

Deciphering the Antifibrotic Property of Metformin

Abstract
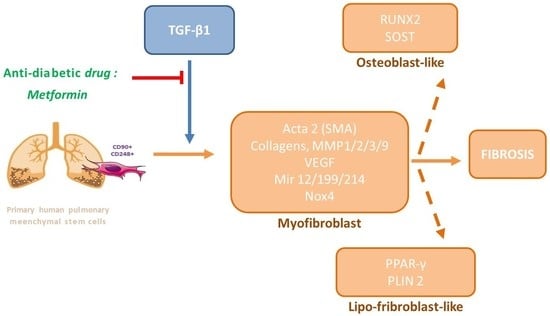
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Cell Culture
2.3. Cytotoxicity Assay
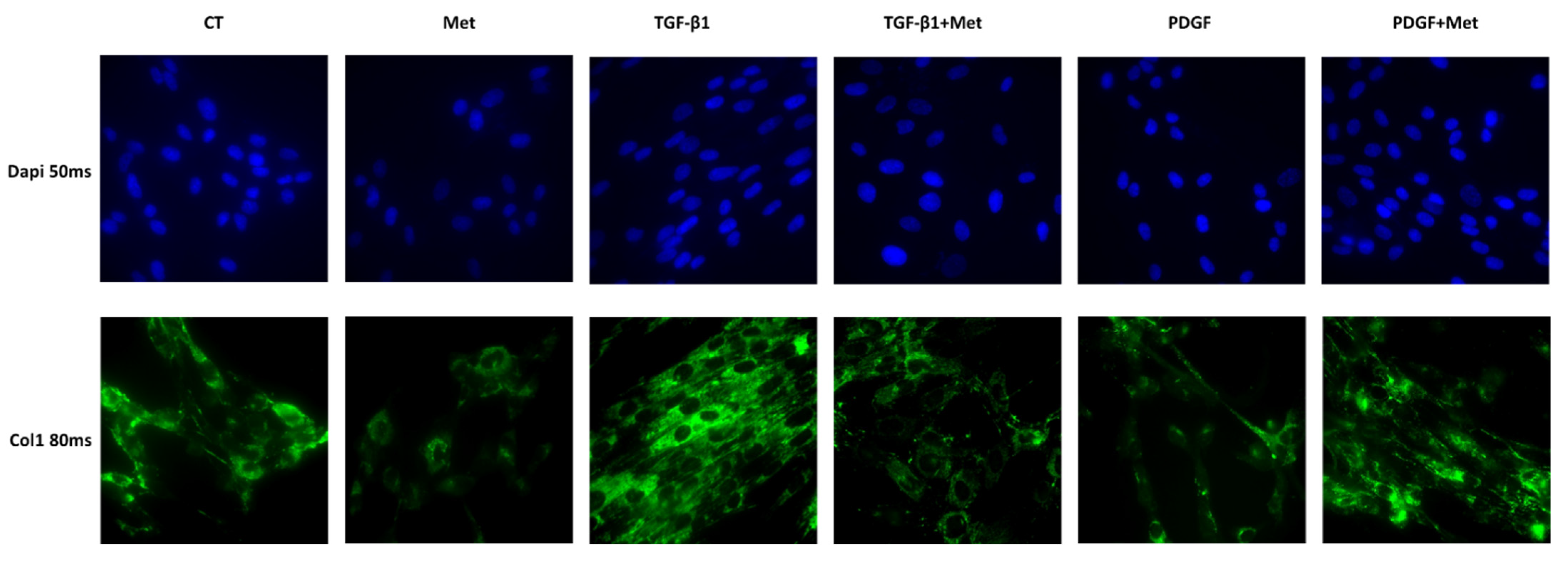
2.4. Immunofluorescence Staining and Microscopy to Assess Cell Marker Expression
2.5. Histo-ELISA Assay
2.6. qRT-PCR (Sybergreen) Analyses
2.7. Statistical Analysis
3. Results
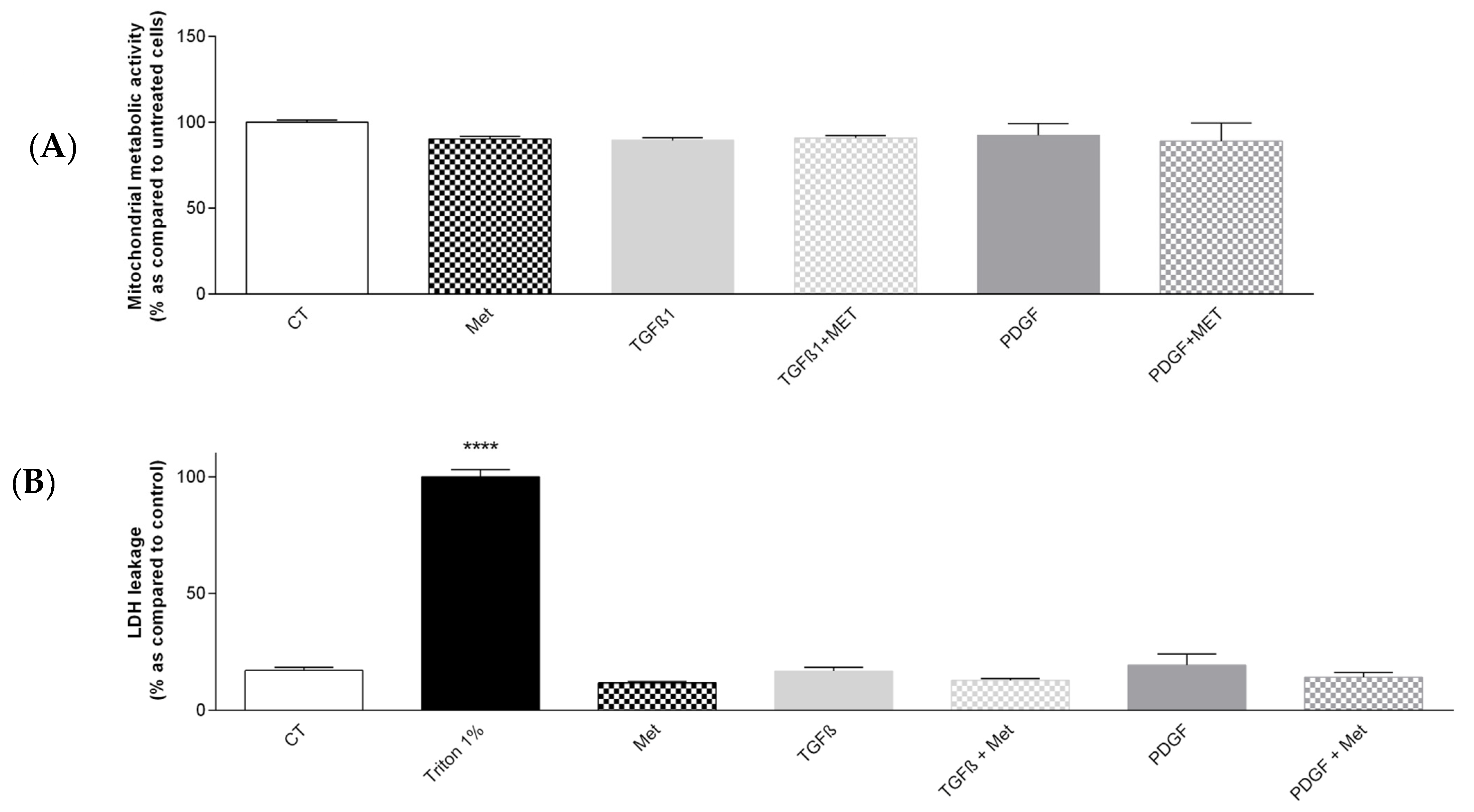
3.1. Study of the Cytotoxicity of the Different Treatments to Induce and Control Fibrosis in Primary Human Pulmonary MSC
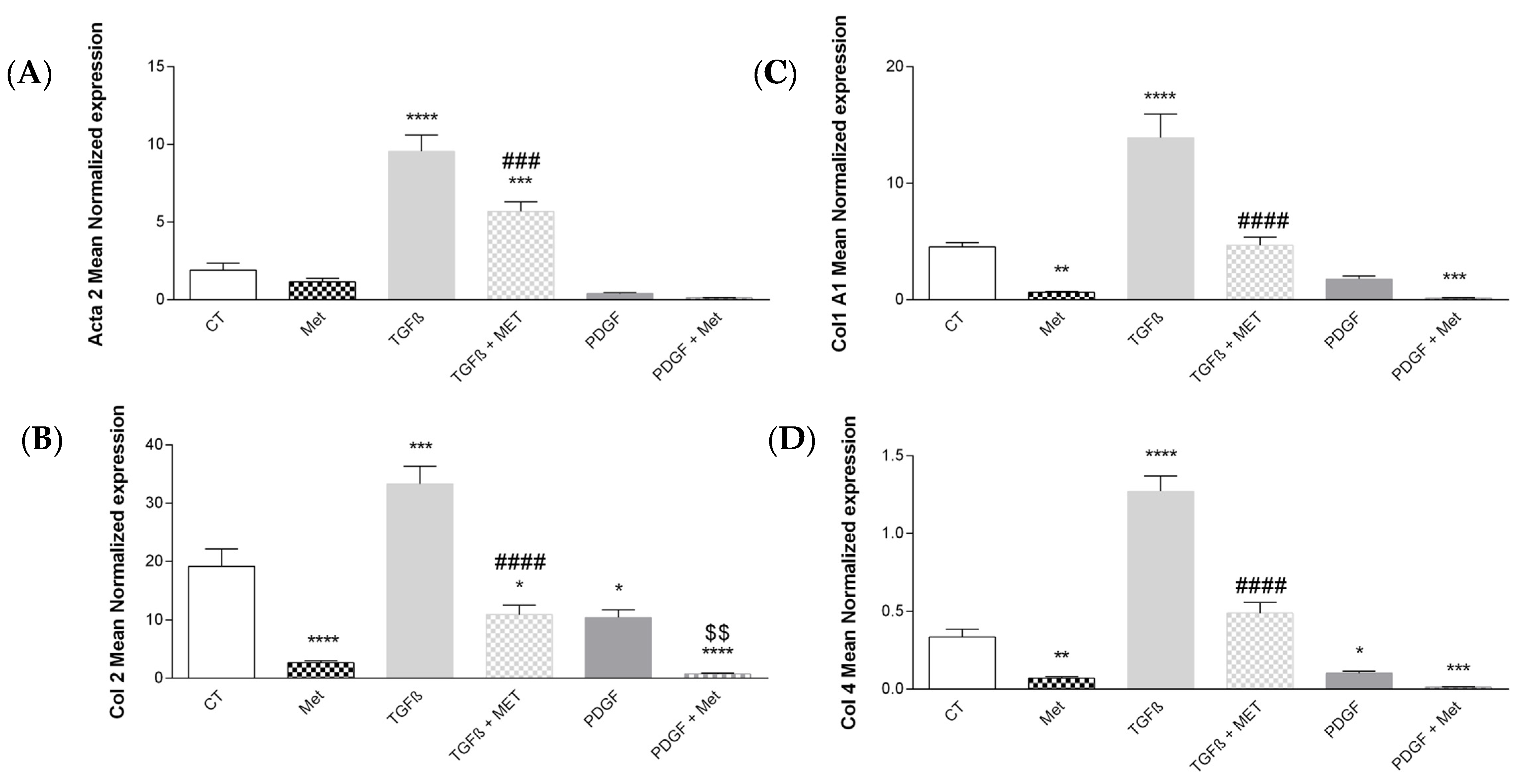
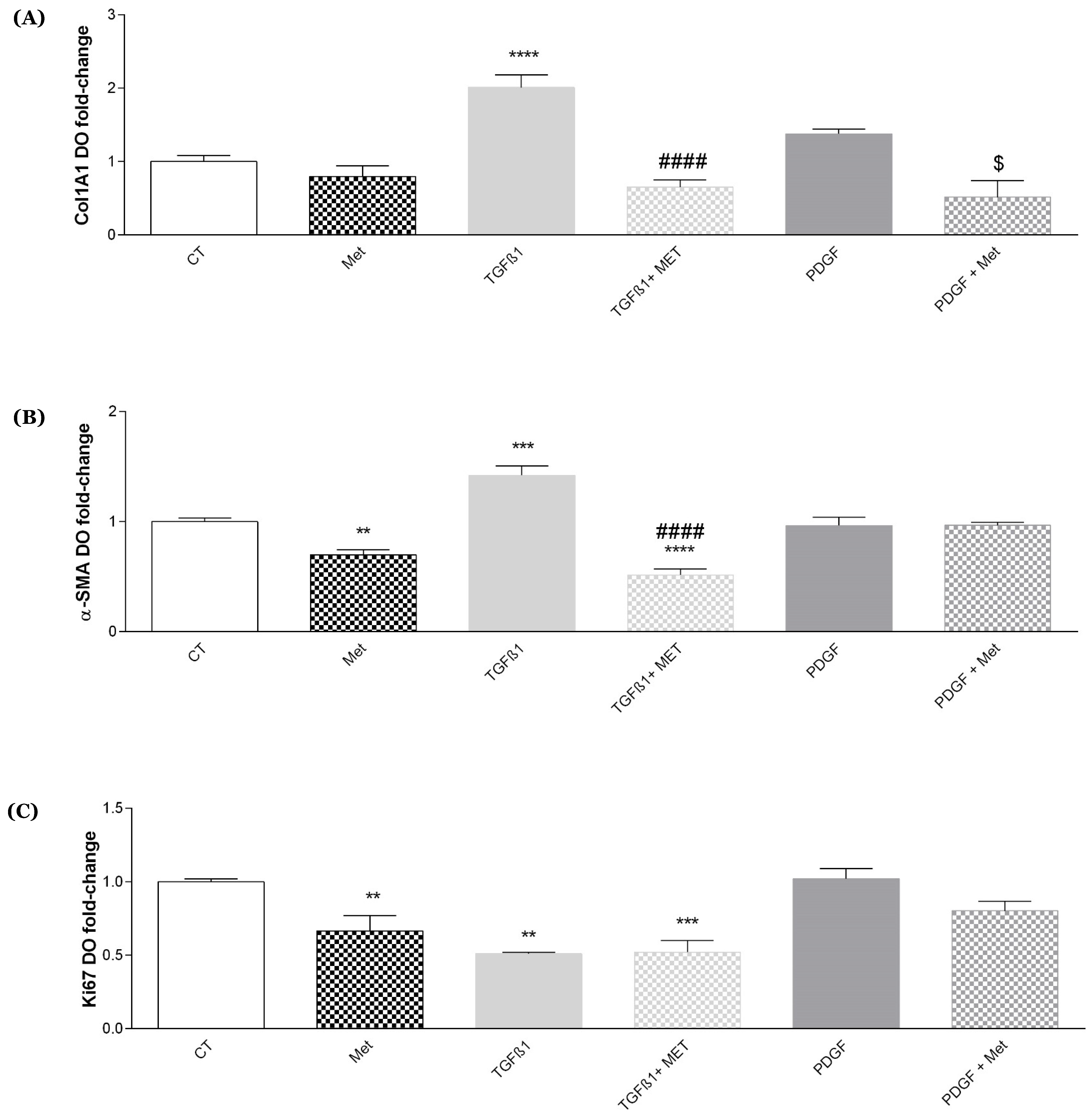
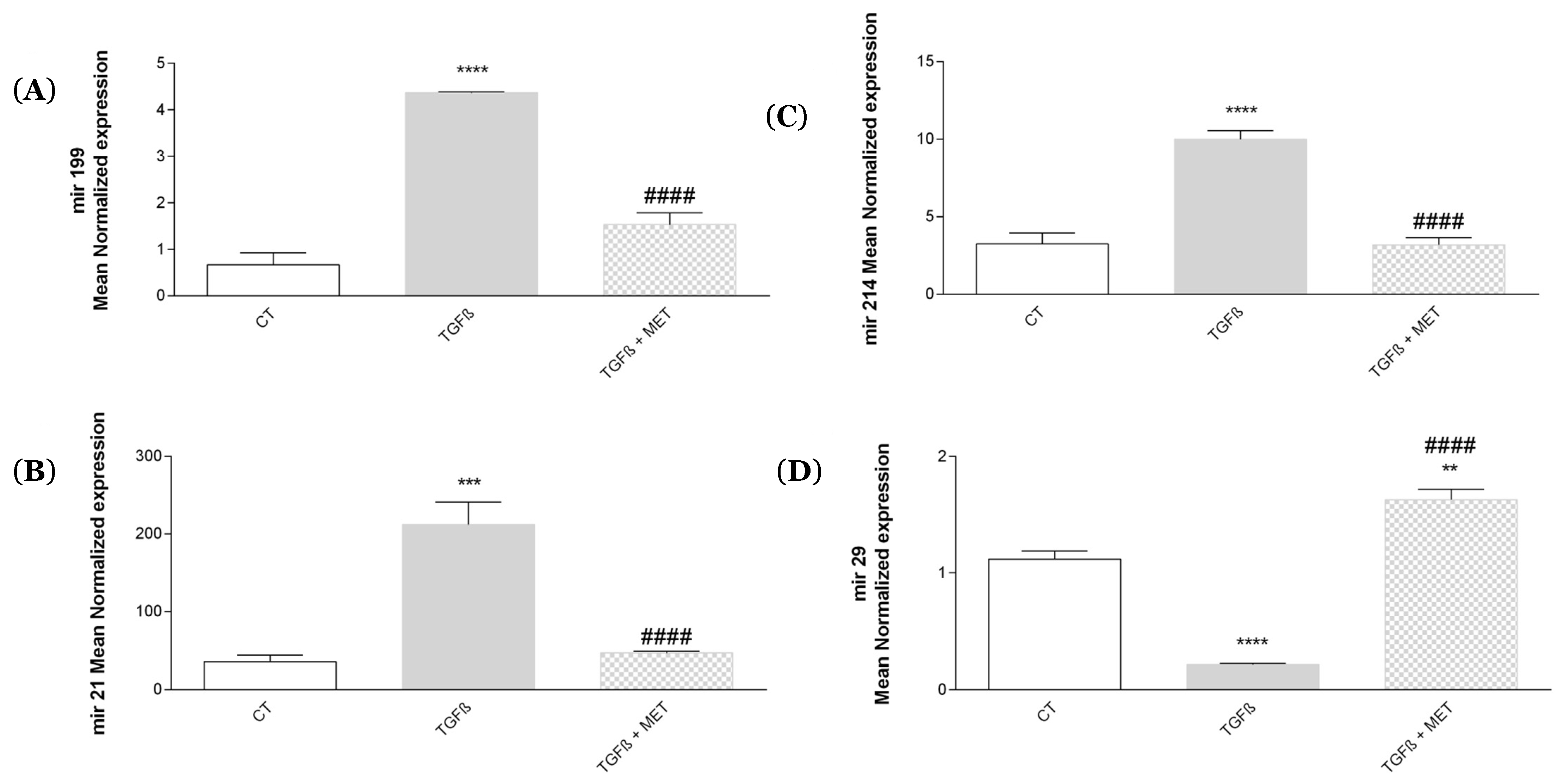
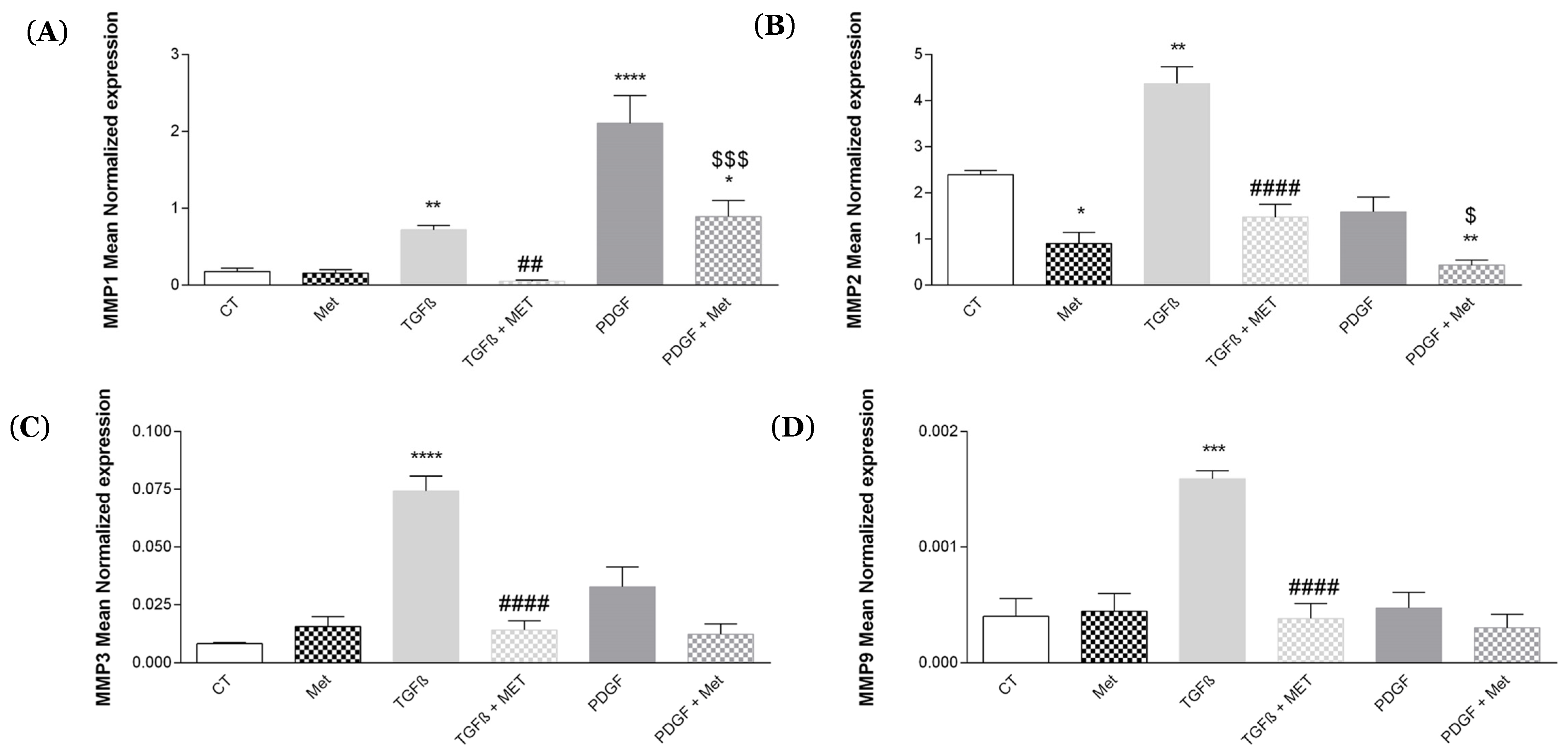
3.2. Study of Metformin Effects on TGF-β1-Mediated MSC Transdifferentiation into Myofibroblasts
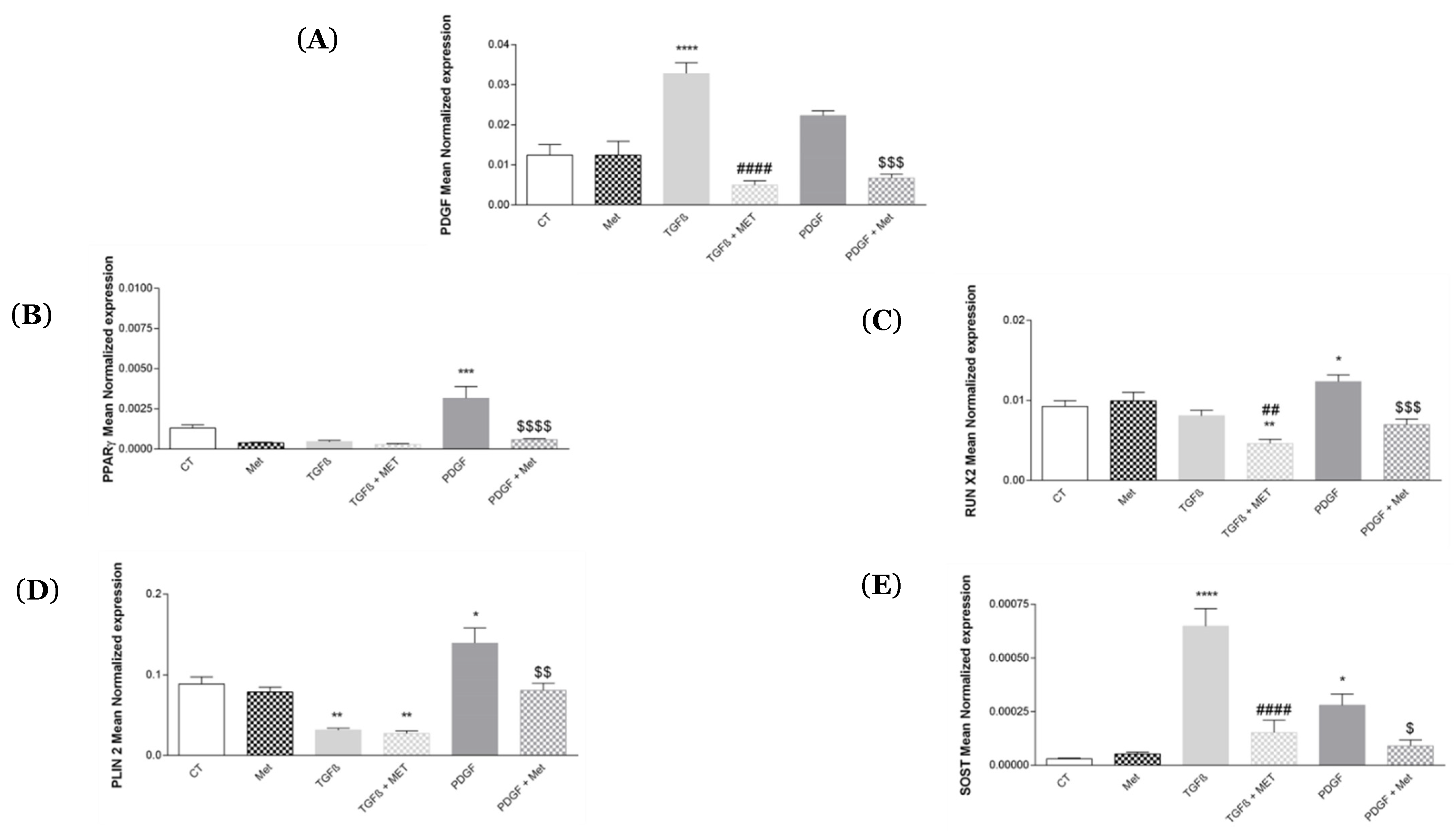
3.3. Study of MSC Transdifferentiation into Lipofibroblasts or Osteoblasts
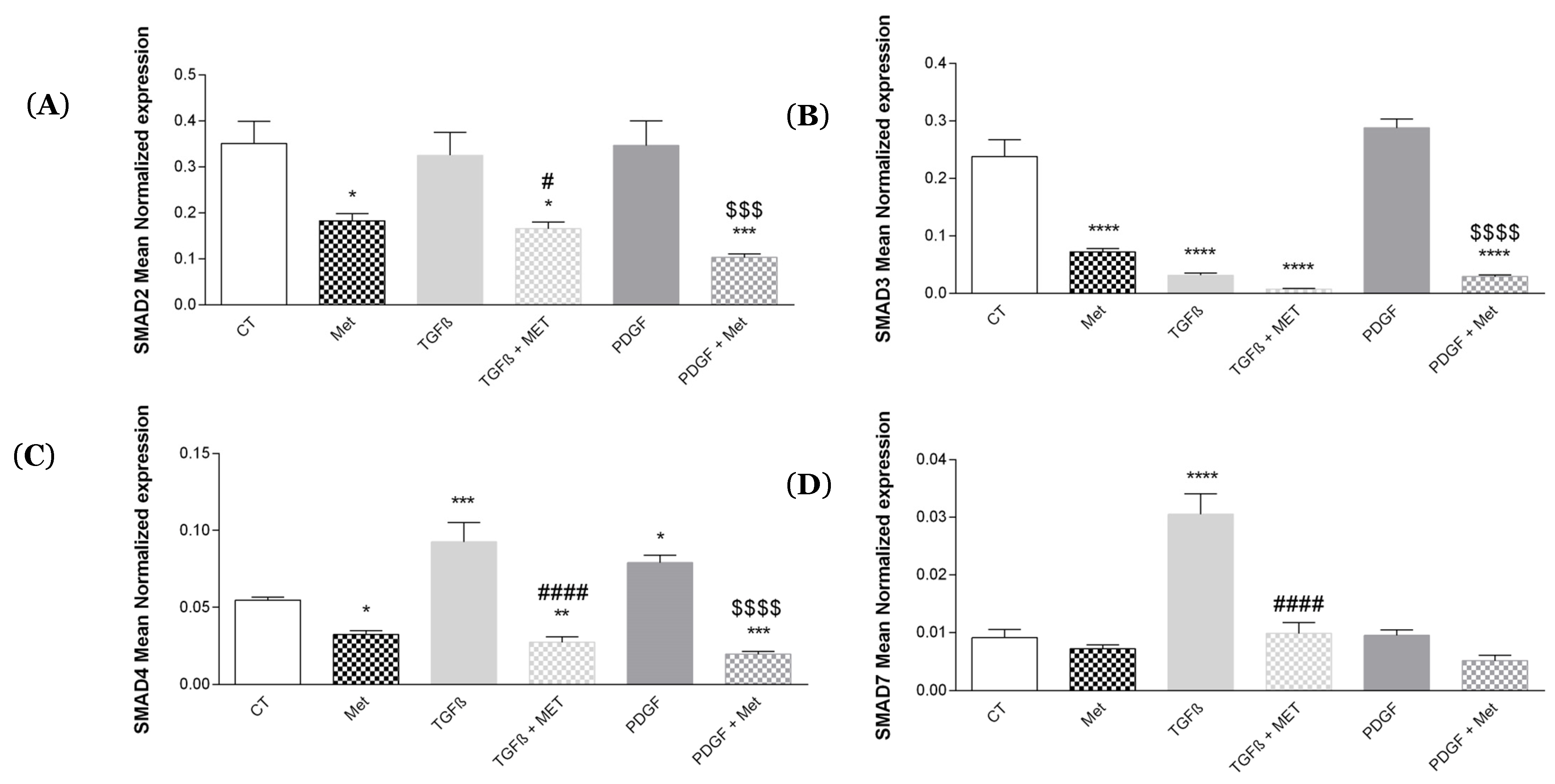
3.4. Study of Met Effects on Canonical Smad mRNA Expression
3.5. Study of Met Effects on the Expression of the Pro-Angiogenic Factor VEGF
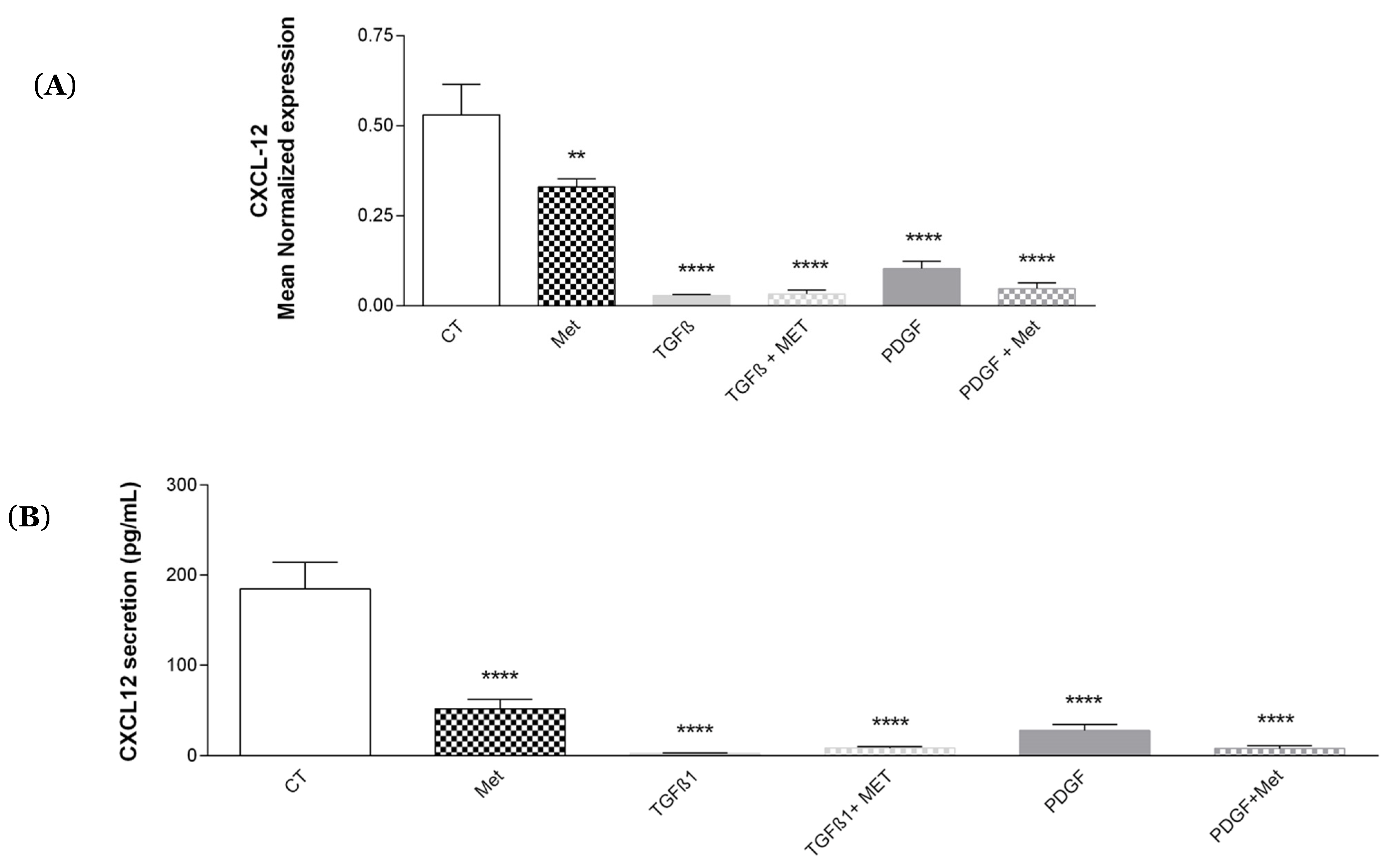
3.6. Study of Met Effects on the Expression of the Growth Factor/Chemokine CXCL-12
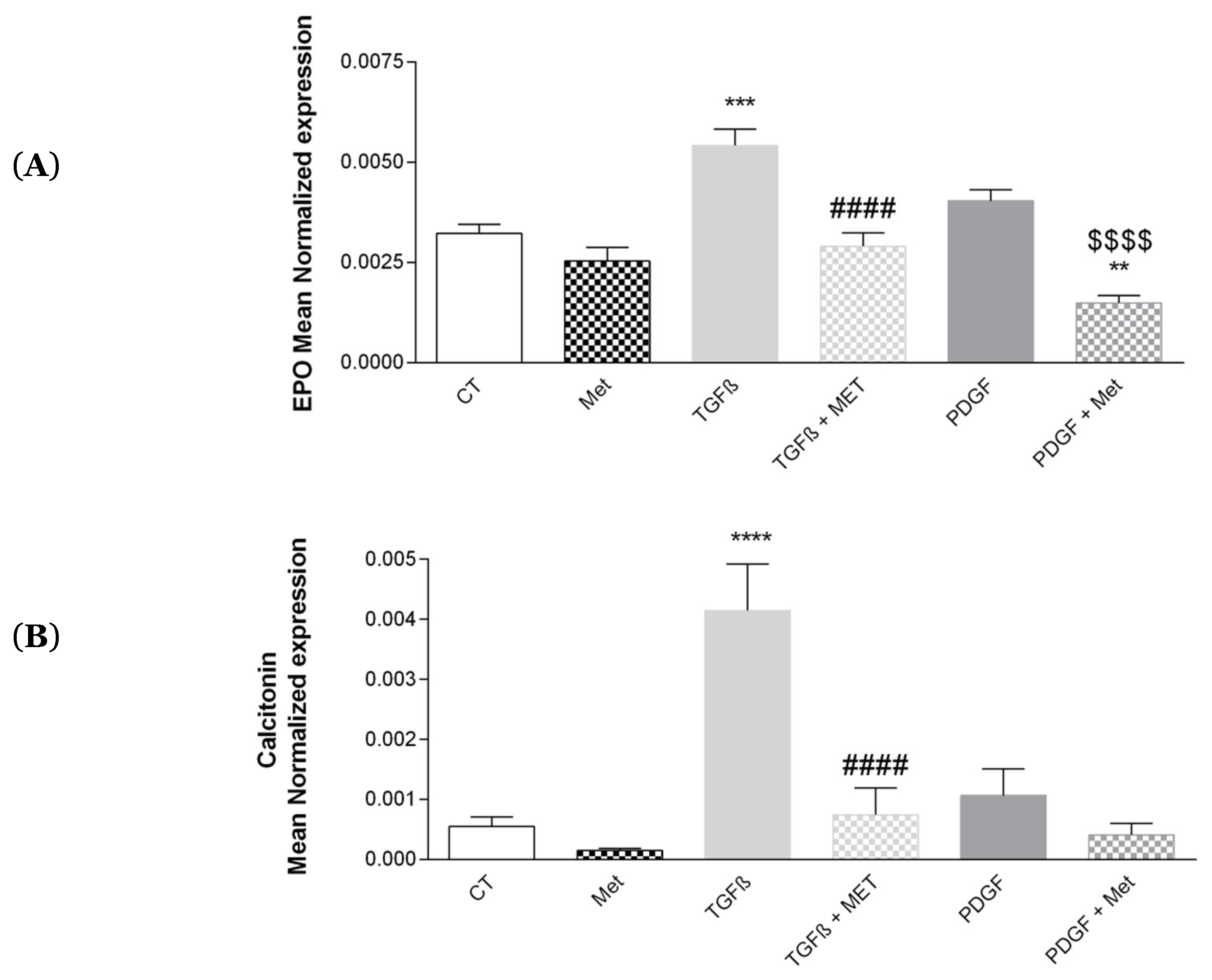
3.7. Study of Met on the Physiological Hormonal Function of MSC
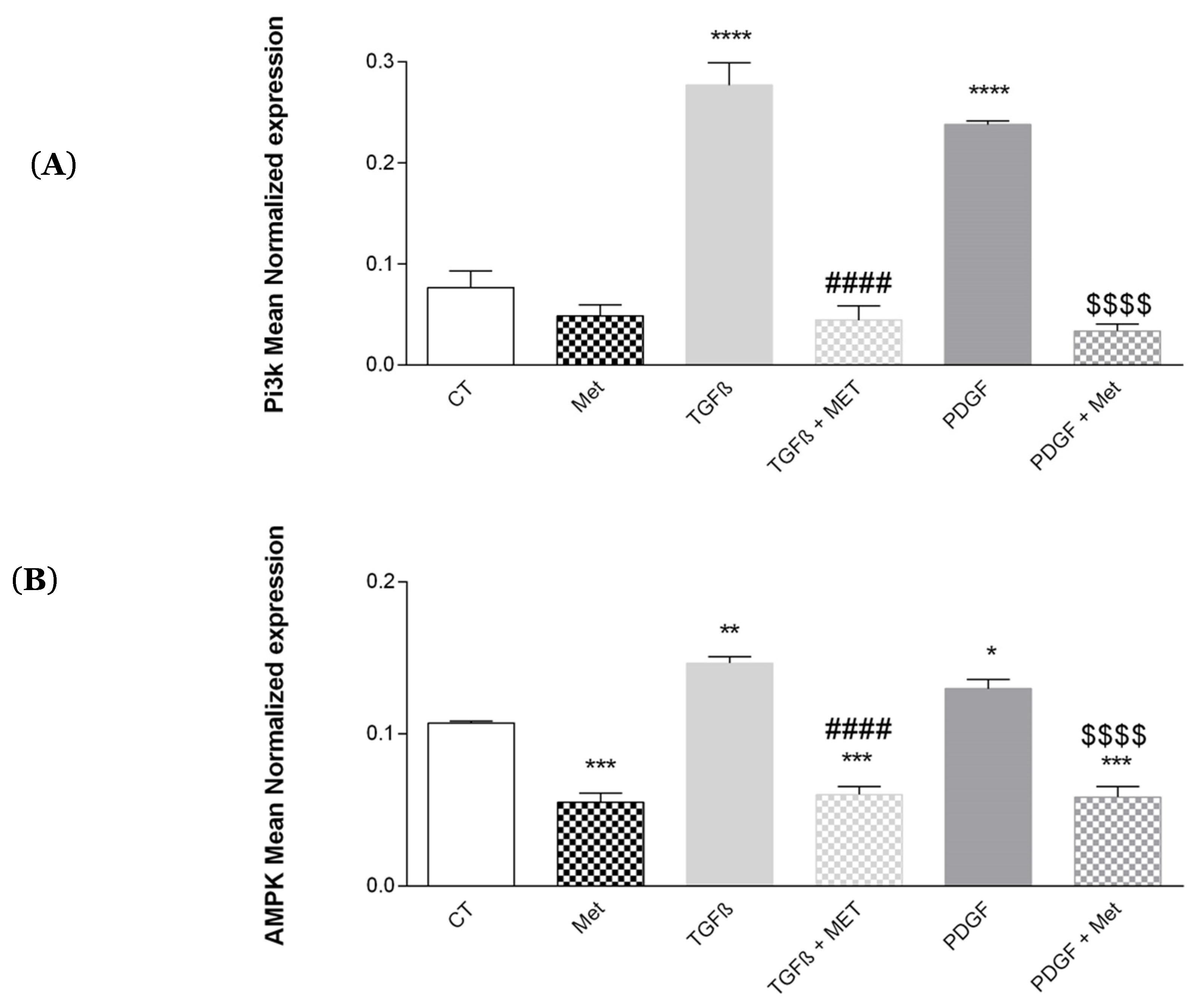
3.8. Study of Met on the Control of AMPK and Pi3K
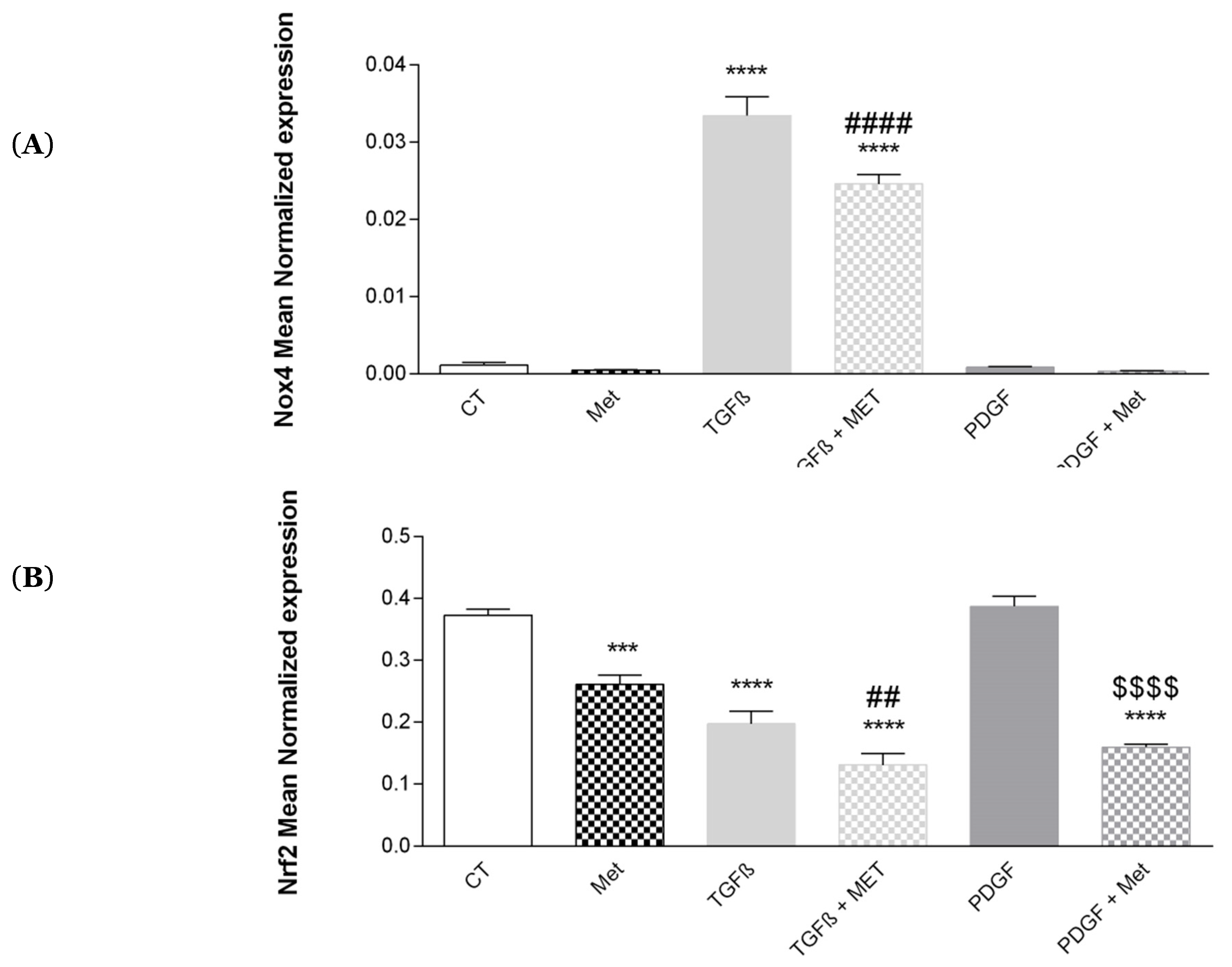
3.9. Study of Oxidative Stress
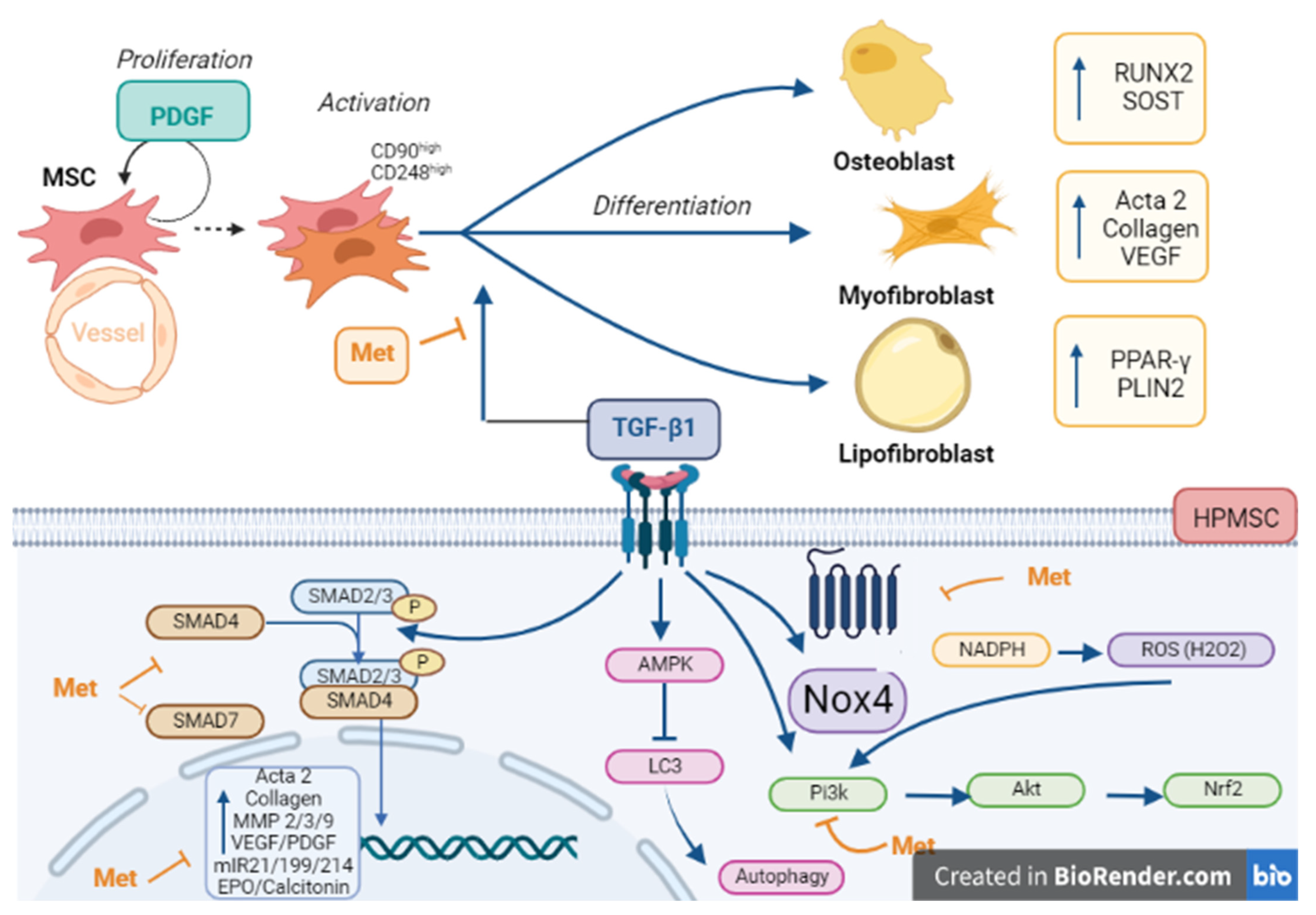
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffield, J.S.; Lupher, M.; Thannickal, V.J.; Wynn, T.A. Host Responses in Tissue Repair and Fibrosis. Annu. Rev. Pathol. 2013, 8, 241–276. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef]
- Gasque, P.; Bandjee, M.C.J.; Reyes, M.M.; Viasus, D. Chikungunya Pathogenesis: From the Clinics to the Bench. J. Infect. Dis 2016, 214, S446–S448. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.J.; Hunninghake, G.W. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef]
- Andersson-Sjöland, A.; de Alba, C.G.; Nihlberg, K.; Becerril, C.; Ramírez, R.; Pardo, A.; Westergren-Thorsson, G.; Selman, M. Fibrocytes Are a Potential Source of Lung Fibroblasts in Idiopathic Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 2129–2140. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and Metabolism in Tissue Repair and Regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Tampe, D.; Zeisberg, M. Potential Approaches to Reverse or Repair Renal Fibrosis. Nat. Rev. Nephrol. 2014, 10, 226–237. [Google Scholar] [CrossRef]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W.; Humphreys, B.D.; Bellusci, S. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef]
- Duffield, J.S. Cellular and Molecular Mechanisms in Kidney Fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef]
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamamura, K.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of Fibroblasts of Extrarenal Origin Underlies Renal Fibrosis and Renal Anemia in Mice. J. Clin. Investig. 2011, 121, 3981–3990. [Google Scholar] [CrossRef]
- Bedoui, Y.; Lebeau, G.; Guillot, X.; Dargai, F.; Guiraud, P.; Neal, J.W.; Ralandison, S.; Gasque, P. Emerging Roles of Perivascular Mesenchymal Stem Cells in Synovial Joint Inflammation. J. Neuroimmune Pharmacol. 2020, 15, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells by Transforming Growth Factor-Β1. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef] [PubMed]
- Swigris, J.J.; Brown, K.K. The Role of Endothelin-1 in the Pathogenesis of Idiopathic Pulmonary Fibrosis. BioDrugs 2010, 24, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Plantier, L.; Cazes, A.; Dinh-Xuan, A.-T.; Bancal, C.; Marchand-Adam, S.; Crestani, B. Physiology of the Lung in Idiopathic Pulmonary Fibrosis. Eur. Respir. Rev. 2018, 27, 170062. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.-H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of Mechanosensitive Signaling in Myofibroblasts Ameliorates Experimental Pulmonary Fibrosis. J. Clin. Investig. 2013, 123, 1096–1108. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis: From Targeting to Biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Desai, O.; Winkler, J.; Minasyan, M.; Herzog, E.L. The Role of Immune and Inflammatory Cells in Idiopathic Pulmonary Fibrosis. Front. Med. 2018, 5, 43. [Google Scholar] [CrossRef]
- Ma, S.; Xie, N.; Li, W.; Yuan, B.; Shi, Y.; Wang, Y. Immunobiology of Mesenchymal Stem Cells. Cell Death Differ. 2014, 21, 216–225. [Google Scholar] [CrossRef]
- Takagi, K.; Yamakuchi, M.; Hashiguchi, T.; Inoue, H. The Role of MiRNAs in Idiopathic Pulmonary Fibrosis; IntechOpen: London, UK, 2019; ISBN 978-1-83881-876-0. [Google Scholar]
- Kumarswamy, R.; Volkmann, I.; Thum, T. Regulation and Function of MiRNA-21 in Health and Disease. RNA Biol. 2011, 8, 706–713. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.-W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA Profiling Reveals a Role for MiR-29 in Human and Murine Liver Fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Kuninty, P.R.; Bojmar, L.; Tjomsland, V.; Larsson, M.; Storm, G.; Ostman, A.; Sandstrom, P.; Prakash, J. MicroRNA-199a and-214 as Potential Therapeutic Targets in Pancreatic Stellate Cells in Pancreatic Tumor. Oncotarget 2016, 7, 16396–16408. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. MiR-21 Mediates Fibrogenic Activation of Pulmonary Fibroblasts and Lung Fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef]
- Pandit, K.V.; Milosevic, J.; Kaminski, N. MicroRNAs in Idiopathic Pulmonary Fibrosis. Transl. Res. 2011, 157, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A Critical Regulator of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.L.L.; Henaoui, I.S.; Courcot, E.; Roderburg, C.; Cauffiez, C.; Aubert, S.; Copin, M.-C.; Wallaert, B.; Glowacki, F.; Dewaeles, E.; et al. MiR-199a-5p Is Upregulated during Fibrogenic Response to Tissue Injury and Mediates TGFbeta-Induced Lung Fibroblast Activation by Targeting Caveolin-1. PLoS Genet. 2013, 9, e1003291. [Google Scholar] [CrossRef]
- Savary, G.; Dewaeles, E.; Diazzi, S.; Buscot, M.; Nottet, N.; Fassy, J.; Courcot, E.; Henaoui, I.-S.; Lemaire, J.; Martis, N.; et al. The Long Noncoding RNA DNM3OS Is a Reservoir of FibromiRs with Major Functions in Lung Fibroblast Response to TGF-β and Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 184–198. [Google Scholar] [CrossRef]
- Henry, T.W.; Mendoza, F.A.; Jimenez, S.A. Role of MicroRNA in the Pathogenesis of Systemic Sclerosis Tissue Fibrosis and Vasculopathy. Autoimmun. Rev. 2019, 18, 102396. [Google Scholar] [CrossRef]
- Nakahira, K.; Pabon Porras, M.A.; Choi, A.M.K. Autophagy in Pulmonary Diseases. Am. J. Respir. Crit. Care Med. 2016, 194, 1196–1207. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, T.; Lei, Y.; Li, X.; Jiang, J.; Lan, J.; Liu, Y.; Chen, H.; Gao, W.; Xie, N.; et al. Identification of ANXA2 (Annexin A2) as a Specific Bleomycin Target to Induce Pulmonary Fibrosis by Impeding TFEB-Mediated Autophagic Flux. Autophagy 2018, 14, 269–282. [Google Scholar] [CrossRef]
- Tu, Y.; Gu, L.; Chen, D.; Wu, W.; Liu, H.; Hu, H.; Wan, Y.; Sun, W. Rhein Inhibits Autophagy in Rat Renal Tubular Cells by Regulation of AMPK/MTOR Signaling. Sci. Rep. 2017, 7, 43790. [Google Scholar] [CrossRef]
- Boutten, A.; Goven, D.; Boczkowski, J.; Bonay, M. Oxidative Stress Targets in Pulmonary Emphysema: Focus on the Nrf2 Pathway. Expert Opin. Ther. Targets 2010, 14, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Sakaguchi, N.; Hachiya, M.; Nakayama, F.; Yamakawa, M.; Akashi, M. Role of Catalase in Monocytic Differentiation of U937 Cells by TPA: Hydrogen Peroxide as a Second Messenger. Leukemia 2009, 23, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Mehrad, B. New Mechanisms of Pulmonary Fibrosis. Chest 2009, 136, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Mazur, W.; Lindholm, P.; Vuorinen, K.; Myllärniemi, M.; Salmenkivi, K.; Kinnula, V.L. Cell-Specific Elevation of NRF2 and Sulfiredoxin-1 as Markers of Oxidative Stress in the Lungs of Idiopathic Pulmonary Fibrosis and Non-Specific Interstitial Pneumonia. APMIS 2010, 118, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Sgalla, G.; Iovene, B.; Calvello, M.; Ori, M.; Varone, F.; Richeldi, L. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respir. Res. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef]
- Samah, M.; El-Aidy, A.E.-R.; Tawfik, M.K.; Ewais, M.M.S. Evaluation of the Antifibrotic Effect of Fenofibrate and Rosiglitazone on Bleomycin-Induced Pulmonary Fibrosis in Rats. Eur. J. Pharmacol. 2012, 689, 186–193. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis (CAPACITY): Two Randomised Trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Rangarajan, S.; Bone, N.B.; Zmijewska, A.A.; Jiang, S.; Park, D.W.; Bernard, K.; Locy, M.L.; Ravi, S.; Deshane, J.; Mannon, R.B.; et al. Metformin Reverses Established Lung Fibrosis in a Bleomycin Model. Nat. Med. 2018, 24, 1121–1127. [Google Scholar] [CrossRef]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.-S.; Kwapiszewska, G.; et al. Metformin Induces Lipogenic Differentiation in Myofibroblasts to Reverse Lung Fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Jang, A.-H.; Kim, H.; Lee, K.H.; Kim, Y.W. Metformin Reduces Bleomycin-Induced Pulmonary Fibrosis in Mice. J. Korean Med. Sci. 2016, 31, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- da Silva-Neto, P.V.; do Valle, V.B.; Fuzo, C.A.; Fernandes, T.M.; Toro, D.M.; Fraga-Silva, T.F.C.; Basile, P.A.; de Carvalho, J.C.S.; Pimentel, V.E.; Pérez, M.M.; et al. Matrix Metalloproteinases on Severe COVID-19 Lung Disease Pathogenesis: Cooperative Actions of MMP-8/MMP-2 Axis on Immune Response through HLA-G Shedding and Oxidative Stress. Biomolecules 2022, 12, 604. [Google Scholar] [CrossRef] [PubMed]
- Tomos, I.P.; Tzouvelekis, A.; Aidinis, V.; Manali, E.D.; Bouros, E.; Bouros, D.; Papiris, S.A. Extracellular Matrix Remodeling in Idiopathic Pulmonary Fibrosis. It Is the “bed” That Counts and Not “the Sleepers”. Expert Rev. Respir. Med. 2017, 11, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, C.M.; Dolgonos, L.; Zemans, R.L.; Young, S.K.; Robertson, J.; Briones, N.; Suzuki, T.; Campbell, M.N.; Gauldie, J.; Radisky, D.C.; et al. Matrix Metalloproteinase 3 Is a Mediator of Pulmonary Fibrosis. Am. J. Pathol. 2011, 179, 1733–1745. [Google Scholar] [CrossRef]
- Zuo, F.; Kaminski, N.; Eugui, E.; Allard, J.; Yakhini, Z.; Ben-Dor, A.; Lollini, L.; Morris, D.; Kim, Y.; DeLustro, B.; et al. Gene Expression Analysis Reveals Matrilysin as a Key Regulator of Pulmonary Fibrosis in Mice and Humans. Proc. Natl. Acad. Sci. USA 2002, 99, 6292–6297. [Google Scholar] [CrossRef]
- Willems, S.; Stijn, W.; Verleden, S.E.; Vanaudenaerde, B.M.; Wynants, M.; Marijke, W.; Dooms, C.; Christophe, D.; Yserbyt, J.; Jonas, Y.; et al. Multiplex Protein Profiling of Bronchoalveolar Lavage in Idiopathic Pulmonary Fibrosis and Hypersensitivity Pneumonitis. Ann. Thorac. Med. 2013, 8, 38–45. [Google Scholar] [CrossRef]
- Chen, Q.; Shou, P.; Zheng, C.; Jiang, M.; Cao, G.; Yang, Q.; Cao, J.; Xie, N.; Velletri, T.; Zhang, X.; et al. Fate Decision of Mesenchymal Stem Cells: Adipocytes or Osteoblasts? Cell Death Differ 2016, 23, 1128–1139. [Google Scholar] [CrossRef]
- Kim, S.; Han, J.-H.; Kim, S.; Lee, H.; Kim, J.-R.; Lim, J.H.; Woo, C.-H. P90RSK Inhibition Ameliorates TGF-Β1 Signaling and Pulmonary Fibrosis by Inhibiting Smad3 Transcriptional Activity. Cell. Physiol. Biochem. 2020, 54, 195–210. [Google Scholar]
- Corvol, P. VEGF, anti-VEGF et pathologies—Académie nationale de médecine | Une institution dans son temps. Bull. Acad. Natl. Med. 2008, 192, 289–302. [Google Scholar] [PubMed]
- Strieter, R.M.; Gomperts, B.N.; Keane, M.P. The Role of CXC Chemokines in Pulmonary Fibrosis. J. Clin. Investig. 2007, 117, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Mehrad, B.; Burdick, M.D.; Strieter, R.M. Fibrocyte CXCR4 Regulation as a Therapeutic Target in Pulmonary Fibrosis. Int. J. Biochem. Cell Biol. 2009, 41, 1708–1718. [Google Scholar] [CrossRef]
- Moreira, L.M.; Takawale, A.; Hulsurkar, M.; Menassa, D.A.; Antanaviciute, A.; Lahiri, S.K.; Mehta, N.; Evans, N.; Psarros, C.; Robinson, P.; et al. Paracrine Signalling by Cardiac Calcitonin Controls Atrial Fibrogenesis and Arrhythmia. Nature 2020, 587, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Horowitz, J.C. Evolving Concepts of Apoptosis in Idiopathic Pulmonary Fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Xia, H.; Nho, R.S.; Kahm, J.; Kleidon, J.; Henke, C.A. Focal Adhesion Kinase Is Upstream of Phosphatidylinositol 3-Kinase/Akt in Regulating Fibroblast Survival in Response to Contraction of Type I Collagen Matrices via a Beta 1 Integrin Viability Signaling Pathway. J. Biol. Chem. 2004, 279, 33024–33034. [Google Scholar] [CrossRef]
- Audousset, C.; McGovern, T.; Martin, J.G. Role of Nrf2 in Disease: Novel Molecular Mechanisms and Therapeutic Approaches—Pulmonary Disease/Asthma. Front. Physiol. 2021, 12, 727806. [Google Scholar] [CrossRef]
- Braun, S.; Hanselmann, C.; Gassmann, M.G.; auf dem Keller, U.; Born-Berclaz, C.; Chan, K.; Wai Kan, Y.; Werner, S. Nrf2 Transcription Factor, a Novel Target of Keratinocyte Growth Factor Action Which Regulates Gene Expression and Inflammation in the Healing Skin Wound. Mol. Cell. Biol. 2002, 22, 5492–5505. [Google Scholar] [CrossRef]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β Signaling in Tissue Fibrosis: Redox Controls, Target Genes and Therapeutic Opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef]
- Scruggs, A.M.; Grabauskas, G.; Huang, S.K. The Role of KCNMB1 and BK Channels in Myofibroblast Differentiation and Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 62, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Dou, Y.; Zhao, Q.; Zhang, J.; Yang, Y.; Wang, T.; Xia, Y.; Dai, Y.; Wei, Z. Paeoniflorin Suppresses TGF-β Mediated Epithelial-Mesenchymal Transition in Pulmonary Fibrosis through a Smad-Dependent Pathway. Acta Pharmacol. Sin. 2016, 37, 794–804. [Google Scholar] [CrossRef]
- Spagnolo, P.; Maher, T.M. Clinical Trial Research in Focus: Why Do so Many Clinical Trials Fail in IPF? Lancet Respir. Med. 2017, 5, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Regulation of AMP-Activated Protein Kinase by Natural and Synthetic Activators. Acta Pharm. Sin. B 2016, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, G.R.; Carling, D. AMP-Activated Protein Kinase: The Current Landscape for Drug Development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef] [PubMed]
- da Silva Meirelles, L.; Caplan, A.I.; Nardi, N.B. Search of the In Vivo Identity of Mesenchymal Stem Cells. Stem Cells 2008, 26, 2287–2299. [Google Scholar] [CrossRef]
- Antoniou, K.M.; Pataka, A.; Bouros, D.; Siafakas, N.M. Pathogenetic Pathways and Novel Pharmacotherapeutic Targets in Idiopathic Pulmonary Fibrosis. Pulm. Pharmacol. Ther. 2007, 20, 453–461. [Google Scholar] [CrossRef]
- Xin, X.; Yao, D.; Zhang, K.; Han, S.; Liu, D.; Wang, H.; Liu, X.; Li, G.; Huang, J.; Wang, J. Protective Effects of Rosavin on Bleomycin-Induced Pulmonary Fibrosis via Suppressing Fibrotic and Inflammatory Signaling Pathways in Mice. Biomed. Pharmacother. 2019, 115, 108870. [Google Scholar] [CrossRef]
- Crunkhorn, S. MiR-21 Antagomir Reverses COPD Pathology. Nat. Rev. Drug Discov. 2021, 21, 20. [Google Scholar] [CrossRef]
- Savary, G.; Buscot, M.; Dewaeles, E.; Henaoui, I.S.; Quarré, S.; Courcot, E.; Cauffiez, C.; Barbry, P.; Perrais, M.; Mari, B.; et al. MiR-214-3p, a New FibromiR Involved in the Pathogenesis of Idiopathic Pulmonary Fibrosis. Eur. Respir. J. 2014, 44, 1731. [Google Scholar]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lü, J. MiR-29 Is a Major Regulator of Genes Associated with Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Duan, L.; Wei, S.; Liu, J.; Tian, L.; Quan, J.; Zhang, Q.; Liu, J.; Yang, J. Metformin Ameliorates Skeletal Muscle Insulin Resistance by Inhibiting MiR-21 Expression in a High-Fat Dietary Rat Model. Oncotarget 2017, 8, 98029–98039. [Google Scholar] [CrossRef]
- Giannandrea, M.; Parks, W.C. Diverse Functions of Matrix Metalloproteinases during Fibrosis. Dis. Model. Mech. 2014, 7, 193–203. [Google Scholar] [CrossRef]
- Lim, D.H.; Cho, J.Y.; Miller, M.; McElwain, K.; McElwain, S.; Broide, D.H. Reduced Peribronchial Fibrosis in Allergen-Challenged MMP-9-Deficient Mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L265–L271. [Google Scholar] [CrossRef]
- Lakshmi, S.P.; Reddy, A.T.; Reddy, R.C. Transforming Growth Factor β Suppresses Peroxisome Proliferator–Activated Receptor γ Expression via Both SMAD Binding and Novel TGF-β Inhibitory Elements. Biochem. J. 2017, 474, 1531–1546. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.; Yu, L.; Deng, Y.; Li, D.; Yu, X.; Chen, D.; Lu, Y.; Liu, S.; Chen, R. Pemafibrate Attenuates Pulmonary Fibrosis by Inhibiting Myofibroblast Differentiation. Int. Immunopharmacol. 2022, 108, 108728. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Akbulut, A.C.; Kaczor, D.M.; Halder, M.; Koenen, R.R.; Kramann, R. Initiation and Propagation of Vascular Calcification Is Regulated by a Concert of Platelet- and Smooth Muscle Cell-Derived Extracellular Vesicles. Front. Cardiovasc. Med. 2018, 5, 36. [Google Scholar] [CrossRef]
- Meng, X.-M.; Tang, P.M.-K.; Li, J.; Lan, H.Y. TGF-Beta/Smad Signaling in Renal Fibrosis. Front. Physiol. 2015, 6, 82. [Google Scholar] [CrossRef]
- Wu, B.; Tang, L.; Kapoor, M. Fibroblasts and Their Responses to Chronic Injury in Pulmonary Fibrosis. Semin. Arthritis Rheum. 2021, 51, 310–317. [Google Scholar] [CrossRef]
- Amano, H.; Mastui, Y.; Ito, Y.; Shibata, Y.; Betto, T.; Eshima, K.; Ogawa, F.; Satoh, Y.; Shibuya, M.; Majima, M. The Role of Vascular Endothelial Growth Factor Receptor 1 Tyrosine Kinase Signaling in Bleomycin-Induced Pulmonary Fibrosis. Biomed. Pharmacother. 2019, 117, 109067. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor Cell Trafficking Is Regulated by Hypoxic Gradients through HIF-1 Induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef]
- Yi, J.Y.; Shin, I.; Arteaga, C.L. Type I Transforming Growth Factor Beta Receptor Binds to and Activates Phosphatidylinositol 3-Kinase. J. Biol. Chem. 2005, 280, 10870–10876. [Google Scholar] [CrossRef]
- Lim, J.-Y.; Oh, M.-A.; Kim, W.H.; Sohn, H.-Y.; Park, S.I. AMP-Activated Protein Kinase Inhibits TGF-Beta-Induced Fibrogenic Responses of Hepatic Stellate Cells by Targeting Transcriptional Coactivator P300. J. Cell. Physiol. 2012, 227, 1081–1089. [Google Scholar] [CrossRef]
- Waghray, M.; Cui, Z.B.; Horowitz, J.C.; Subramanian, I.M.; Martinez, F.J.; Toews, G.B.; Thannickal, V.J. Hydrogen Peroxide Is a Diffusible Paracrine Signal for the Induction of Epithelial Cell Death by Activated Myofibroblasts. Faseb J. 2005, 19, 1–16. [Google Scholar] [CrossRef]
- Hecker, L.; Vittal, R.; Jones, T.; Jagirdar, R.; Luckhardt, T.R.; Horowitz, J.C.; Pennathur, S.; Martinez, F.J.; Thannickal, V.J. NADPH Oxidase-4 Mediates Myofibroblast Activation and Fibrogenic Responses to Lung Injury. Nat. Med. 2009, 15, 1077–1081. [Google Scholar] [CrossRef]
- Sato, N.; Takasaka, N.; Yoshida, M.; Tsubouchi, K.; Minagawa, S.; Araya, J.; Saito, N.; Fujita, Y.; Kurita, Y.; Kobayashi, K.; et al. Metformin Attenuates Lung Fibrosis Development via NOX4 Suppression. Respir. Res. 2016, 17, 107. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of Persistent Fibrosis in Aging by Targeting Nox4-Nrf2 Redox Imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Forward | Reverse | Detection |
|---|---|---|---|
| GAPDH | TGCGTCGCCAGCCGAG | AGTTAAAAGCAGCCCTGGTG | SYBR Green |
| Acta2 | TTCATCGGGATGGAGTCTGCTGG | TCGGTCGGCAATGCCAGGGT | SYBR Green |
| Col1 A1 | TCTAGACATGTTCAGCTTTGTGGAC | TCTGTACGCAGGTGATTGGT | SYBR Green |
| Col 2 | GAGGGCAACAGCAGGTTCACTTA | TCAGCACCACCGATGTCCA | SYBR Green |
| Col 4 | GGACGGCGTAGGCTTCTTG | GCAAACGCTTCAGCTTTTGG | SYBR Green |
| PPAR-γ | GGTGACCAGAAGCCTGCATT | TGTCAACCATGGTCATTCGTT | SYBR Green |
| PLIN 2 | GCTGCAGTCCGTCGATTTCT | TCACACCGTTCTCTGCCATC | SYBR Green |
| RUNX 2 | TAGGCGCATTTCAGGTGCTT | TGCATTCGTGGGTTGGAGAA | SYBR Green |
| PDGF | TCCTGTCTCTCTGCTGCTAC | ATCAAAGGAGCGGATCGAGT | SYBR Green |
| SOST | TTCCCCGGATGTTTGGCTAC | AGTTGGGGCGGATGTGATTT | SYBR Green |
| SMAD2 | AACAGAACTTCCGCCTCTGG | GGAGGTGGCGTTTCTGGAAT | SYBR Green |
| SMAD3 | TGGACGCAGGTTCTCCAAAC | CCGGCTCGCAGTAGGTAAC | SYBR Green |
| SMAD4 | GGACTGTTGCAGATAGCATC | GCTGGAATGCAAGCTCATTG | SYBR Green |
| SMAD7 | CCATCGGGTATCTGGAGTAAGGA | TGCTGTGCAAAGTGTTCAGGTG | SYBR Green |
| VEGF | ACAACAAATGTGAATGCAGACCA | GAGGCTCCAGGGCATTAGAC | SYBR Green |
| CXCL-12 | CTACAGATGCCCATGCCGAT | CAGCCGGGCTACAATCTGAA | SYBR Green |
| EPO | CGAGAATATCACGACGGGCT | CAGACTTCTACGGCCTGCTG | SYBR Green |
| Calcitonin | ATCAGAGACACTGCCCAGC | CCAGGGCAGACCTGAATGG | SYBR Green |
| MMP1 | TTTGTCAGGGGAGATCATCGG | TCCAAGAGAATGGCCGAGTT | SYBR Green |
| MMP 2 | CCCTGATGTCCAGCGAGTG | ACGACGGCATCCAGGTTATC | SYBR Green |
| MMP 3 | TCAGTCCCTCTATGGACCTCCC | GGTTCAAGCTTCCGAGGGAT | SYBR Green |
| MMP 9 | TGCCCGGACCAAGGATACAGTTT | GTTCAGGGCGAGGACCATAGAGG | SYBR Green |
| Pi3k | TCTTTGTGCAACCTACGTGA | AGCCATTCATTCCACCTGGG | SYBR Green |
| AMPK | TGTCACAGGCATATGGTGGTC | GGGCCTGCATACAATCTTCC | SYBR Green |
| Nrf2 | GCTATGGAGACACACTACTTGG | CCAGGACTTCAGGCAATTCT | SYBR Green |
| Nox4 | TCGCCAACGAAGGGGTTAAA | GACACAATCTAGCCCCAACA | SYBR Green |
| CD 90 | TGAAAACTGCGGGGTCCGA | TGCAAGACTGTAGCAGGGAG | SYBR Green |
| CD 248 | TTGCACTGGGCATCGTGTA | TTGCTCCCAGCATGGATGAC | SYBR Green |
| Target Gene | Forward | Detection |
|---|---|---|
| hsa miR-21-5p | GCAGTAGCTTATCAGACTGATG | SYBR Green |
| hsa miR-214-3p | ACAGCAGGCACAGACA | SYBR Green |
| hsa miR-29b-3p | CAGTAGCACCATTTGAAATCAG | SYBR Green |
| hsa miR-199a-5p | GCCCAGTGTTCAGACTAC | SYBR Green |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Septembre-Malaterre, A.; Boina, C.; Douanier, A.; Gasque, P. Deciphering the Antifibrotic Property of Metformin. Cells 2022, 11, 4090. https://doi.org/10.3390/cells11244090
Septembre-Malaterre A, Boina C, Douanier A, Gasque P. Deciphering the Antifibrotic Property of Metformin. Cells. 2022; 11(24):4090. https://doi.org/10.3390/cells11244090
Chicago/Turabian StyleSeptembre-Malaterre, Axelle, Chailas Boina, Audrey Douanier, and Philippe Gasque. 2022. "Deciphering the Antifibrotic Property of Metformin" Cells 11, no. 24: 4090. https://doi.org/10.3390/cells11244090
APA StyleSeptembre-Malaterre, A., Boina, C., Douanier, A., & Gasque, P. (2022). Deciphering the Antifibrotic Property of Metformin. Cells, 11(24), 4090. https://doi.org/10.3390/cells11244090