
Reassessment of Gene-Elusive Familial Dilated Cardiomyopathy Leading to the Discovery of a Homozygous AARS2 Variant—The Importance of Regular Reassessment of Genetic Findings

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
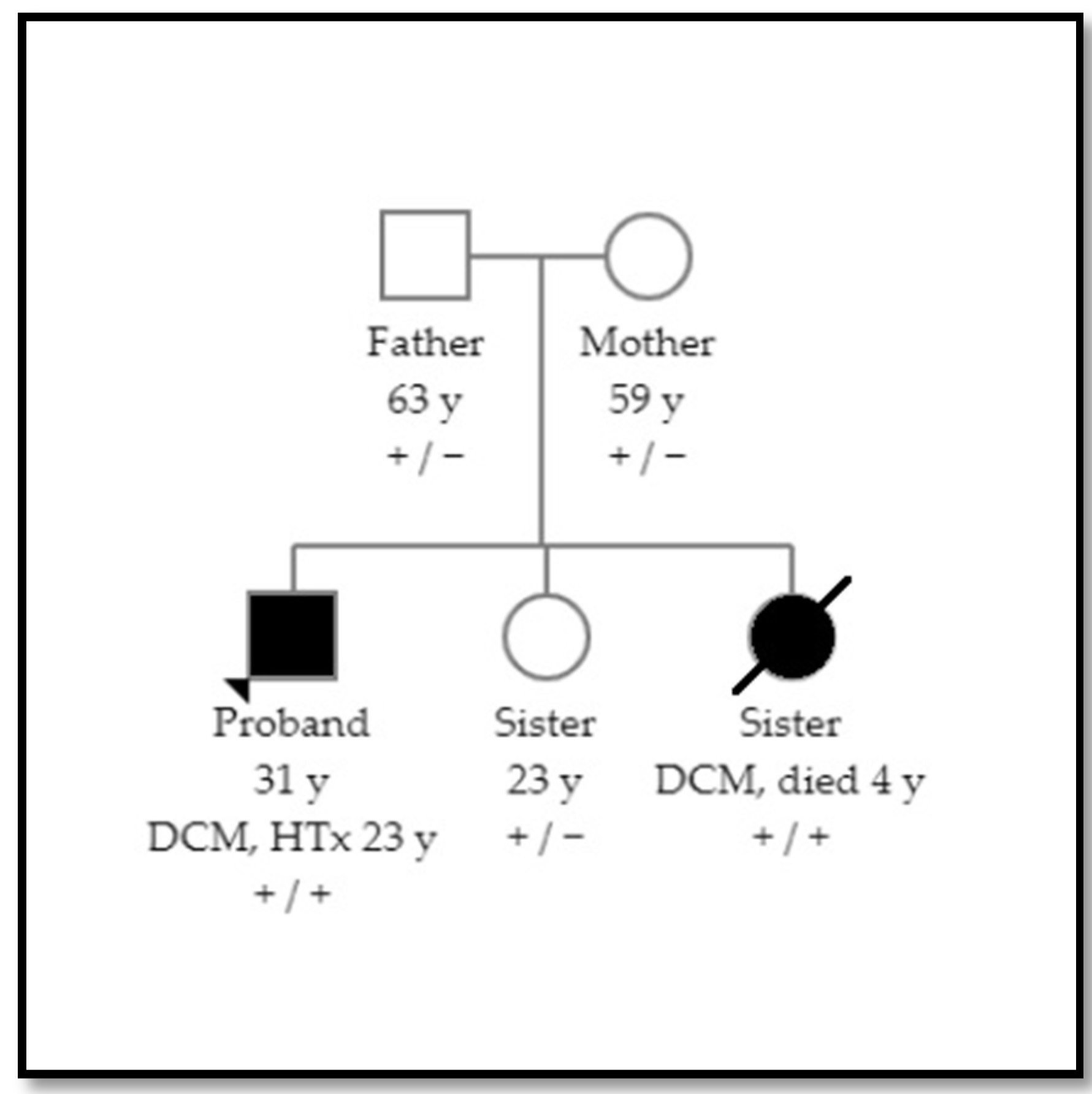
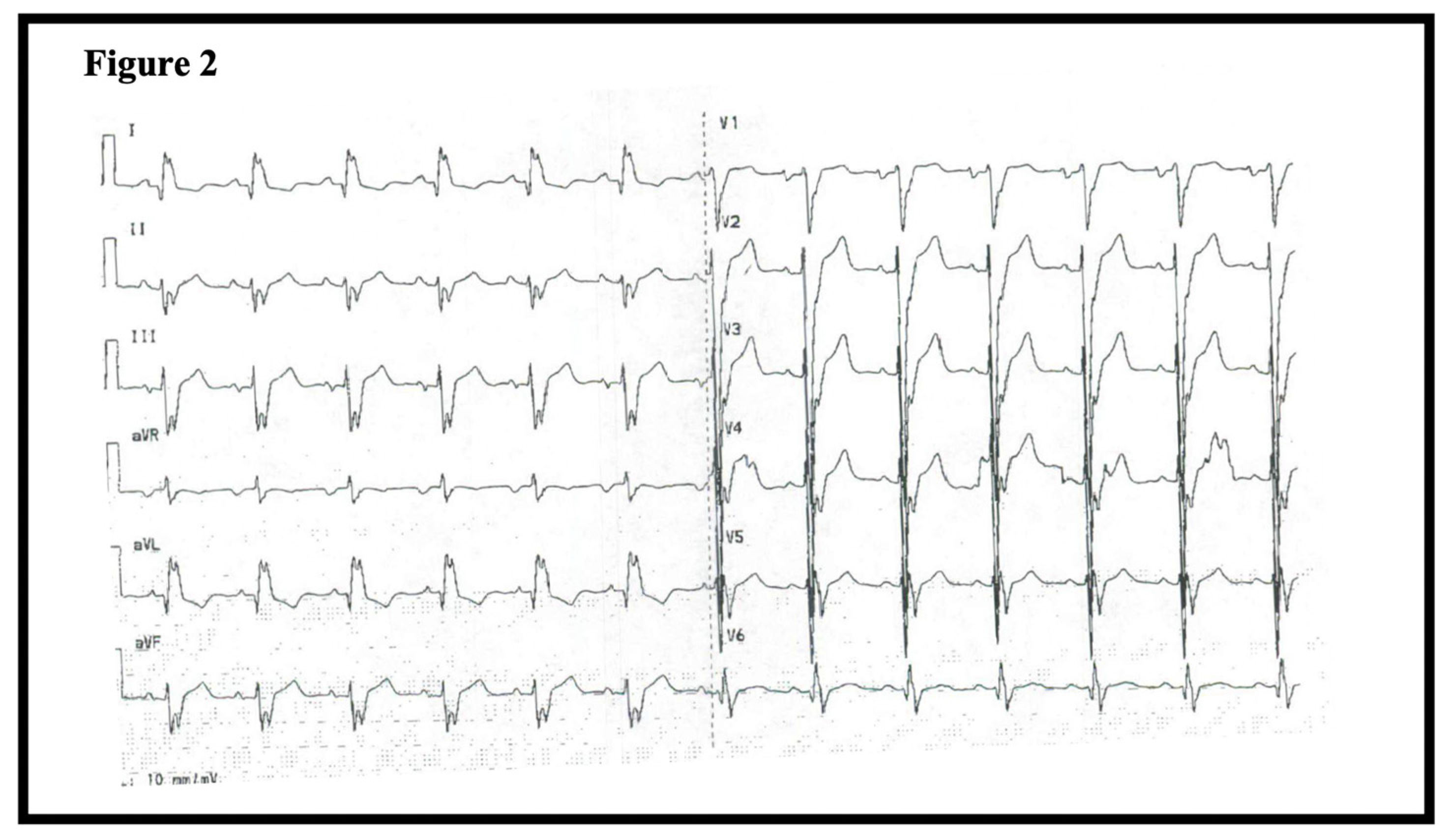
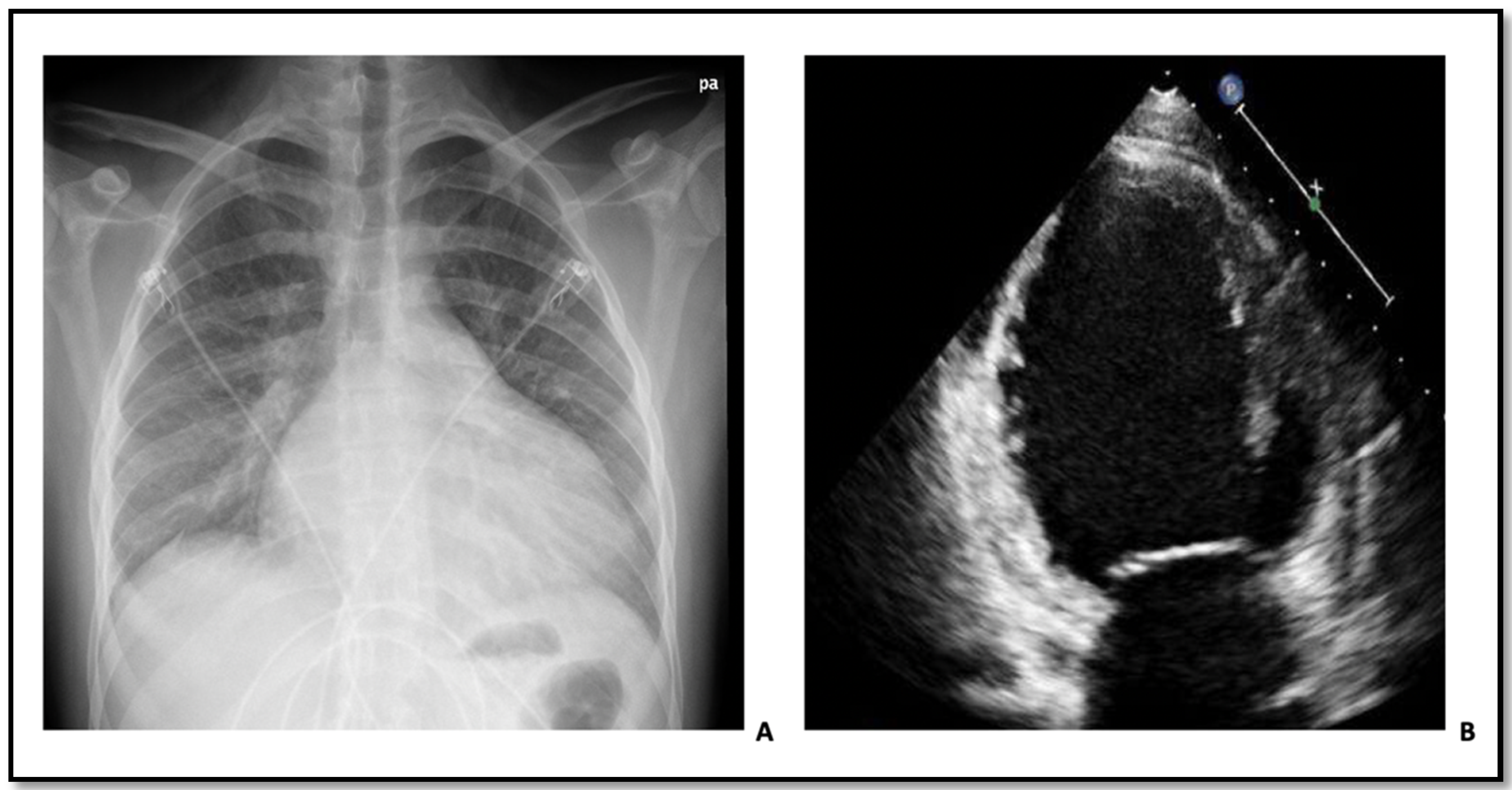
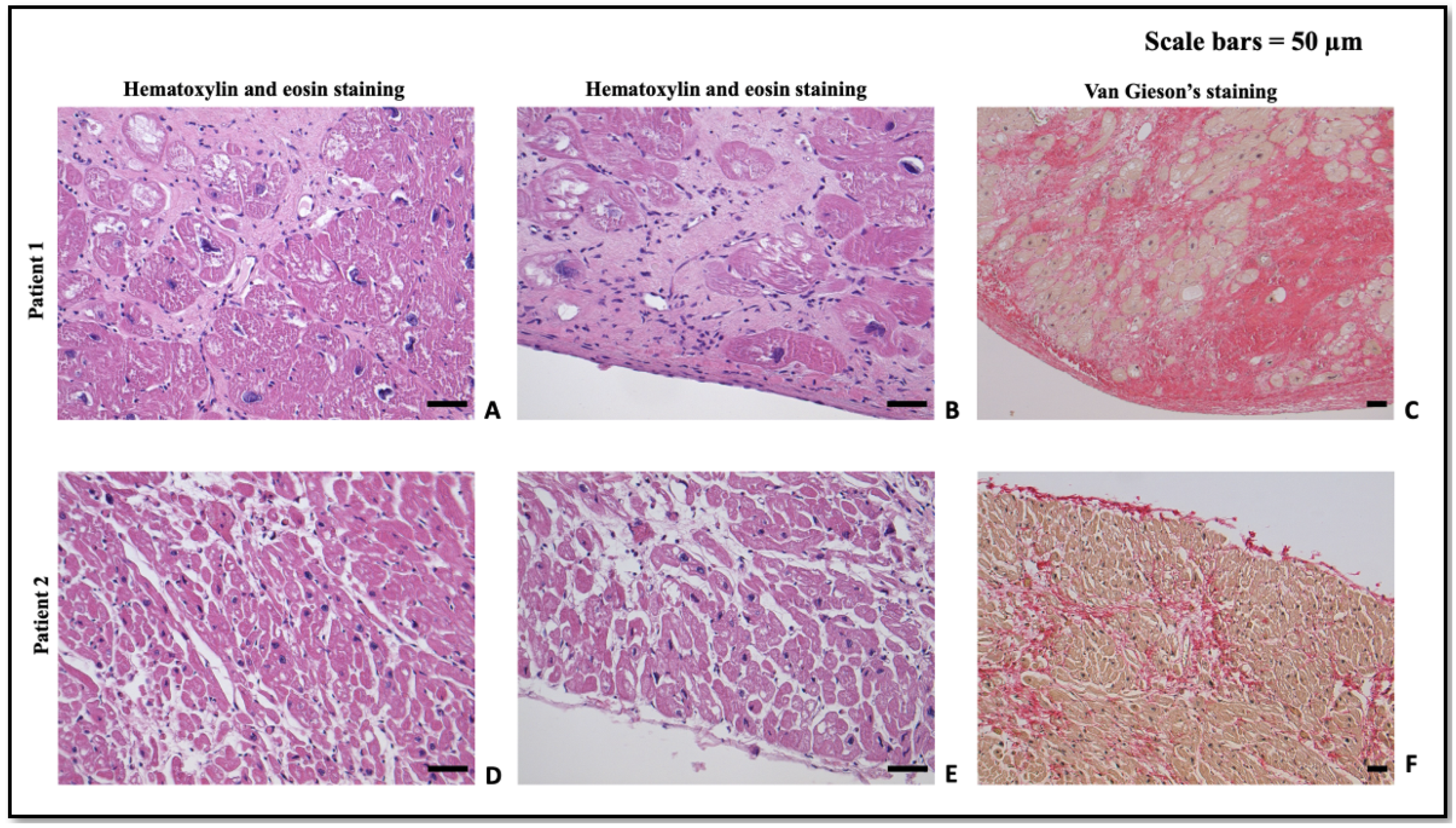
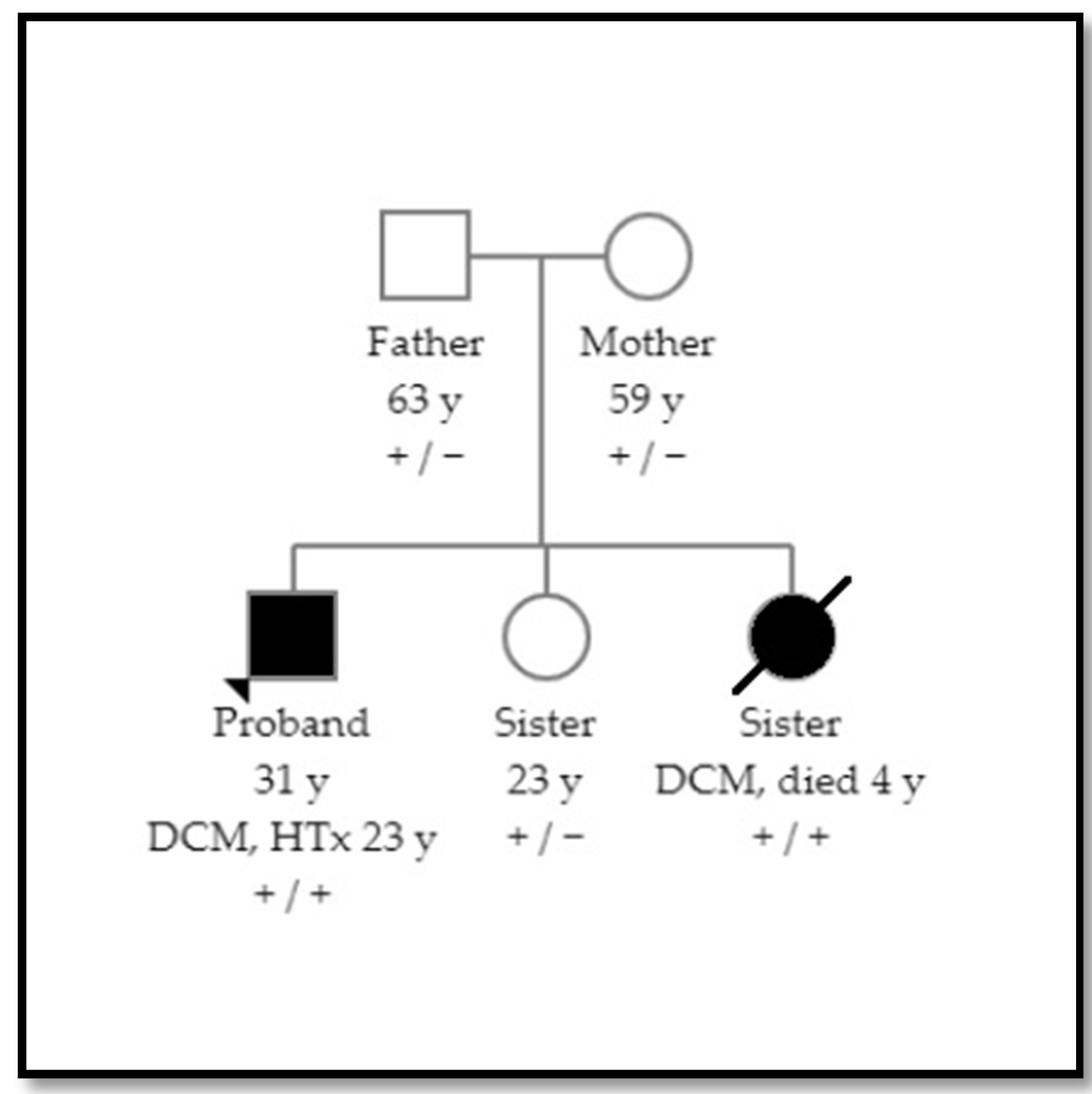
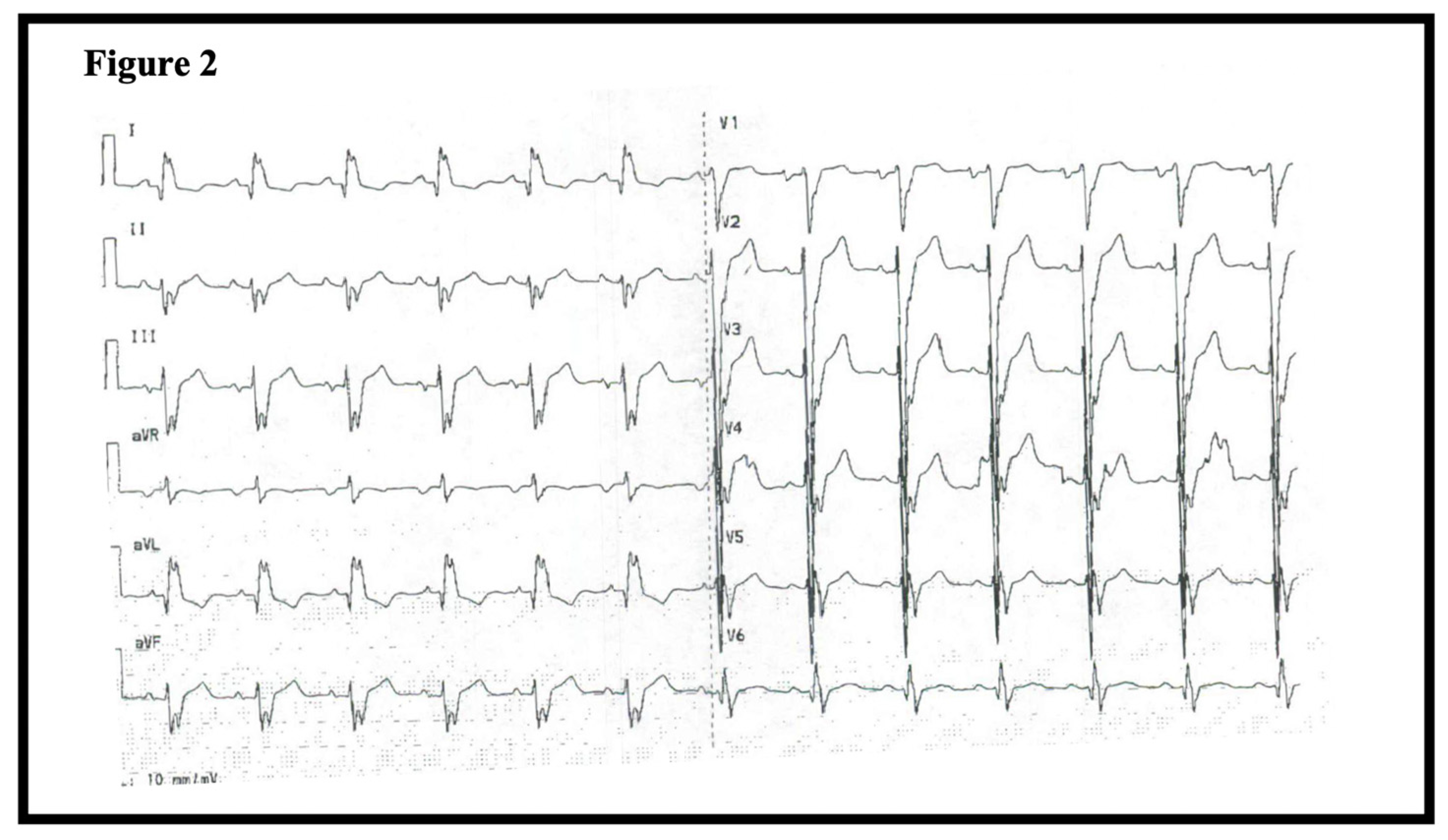
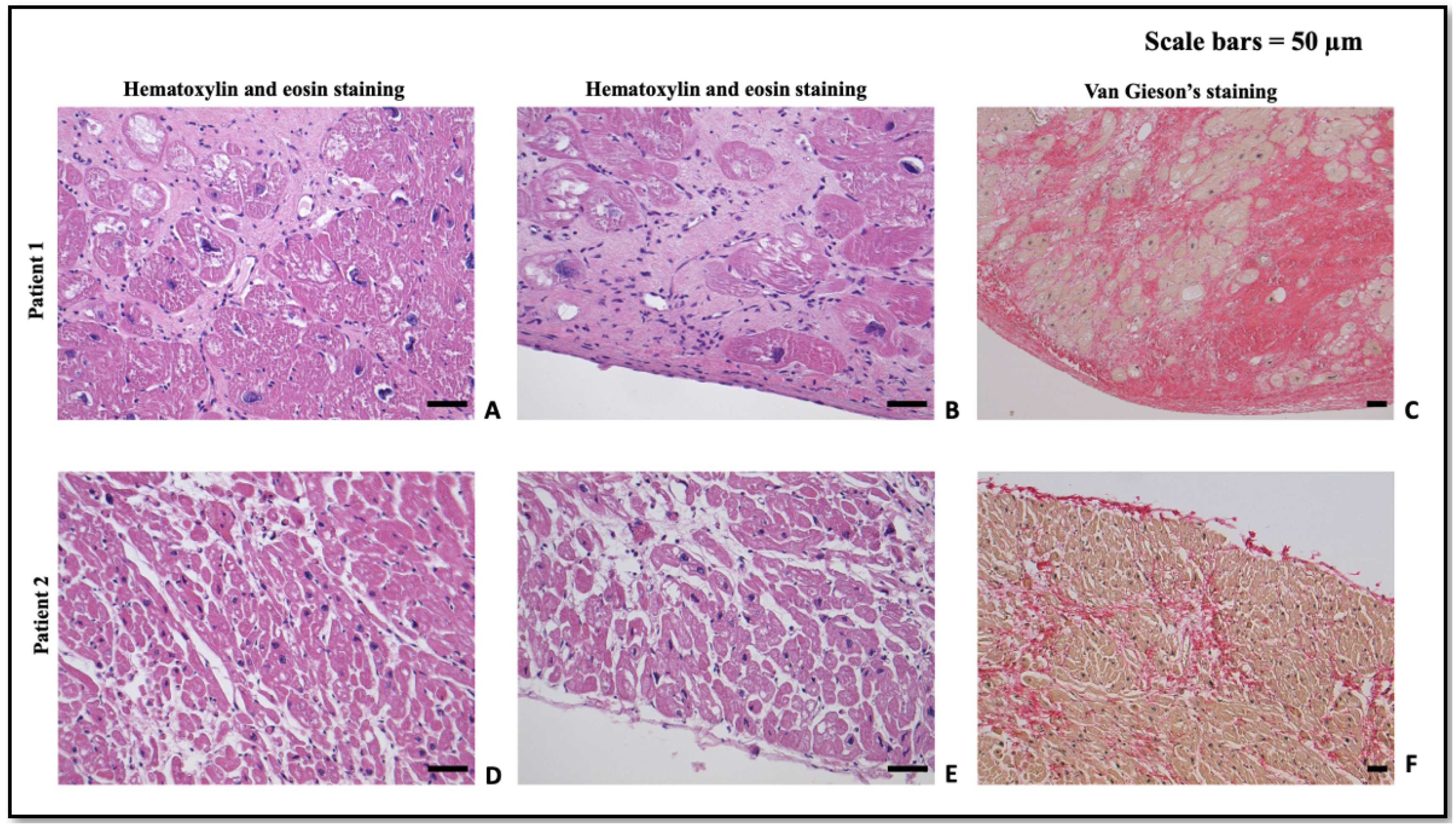
1.1. Patient 1
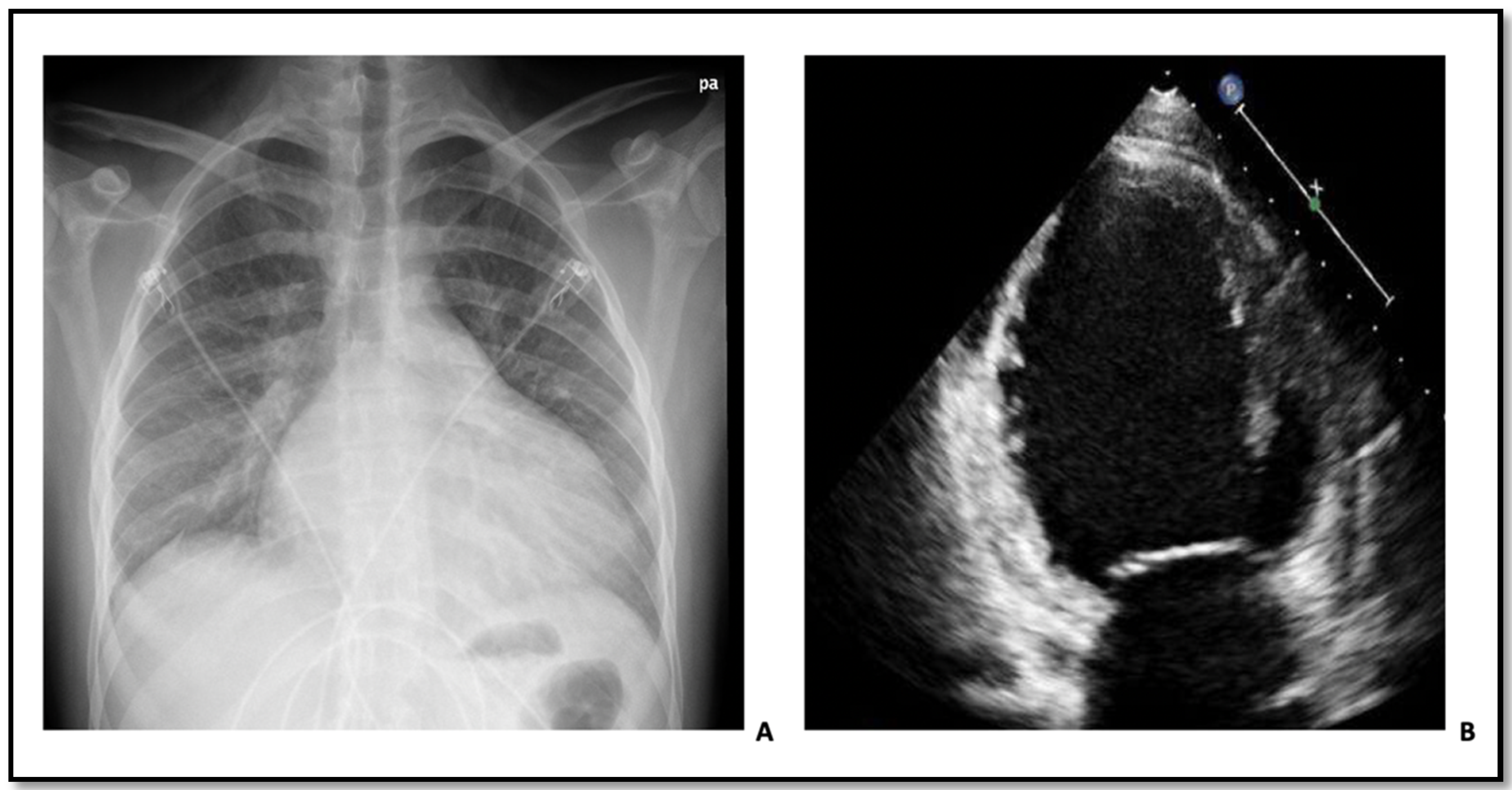
1.2. Patient 2
2. Genetic Investigations
2.1. Patient 1
2.2. Patient 2
2.3. Family Screening and Variant Classification
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Götz, A.; Tyynismaa, H.; Euro, L.; Ellonen, P.; Hyötyläinen, T.; Ojala, T.; Hämäläinen, R.H.; Tommiska, J.; Raivio, T.; Oresic, M.; et al. Exome sequencing identifies mitochondrial alanyl-tRNA Synthetase mutations in infantile mitochondrial cardiomyopathy. Am. J. Hum. Genet. 2011, 88, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.K.; Hansen, F.; Schrøder, H.D.; Wibrand, F.; Gustafsson, F.; Mogensen, J. Recessive inheritance of a rare variant in the nuclear mitochondrial gene for AARS2 in late-onset dilated cardiomyopathy. Circ. Genom. Precis. Med. 2020, 13, 560–562. [Google Scholar] [CrossRef] [PubMed]
- Sommerville, E.W.; Zhou, X.-L.; Oláhová, M.; Jenkins, J.; Euro, L.; Konovalova, S.; Hilander, T.; Pyle, A.; He, L.; Habeebu, S.; et al. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Hum. Mol. Genet. 2018, 28, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Pyle, A.; Griffin, H.R.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euro, L.; Konovalova, S.; Asin-Cayuela, J.; Tulinius, M.; Griffin, H.R.; Horvath, R.; Taylor, R.W.; Chinnery, P.F.; Schara, U.; Thorburn, D.R.; et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Front. Genet. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkel, B.G.; Larsen, M.K.; Berge, K.E.; Leren, T.P.; Nissen, P.H.; Olesen, M.S.; Hollegaard, M.V.; Jespersen, T.; Yuan, L.; Nielsen, N.; et al. The prevalence of mutations inKCNQ1, KCNH2, and SCN5A in an unselected national cohort of young sudden unexplained death cases. J. Cardiovasc. Electrophysiol. 2012, 23, 1092–1098. [Google Scholar] [CrossRef]
- Calvo, S.E.; Compton, A.; Hershman, S.; Lim, S.C.; Lieber, D.S.; Tucker, E.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [Green Version]
- Mazurova, S.; Magner, M.; Vidrová, V.K.; Vondráčková, A.; Stranecky, V.; Pristoupilova, A.; Zamecnik, J.; Hansikova, H.; Zeman, J.; Tesarova, M.; et al. Thymidine kinase 2 and alanyl-tRNA synthetase 2 deficiencies cause lethal mitochondrial cardiomyopathy: Case reports and review of the literature. Cardiol. Young 2016, 27, 936–944. [Google Scholar] [CrossRef]
- Kamps, R.; Szklarczyk, R.; Theunissen, T.E.; Hellebrekers, D.; Sallevelt, S.; Boesten, I.B.; De Koning, B.; van den Bosch, B.J.; Salomons, G.S.; Simas-Mendes, M.; et al. Genetic defects in mtDNA-encoded protein translation cause pediatric, mitochondrial cardiomyopathy with early-onset brain disease. Eur. J. Hum. Genet. 2018, 26, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.; Johnson, R.; Birch, S.; Zentner, D.; Hershberger, R.E.; Fatkin, D. Familial dilated cardiomyopathy. Heart Lung Circ. 2020, 29, 566–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, R.E.; on behalf of the ACMG Professional Practice and Guidelines Committee; Givertz, M.M.; Ho, C.Y.; Judge, D.; Kantor, P.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; et al. Genetic evaluation of cardiomyopathy: A clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2018, 20, 899–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Genga, M.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Aranda, R.S.; Climent, V.; Padron-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Hey, T.; Rasmussen, T.B.; Madsen, T.; Aagaard, M.M.; Harbo, M.; Mølgaard, H.; Møller, J.E.; Eiskjær, H.; Mogensen, J. Pathogenic RBM20—Variants are associated with a severe disease expression in male patients with dilated cardiomyopathy. Circ. Heart Fail. 2019, 12, e005700. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhardwaj, P.; Vissing, C.R.; Stampe, N.K.; Rossing, K.; Christensen, A.H.; Jensen, T.H.L.; Winkel, B.G. Reassessment of Gene-Elusive Familial Dilated Cardiomyopathy Leading to the Discovery of a Homozygous AARS2 Variant—The Importance of Regular Reassessment of Genetic Findings. Cardiogenetics 2021, 11, 122-128. https://doi.org/10.3390/cardiogenetics11030013
Bhardwaj P, Vissing CR, Stampe NK, Rossing K, Christensen AH, Jensen THL, Winkel BG. Reassessment of Gene-Elusive Familial Dilated Cardiomyopathy Leading to the Discovery of a Homozygous AARS2 Variant—The Importance of Regular Reassessment of Genetic Findings. Cardiogenetics. 2021; 11(3):122-128. https://doi.org/10.3390/cardiogenetics11030013
Chicago/Turabian StyleBhardwaj, Priya, Christoffer Rasmus Vissing, Niels Kjær Stampe, Kasper Rossing, Alex Hørby Christensen, Thomas Hartvig Lindkær Jensen, and Bo Gregers Winkel. 2021. "Reassessment of Gene-Elusive Familial Dilated Cardiomyopathy Leading to the Discovery of a Homozygous AARS2 Variant—The Importance of Regular Reassessment of Genetic Findings" Cardiogenetics 11, no. 3: 122-128. https://doi.org/10.3390/cardiogenetics11030013
APA StyleBhardwaj, P., Vissing, C. R., Stampe, N. K., Rossing, K., Christensen, A. H., Jensen, T. H. L., & Winkel, B. G. (2021). Reassessment of Gene-Elusive Familial Dilated Cardiomyopathy Leading to the Discovery of a Homozygous AARS2 Variant—The Importance of Regular Reassessment of Genetic Findings. Cardiogenetics, 11(3), 122-128. https://doi.org/10.3390/cardiogenetics11030013