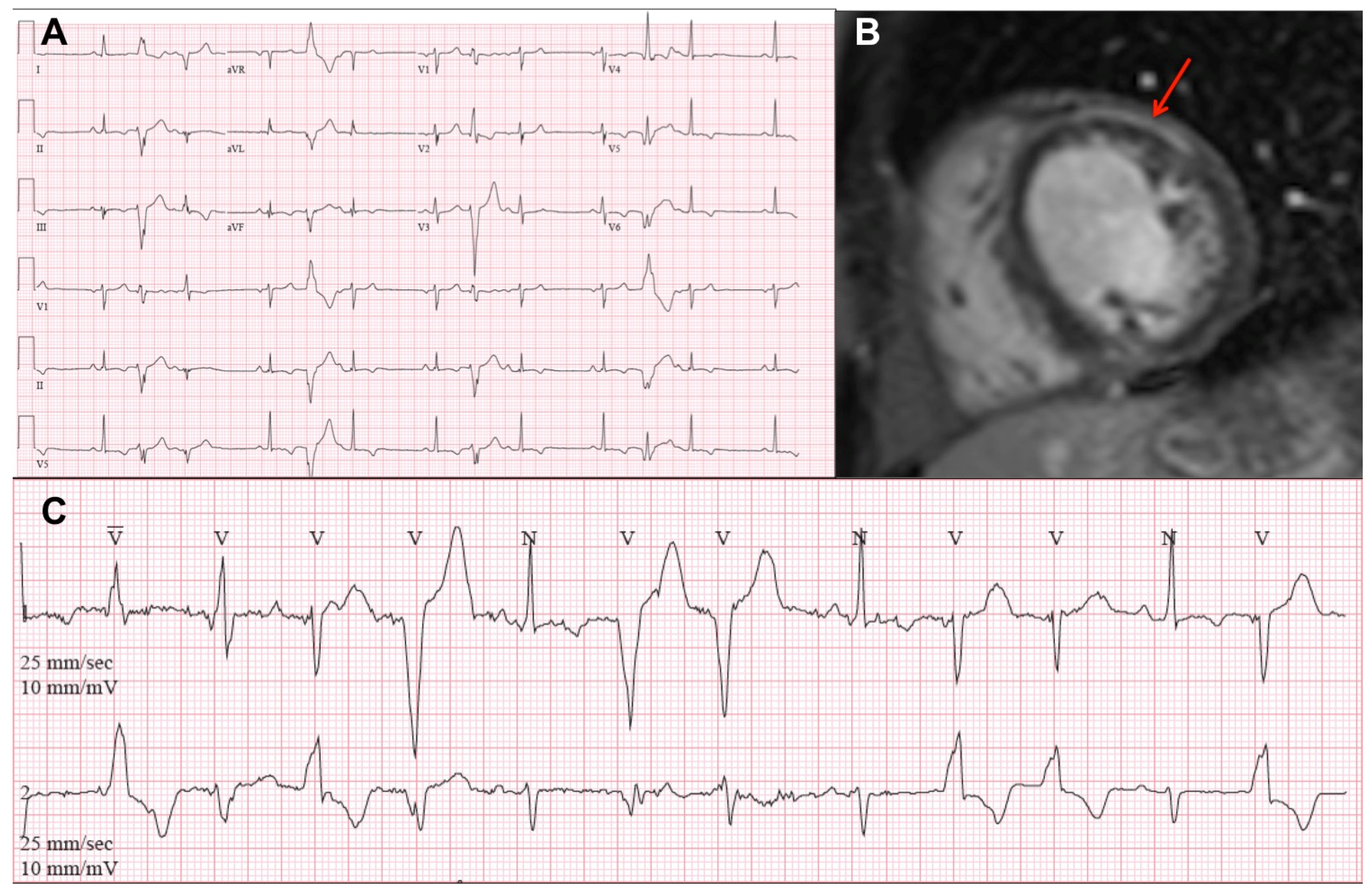
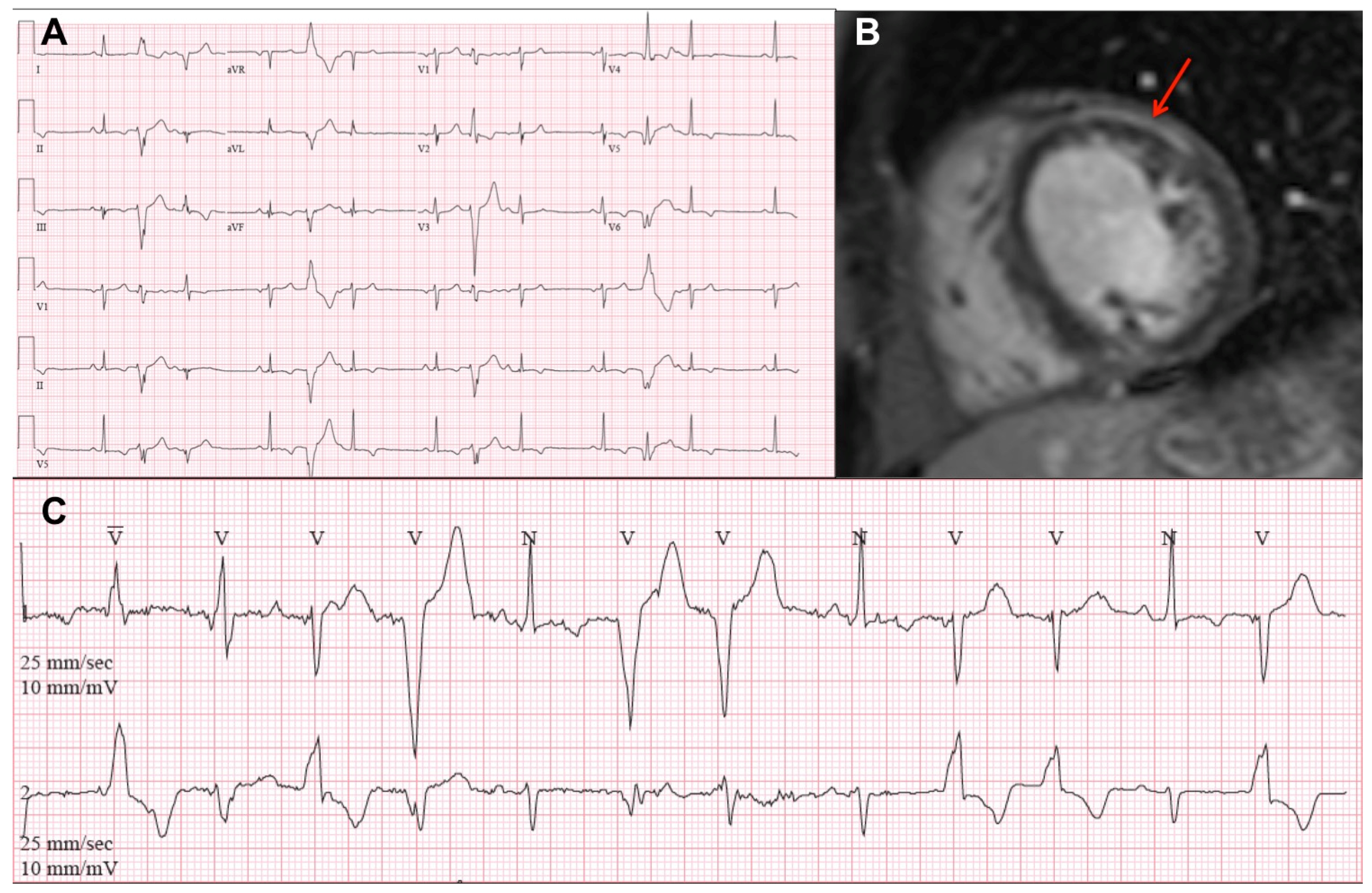{kind=link}
{kind=link}
| Characteristic | Relatives | Probands | p-Value |
|---|---|---|---|
| N = 8 | N = 11 | ||
| Age at Diagnosis (years), median (IQR) | 35.5 (31.0–48.0) | 42.0 (30.0–48.0) | 0.53 |
| Sex (%) | 1.00 | ||
| Female | 5 (62%) | 7 (64%) | |
| Male | 3 (38%) | 4 (36%) | |
| Race/Ethnicity (%) | 1.00 | ||
| Hispanic Latinx/White | 1 (12%) | 1 (9%) | |
| White | 7 (88%) | 9 (82%) | |
| Unknown | 0 (0%) | 1 (9%) | |
| Symptoms at Initial Assessment (%) | |||
| NYHA Class | 0.11 | ||
| I | 5 (62%) | 6 (55%) | |
| II | 1 (12%) | 5 (45%) | |
| III | 2 (25%) | 0 (0%) | |
| Dyspnea | 2 (25%) | 1 (9%) | 0.55 |
| Chest Pain | 2 (25%) | 1 (9%) | 0.55 |
| Palpitations | 5 (62%) | 3 (27%) | 0.18 |
| Presyncope | 1 (12%) | 2 (18%) | 1.00 |
| Syncope | 1 (12%) | 0 (0%) | 0.42 |
| Edema | 0 (0%) | 0 (0%) | 1.00 |
| Family History of SCD (%) | — | 5 (45%) | |
| Family History of DCM (%) | — | 5 (45%) |
| Patient ID | Family Number | Nucleotide Change (c.) | Amino Acid Change (p.) | Variant Type | ClinVar Citations | ACMG Classification |
|---|---|---|---|---|---|---|
| 1 | 1 | 5940dupC | Tyr1981fs | Frameshift | None | Likely pathogenic |
| 2 | 2 | 6767delG | Gly2256Valfs*5 | Frameshift | None | Pathogenic |
| 3 | 2 | 6767delG | Gly2256Valfs*5 | Frameshift | None | Pathogenic |
| 4 | 3 | 5212C > T | Arg1738Ter | Nonsense | PMID: 28492532 PMID: 26314686 PMID: 29759408 PMID: 25616645 | Pathogenic |
| 5 | 4 | 939 + 1G > A | IVS7 + 1 G > A | Splice site | PMID: 24503780 PMID: 20864495 PMID: 19558499 PMID: 19279339 PMID: 19095136 PMID: 10594734 | Pathogenic |
| 6 | 5 | 7491_7492delTG | Cys2497Ter | Truncating | None | Likely pathogenic |
| 7 * | 5 | 7491_7492delTG | Cys2497Ter | Truncating | None | Likely pathogenic |
| 8 | 6 | 3799C > T | Arg1267Ter | Truncating | PMID: 16467215 | Likely pathogenic |
| 9 | 7 | 4999C > T | Gln1667Ter | Nonsense | PMID: 28492532 | Pathogenic |
| 10 | 7 | 4999C > T | Gln1667Ter | Nonsense | PMID: 28492532 | Pathogenic |
| 11 | 7 | 4999C > T | Gln1667Ter | Nonsense | PMID: 28492532 | Pathogenic |
| 12 | 8 | 5851C > T | Arg1951Ter | Truncating | PMID: 28527814 PMID: 26899768 PMID: 21859740 PMID: 11063735 | Pathogenic |
| 13 * | 9 | 888C > G | Tyr296* | Truncating | None | Pathogenic |
| 14 | 10 | 313C > T | Arg105* | Truncating | None | Pathogenic |
| 15 | 11 | 478C > T | Arg160* | Truncating | PMID: 28588093 PMID: 28492532 PMID: 27532257 PMID: 26850880 PMID: 25616645 PMID: 23810894 | Pathogenic |
| 16 | 12 | 3415_3417delinsG | Tyr1139Glyfs*10 | Truncating | None | Pathogenic |
| 17 | 7 | 4999C > T | Gln1667Ter | Nonsense | PMID: 28492532 | Pathogenic |
| 18 | 13 | 7372_7373delAA | Lys2458GlufsX7 | Truncating | None | Likely pathogenic |
| 19 | 14 | 4531C > T | Gln1511* | Truncating | PMID: 28492532 | Pathogenic |
| Characteristic | Relatives | Probands | p-Value |
|---|---|---|---|
| N = 8 | N = 11 | ||
| T wave inversions, II, III, aVF | 2 (25%) | 6 (55%) | 0.35 |
| T wave inversions, V1–V2 | 3 (38%) | 0 (0%) | 0.06 |
| T wave inversions, V1–V3 | 3 (38%) | 0 (0%) | 0.06 |
| T wave inversions, V3–V4 | 2 (25%) | 2 (18%) | 1.00 |
| T wave inversions, V4–V6 | 3 (38%) | 3 (27%) | 1.00 |
| T wave inversions, V5–V6 | 4 (50%) | 5 (45%) | 1.00 |
| Ventricular ectopy (VE) (%) | 5 (62%) | 11 (100%) | 0.058 |
| MCOT VE count, median (IQR) | 933.0 (74.0–3235.5) | 16,460.5 (9319.5–35,926.0) | 0.011 |
| MCOT hours, median (IQR) | 91.5 (36.0–165.5) | 35.5 (24.0–49.0) | 0.33 |
| MCOT VE burden (beats per hour), median (IQR) | 5.9 (1.5–103.4) | 554.1 (194.3–1024.3) | 0.017 |
| Arrhythmia (%) | 6 (75%) | 11 (100%) | 0.16 |
| NSVT (%) | 4 (50%) | 11 (100%) | 0.018 |
| VT (%) | 1 (12%) | 5 (45%) | 0.18 |
| Atrial fibrillation (%) | 1 (12%) | 1 (9%) | 1.00 |
| Supraventricular tachycardia (%) | 4 (50%) | 1 (9%) | 0.11 |
| Sudden cardiac arrest (%) | 1 (12%) | 1 (9%) | 1.00 |
| ICD (%) | 4 (50%) | 9 (82%) | 0.32 |
| Primary prevention ICD | 3 (38%) | 6 (55%) | 0.48 |
| Secondary prevention ICD | 1 (12%) | 3 (27%) | 0.48 |
| Type of ICD (%) | 0.92 | ||
| Single chamber | 1 (12%) | 3 (27%) | |
| Dual chamber | 1 (12%) | 2 (18%) | |
| Subcutaneous | 2 (25%) | 3 (27%) | |
| Unknown | 4 (50%) | 3 (27%) | |
| LVEF (%) at time of ICD implantation, median (IQR) | 32.0 (27.5–44.5) | 36.0 (27.5–47.0) | 0.80 |
| Appropriate ICD shock (%) | 0 (0%) | 3 (27%) | 0.21 |
| Inappropriate ICD shock (%) | 1 (12%) | 0 (0%) | 0.084 |
| VT catheter ablation (%) | 0 (0%) | 2 (18%) | 0.49 |
| PVC catheter ablation (%) | 0 (0%) | 3 (27%) | 0.23 |
| Characteristic | Relatives | Probands | p-Value |
|---|---|---|---|
| N = 8 | N = 11 | ||
| LVEF (%) by TTE at initial assessment, median (IQR) | 46.0 (24.5–62.5) | 30.0 (25.0–45.0) | 0.59 |
| LVEDD (millimeters) by TTE at initial assessment, median (IQR) | 48.5 (45.0–56.5) | 57.0 (50.0–68.0) | 0.13 |
| RV function on TTE at initial assessment (%) | 0.37 | ||
| Normal | 6 (75%) | 6 (55%) | |
| Normal to Mildly Decreased Mildly Decreased | 1 (12%) 0 (0%) | 0 (0%) 1 (9%) | |
| Unknown | 1 (12%) | 4 (36%) | |
| LV LGE location on CMR (%) | 0.15 | ||
| Subepicardial | 1 (12%) | 0 (0%) | |
| Subepicardial + mid-myocardial | 0 (0%) | 4 (36%) | |
| Mid-myocardial | 1 (12%) | 2 (18%) | |
| Transmural | 0 (0%) | 1 (9%) | |
| Epicardial + transmural + mid myocardial | 0 (0%) | 1 (9%) | |
| Unknown | 1 (12%) | 1 (9%) | |
| No CMR | 5 (62%) | 2 (18%) |
| Patient ID | Specimen Type | Myocyte Hypertrophy | Fibrosis | Other Notable Findings |
|---|---|---|---|---|
| 1 | RV | Nonspecific | Mild | Increased number of mitochondria on electron microscopy |
| 7 | Explanted heart | — | Interstitial & subendocardial | No amyloid, parenchymal iron or excess glycogen deposition, granulomas, giant cells or inflammatory infiltrates |
| 9 | Explanted heart | Mild to moderate | Mild interstitial & subendocardial | No amyloid |
| 15 | LV | Severe | Interstitial | Intracellular glycogen present on PAS with and without diastase No amyloid, parenchymal iron deposition, granulomas, giant cells or inflammatory infiltrates |
| 19 | LV | Moderate | Patchy interstitial & subendocardial | — |
| Relatives | Probands | p-Value | |
|---|---|---|---|
| N | 8 | 11 | |
| ECMO (%) | 1 (12%) | 0 (0%) | 0.42 |
| Heart Transplantation (%) | 0 (0%) | 2 (18%) | 0.49 |
| Death (%) | 1 (12%) | 0 (0%) | 0.42 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reza, N.; de Feria, A.; Chowns, J.L.; Hoffman-Andrews, L.; Vann, L.; Kim, J.; Marzolf, A.; Owens, A.T. Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP). Cardiogenetics 2022, 12, 24-36. https://doi.org/10.3390/cardiogenetics12010003
Reza N, de Feria A, Chowns JL, Hoffman-Andrews L, Vann L, Kim J, Marzolf A, Owens AT. Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP). Cardiogenetics. 2022; 12(1):24-36. https://doi.org/10.3390/cardiogenetics12010003
Chicago/Turabian StyleReza, Nosheen, Alejandro de Feria, Jessica L. Chowns, Lily Hoffman-Andrews, Laura Vann, Jessica Kim, Amy Marzolf, and Anjali Tiku Owens. 2022. "Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP)" Cardiogenetics 12, no. 1: 24-36. https://doi.org/10.3390/cardiogenetics12010003
APA StyleReza, N., de Feria, A., Chowns, J. L., Hoffman-Andrews, L., Vann, L., Kim, J., Marzolf, A., & Owens, A. T. (2022). Cardiovascular Characteristics of Patients with Genetic Variation in Desmoplakin (DSP). Cardiogenetics, 12(1), 24-36. https://doi.org/10.3390/cardiogenetics12010003