
Therapeutic Efficacy of Small Extracellular Vesicles Loaded with ROCK Inhibitor in Parkinson’s Disease
 , , , , , and
, , , , , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animal Model and Treatments with EV-SR3677
2.2. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
2.3. Western Blotting
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Hematoxylin and Eosin (H&E) Staining
2.6. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight (MALDI-TOF) Mass Spectrometry (MS)
2.7. Mitochondria Bioenergetics
2.8. Statistical Analysis
3. Results
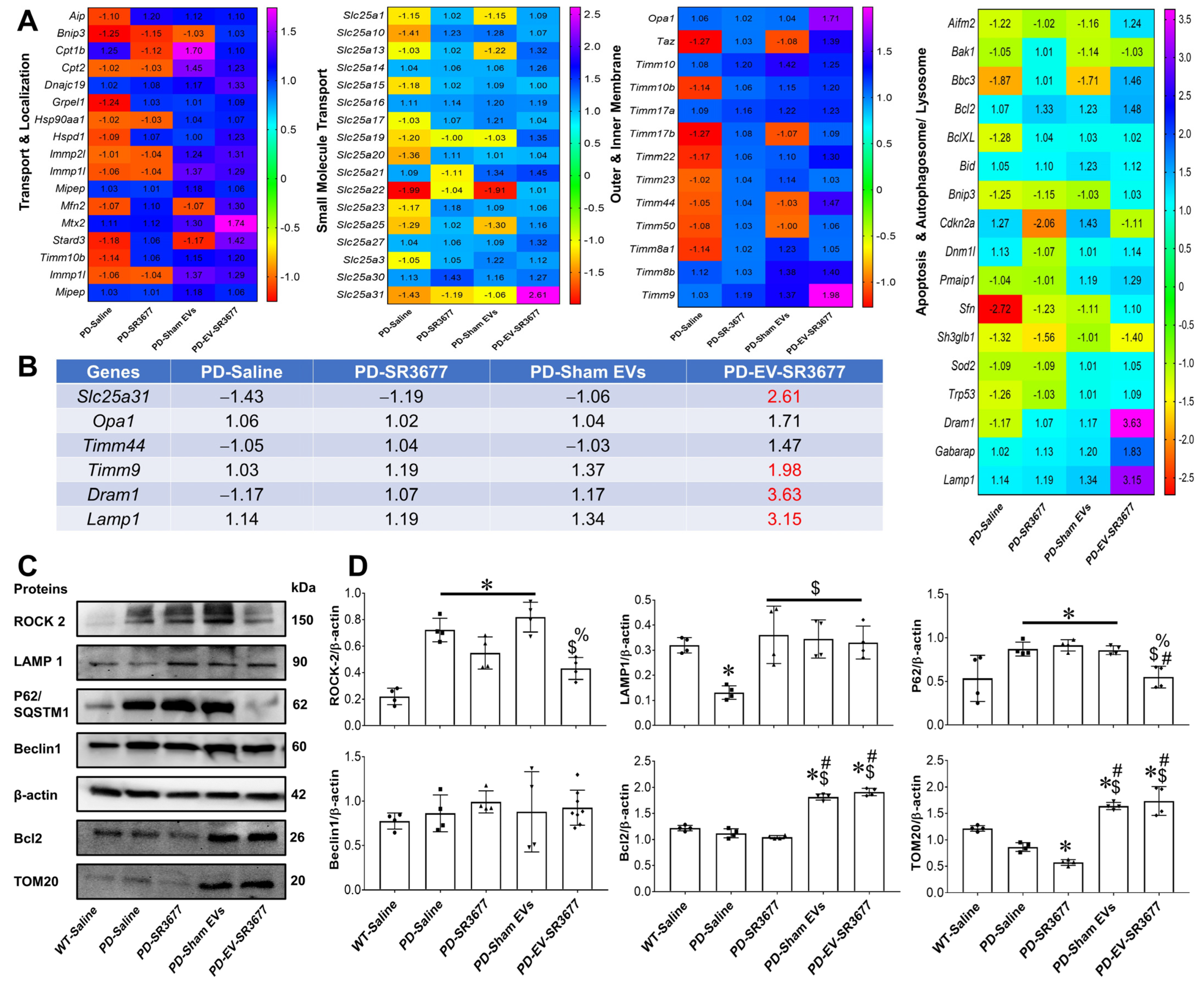
3.1. sEVs-SR3677 Upregulated the Transcription of Genes and Proteins Responsible for Mitochondrial Function and the Mitophagy Pathway in the PD Mouse Brain
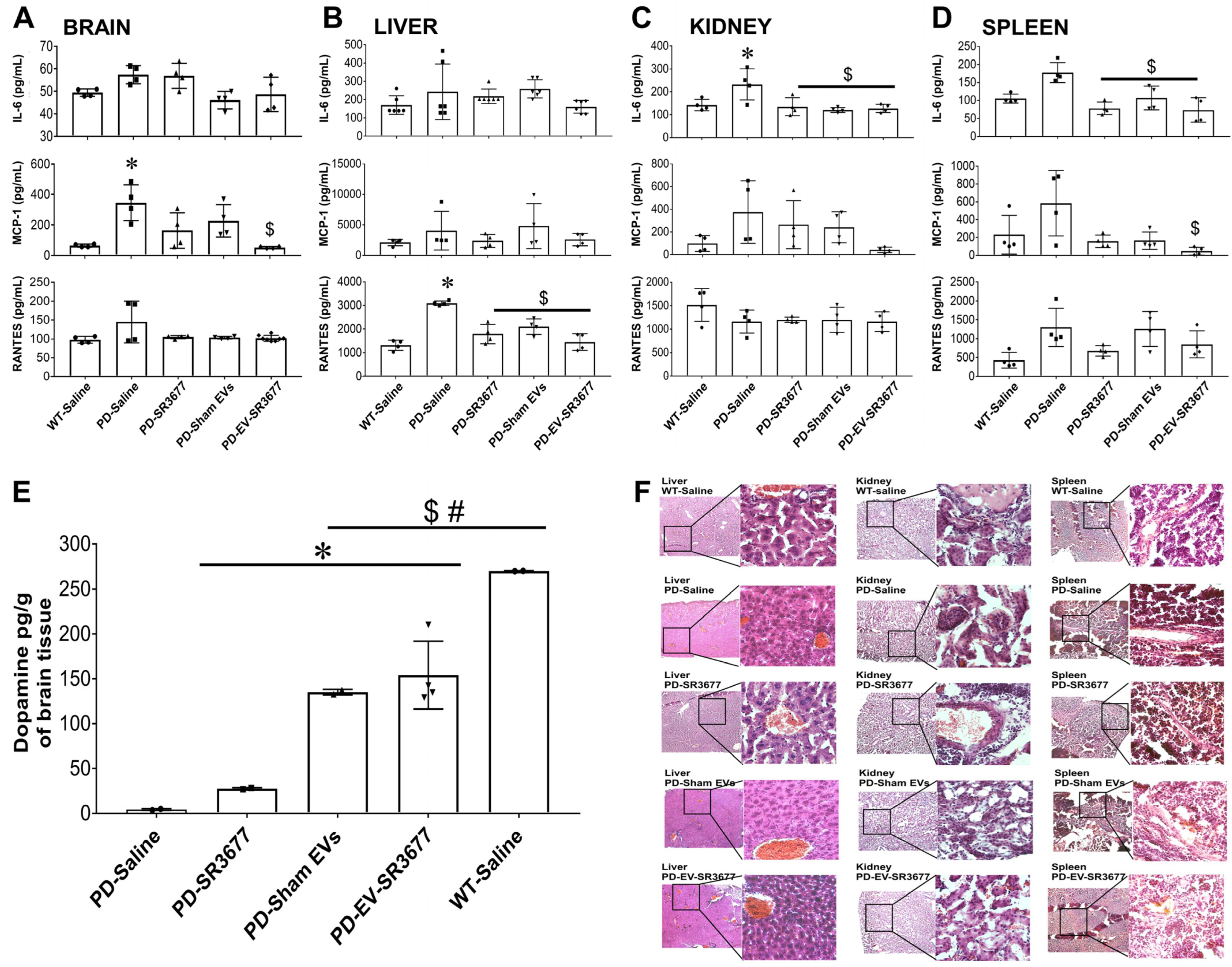
3.2. sEVs-SR3677 Does Not Affect Inflammatory Factors in the Brain or Other Organs of the PD Mouse
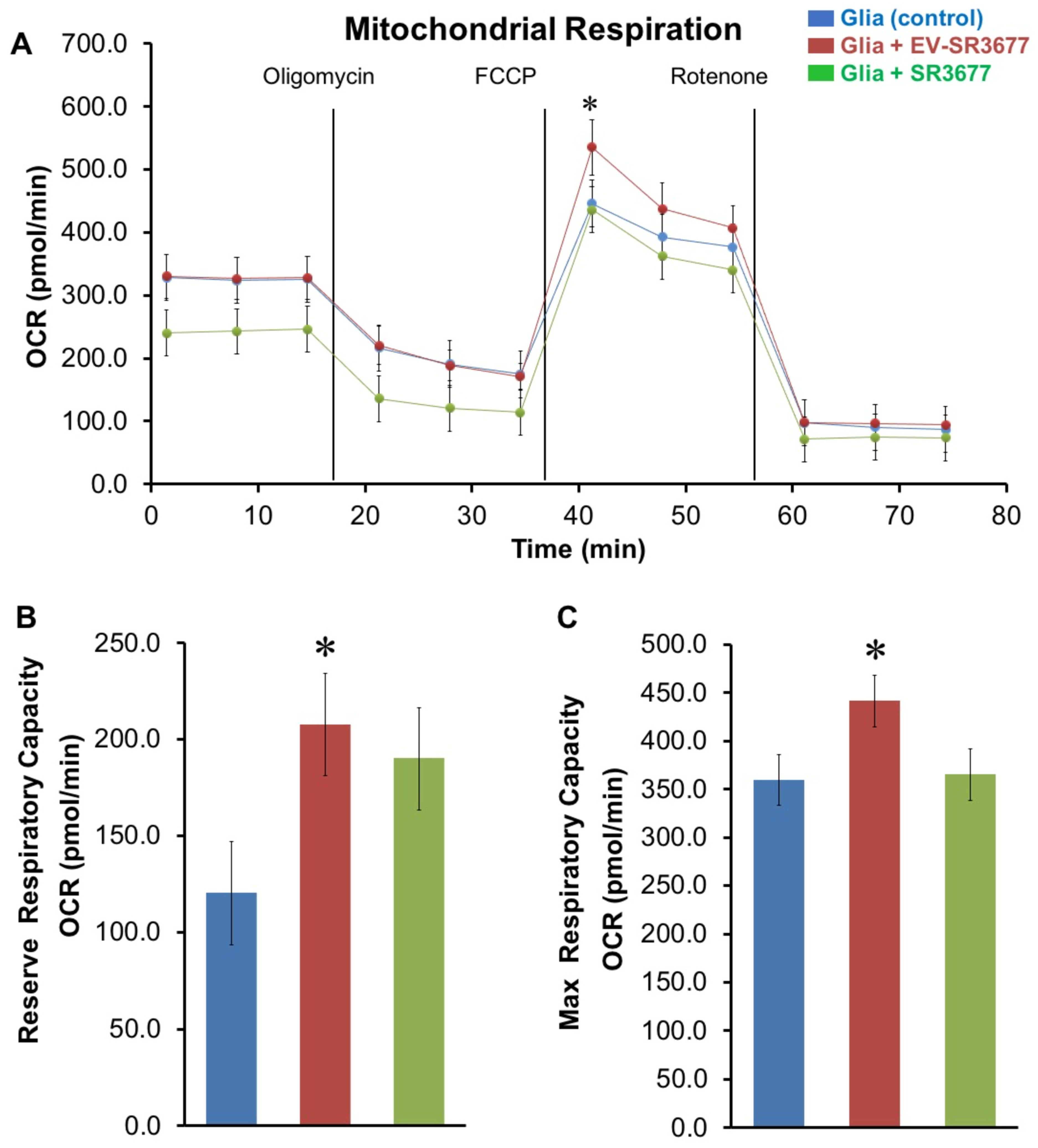
3.3. sEV-SR3677 Stimulates Mitochondrial Respiration Capacity in Ex Vivo Glial Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Girigoswami, K.; Pallavi, P.; Girigoswami, A. Intricate subcellular journey of nanoparticles to the enigmatic domains of endoplasmic reticulum. Drug Deliv. 2023, 30, 2284684. [Google Scholar] [CrossRef] [PubMed]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar] [PubMed]
- Ishizaki, T.; Maekawa, M.; Fujisawa, K.; Okawa, K.; Iwamatsu, A.; Fujita, A.; Watanabe, N.; Saito, Y.; Kakizuka, A.; Morii, N.; et al. The small GTP-binding protein Rho binds to and activates a 160 kDa Ser/Thr protein kinase homologous to myotonic dystrophy kinase. EMBO J. 1996, 15, 1885–1893. [Google Scholar] [CrossRef]
- Liao, J.K.; Seto, M.; Noma, K. Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharmacol. 2007, 50, 17–24. [Google Scholar] [CrossRef]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell. Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, C.; Huang, J.; Liu, W.; Lai, W.; Leng, F.; Tang, Q.; Liu, Y.; Wang, Q.; Zhou, M.; et al. ROCK1 induces dopaminergic nerve cell apoptosis via the activation of Drp1-mediated aberrant mitochondrial fission in Parkinson’s disease. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, C.; Huang, W.; Zhang, P.; Shi, J.; Yu, T.; Wang, J.; Hu, Y.; Zhao, L.; Zhang, R.; Wang, G.; et al. Critical role of ROCK1 in AD pathogenesis via controlling lysosomal biogenesis and acidification. Transl. Neurodegener. 2024, 13, 54. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nakagawa, O.; Fujisawa, K.; Ishizaki, T.; Saito, Y.; Nakao, K.; Narumiya, S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996, 392, 189–193. [Google Scholar] [CrossRef]
- Zhou, Q.; Gensch, C.; Liao, J.K. Rho-associated coiled-coil-forming kinases (ROCKs): Potential targets for the treatment of atherosclerosis and vascular disease. Trends Pharmacol. Sci. 2011, 32, 167–173. [Google Scholar] [CrossRef]
- Matoba, K.; Takeda, Y.; Nagai, Y.; Sekiguchi, K.; Yokota, T.; Utsunomiya, K.; Nishimura, R. The Physiology, Pathology, and Therapeutic Interventions for ROCK Isoforms in Diabetic Kidney Disease. Front. Pharmacol. 2020, 11, 585633. [Google Scholar] [CrossRef] [PubMed]
- Niklander, S.; Bandaru, D.; Lambert, D.W.; Hunter, K.D. ROCK inhibition modulates the senescence-associated secretory phenotype (SASP) in oral keratinocytes. FEBS Open Bio 2020, 10, 2740–2749. [Google Scholar] [CrossRef]
- Dong, Y.; Zhuang, X.-X.; Wang, Y.-T.; Tan, J.; Feng, D.; Li, M.; Zhong, Q.; Song, Z.; Shen, H.-M.; Fang, E.F.; et al. Chemical mitophagy modulators: Drug development strategies and novel regulatory mechanisms. Pharmacol. Res. 2023, 194, 106835. [Google Scholar] [CrossRef]
- Saal, K.A.; Koch, J.C.; Tatenhorst, L.; Szegő, É.; Ribas, V.T.; Michel, U.; Bähr, M.; Tönges, L.; Lingor, P. AAV.shRNA-mediated downregulation of ROCK2 attenuates degeneration of dopaminergic neurons in toxin-induced models of Parkinson’s disease in vitro and in vivo. Neurobiol. Dis. 2015, 73, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Weihe, E.; Depboylu, C.; Schütz, B.; Schäfer, M.K.-H.; Eiden, L.E. Three types of tyrosine hydroxylase-positive CNS neurons distinguished by dopa decarboxylase and VMAT2 co-expression. Cell. Mol. Neurobiol. 2006, 26, 659–678. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Pandit, R.; Chen, L.; Götz, J. The blood-brain barrier: Physiology and strategies for drug delivery. Adv. Drug Deliv. Rev. 2020, 165–166, 1–14. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Fay, J.; Duhyeong, H.; Wang, M.; Wang, H.; Li, Z.; Lee, Y.Z.; Karuppan, M.K.; El-Hage, N.; et al. Genetically modified macrophages accomplish targeted gene delivery to the inflamed brain in transgenic Parkin Q311X(A) mice: Importance of administration routes. Sci. Rep. 2020, 10, 11818. [Google Scholar] [CrossRef]
- Zhao, Y.; Haney, M.J.; Fallon, J.K.; Rodriguez, M.; Swain, C.J.; Arzt, C.J.; Smith, P.C.; Loop, M.S.; Harrison, E.B.; El-Hage, N.; et al. Using Extracellular Vesicles Released by GDNF-Transfected Macrophages for Therapy of Parkinson Disease. Cells 2022, 11, 1933. [Google Scholar] [CrossRef]
- Pedrosa, M.A.; Labandeira, C.M.; Lago-Baameiro, N.; Valenzuela, R.; Pardo, M.; Labandeira-Garcia, J.L.; Rodriguez-Perez, A.I. Extracellular Vesicles and Their Renin–Angiotensin Cargo as a Link between Metabolic Syndrome and Parkinson’s Disease. Antioxidants 2023, 12, 2045. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Clayton, A.; Amigorena, S.; Raposo, G. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3.22.1–3.22.29. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Elsharkasy, O.M.; Nordin, J.Z.; Hagey, D.W.; de Jong, O.G.; Schiffelers, R.M.; Andaloussi, S.E.; Vader, P. Extracellular vesicles as drug delivery systems: Why and how? Adv. Drug Deliv. Rev. 2020, 159, 332–343. [Google Scholar] [CrossRef]
- Herrmann, I.K.; Wood, M.J.A.; Fuhrmann, G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021, 16, 748–759. [Google Scholar] [CrossRef]
- Lau, S.Y.; Kang, M.; Hisey, C.L.; Chamley, L.W. Studying exogenous extracellular vesicle biodistribution by in vivo fluorescence microscopy. Dis. Model. Mech. 2023, 16, dmm050074. [Google Scholar] [CrossRef]
- Haney, M.J.; Yuan, H.; Shipley, S.T.; Wu, Z.; Zhao, Y.; Pate, K.; Frank, J.E.; Massoud, N.; Stewart, P.W.; Perlmutter, J.S.; et al. Biodistribution of Biomimetic Drug Carriers, Mononuclear Cells, and Extracellular Vesicles, in Nonhuman Primates. Adv. Biol. 2022, 6, e2101293. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Jin, Y.S.; Batrakova, E.V. Extracellular Vesicles as Drug Carriers for Enzyme Replacement Therapy to Treat CLN2 Batten Disease: Optimization of Drug Administration Routes. Cells 2020, 9, 1273. [Google Scholar] [CrossRef]
- Zhang, Q.; Higginbotham, J.N.; Jeppesen, D.K.; Yang, Y.-P.; Li, W.; McKinley, E.T.; Graves-Deal, R.; Ping, J.; Britain, C.M.; Dorsett, K.A.; et al. Transfer of Functional Cargo in Exomeres. Cell Rep. 2019, 27, 940–954.e6. [Google Scholar] [CrossRef]
- El-Hage, N.; Haney, M.J.; Zhao, Y.; Rodriguez, M.; Wu, Z.; Liu, M.; Swain, C.J.; Yuan, H.; Batrakova, E.V. Extracellular Vesicles Released by Genetically Modified Macrophages Activate Autophagy and Produce Potent Neuroprotection in Mouse Model of Lysosomal Storage Disorder, Batten Disease. Cells 2023, 12, 1497. [Google Scholar] [CrossRef]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control Release 2015, 207, 18–30. [Google Scholar] [CrossRef]
- Rodriguez, M.; Lapierre, J.; Ojha, C.R.; Kaushik, A.; Batrakova, E.; Kashanchi, F.; Dever, S.M.; Nair, M.; El-Hage, N. Intranasal drug delivery of small interfering RNA targeting Beclin1 encapsulated with polyethylenimine (PEI) in mouse brain to achieve HIV attenuation. Sci. Rep. 2017, 7, 1862. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Haney, M.J.; Jin, Y.S.; Uvarov, O.; Vinod, N.; Lee, Y.Z.; Langworthy, B.; Fine, J.P.; Rodriguez, M.; El-Hage, N.; et al. GDNF-expressing macrophages restore motor functions at a severe late-stage, and produce long-term neuroprotective effects at an early-stage of Parkinson’s disease in transgenic Parkin Q311X(A) mice. J. Control Release 2019, 315, 139–149. [Google Scholar] [CrossRef]
- Lapierre, J.; Karuppan, M.K.M.; Perry, M.; Rodriguez, M.; El-Hage, N. Different Roles of Beclin1 in the Interaction Between Glia and Neurons after Exposure to Morphine and the HIV- Trans-Activator of Transcription (Tat) Protein. J. Neuroimmune Pharmacol. 2022, 17, 470–486. [Google Scholar] [CrossRef]
- Lapierre, J.; Rodriguez, M.; Ojha, C.R.; El-Hage, N. Critical Role of Beclin1 in HIV Tat and Morphine-Induced Inflammation and Calcium Release in Glial Cells from Autophagy Deficient Mouse. J. Neuroimmune Pharmacol. 2018, 13, 355–370. [Google Scholar] [CrossRef] [PubMed]
- Plitzko, B.; Loesgen, S. Measurement of Oxygen Consumption Rate (OCR) and Extracellular Acidification Rate (ECAR) in Culture Cells for Assessment of the Energy Metabolism. Bio Protoc. 2018, 8, e2850. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef]
- Dutta, S.; Sengupta, P. Men and mice: Relating their ages. Life Sci. 2016, 152, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Lynch-Day, M.A.; Mao, K.; Wang, K.; Zhao, M.; Klionsky, D.J. The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009357. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.I.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s Disease: Mechanisms and Therapeutic Implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Neilson, A.; Swift, A.L.; Moran, R.; Tamagnine, J.; Parslow, D.; Armistead, S.; Lemire, K.; Orrell, J.; Teich, J.; et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007, 292, C125–C136. [Google Scholar] [CrossRef] [PubMed]
- Pfleger, J.; He, M.; Abdellatif, M. Mitochondrial complex II is a source of the reserve respiratory capacity that is regulated by metabolic sensors and promotes cell survival. Cell Death Dis. 2015, 6, e1835. [Google Scholar] [CrossRef] [PubMed]
- Watson, D.C.; Bayik, D.; Storevik, S.; Moreino, S.S.; Sprowls, S.A.; Han, J.; Augustsson, M.T.; Lauko, A.; Sravya, P.; Røsland, G.V.; et al. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat. Cancer 2023, 4, 648–664. [Google Scholar] [CrossRef]
- Zhang, T.; Gillies, M.C.; Madigan, M.C.; Shen, W.; Du, J.; Grünert, U.; Zhou, F.; Yam, M.; Zhu, L. Disruption of De Novo Serine Synthesis in Müller Cells Induced Mitochondrial Dysfunction and Aggravated Oxidative Damage. Mol. Neurobiol. 2018, 55, 7025–7037. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.-E.; Saal, K.-A.; Tönges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacol. Ther. 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Pan, J.; Yin, Y.; Zhao, L.; Feng, Y. Discovery of (S)-6-methoxy-chroman-3-carboxylic acid (4-pyridin-4-yl-phenyl)-amide as potent and isoform selective ROCK2 inhibitors. Bioorg. Med. Chem. 2019, 27, 1382–1390. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Li, J.; Hu, C.; Zhang, L.; Yan, J.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside alleviated inflammatory damage by mitophagy via AMPK related PINK1/Parkin signaling pathway. Biochem. Pharmacol. 2020, 177, 113997. [Google Scholar] [CrossRef]
- Moskal, N.; Riccio, V.; Bashkurov, M.; Taddese, R.; Datti, A.; Lewis, P.N.; McQuibban, G.A. ROCK inhibitors upregulate the neuroprotective Parkin-mediated mitophagy pathway. Nat. Commun. 2020, 11, 88. [Google Scholar] [CrossRef]
- Herskowitz, J.H.; Feng, Y.; Mattheyses, A.L.; Hales, C.M.; Higginbotham, L.A.; Duong, D.M.; Montine, T.J.; Troncoso, J.C.; Thambisetty, M.; Seyfried, N.T.; et al. Pharmacologic inhibition of ROCK2 suppresses amyloid-β production in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 19086–19098. [Google Scholar] [CrossRef]
- Kang, B.H.; Xia, F.; Pop, R.; Dohi, T.; Socolovsky, M.; Altieri, D.C. Developmental control of apoptosis by the immunophilin aryl hydrocarbon receptor-interacting protein (AIP) involves mitochondrial import of the survivin protein. J. Biol. Chem. 2011, 286, 16758–16767. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Han, J.W.; Flemington, C.; Houghton, A.B.; Gu, Z.; Zambetti, G.P.; Lutz, R.J.; Zhu, L.; Chittenden, T. Expression of bbc3, a pro-apoptotic BH3-only gene, is regulated by diverse cell death and survival signals. Proc. Natl. Acad. Sci. USA 2001, 98, 11318–11323. [Google Scholar] [CrossRef]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116 Pt 13, 2763–2774. [Google Scholar] [CrossRef]
- Cipolat, S.; de Brito, O.M.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed]
- Varanita, T.; Soriano, M.E.; Romanello, V.; Zaglia, T.; Quintana-Cabrera, R.; Semenzato, M.; Menabò, R.; Costa, V.; Civiletto, G.; Pesce, P.; et al. The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015, 21, 834–844. [Google Scholar] [CrossRef]
- Ko, A.; Han, S.Y.; Song, J. Dynamics of ARF regulation that control senescence and cancer. BMB Rep. 2016, 49, 598–606. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, Q.; Tang, K.; Markby, G.R.; Parys, M.; Phadwal, K.; MacRae, V.E.; Corcoran, B.M. Autophagy Regulates Cellular Senescence by Mediating the Degradation of CDKN1A/P21 and CDKN2A/P16 through SQSTM1/P62-Mediated Selective Autophagy in Myxomatous Mitral Valve Degeneration. Autophagy 2025, 1–23. [Google Scholar] [CrossRef]
- Joseph, C.; Mangani, A.S.; Gupta, V.; Chitranshi, N.; Shen, T.; Dheer, Y.; Kb, D.; Mirzaei, M.; You, Y.; Graham, S.L.; et al. Cell Cycle Deficits in Neurodegenerative Disorders: Uncovering Molecular Mechanisms to Drive Innovative Therapeutic Development. Aging Dis. 2020, 11, 946–966. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- O’Gorman Tuura, R.L.; Baumann, C.R.; Baumann-Vogel, H. Beyond Dopamine: GABA, Glutamate, and the Axial Symptoms of Parkinson Disease. Front. Neurol. 2018, 9, 806. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, B.; Al-Kuraishy, H.M.; Al-Gareeb, A.I.; Elekhnawy, E.; Alharbi, H.; Alexiou, A.; Papadakis, M.; Batiha, G.E. Role of GABA pathway in motor and non-motor symptoms in Parkinson’s disease: A bidirectional circuit. Eur. J. Med. Res. 2024, 29, 205. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rahmani, Z.; Surabhi, S.; Rojo-Cortés, F.; Dulac, A.; Jenny, A.; Birman, S. Lamp1 Deficiency Enhances Sensitivity to α-Synuclein and Oxidative Stress in Drosophila Models of Parkinson Disease. Int. J. Mol. Sci. 2022, 23, 13078. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nagano, Y.; Taylor, J.P.; Lim, K.L.; Yao, T.-P. Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- van Horssen, J.; van Schaik, P.; Witte, M. Inflammation and mitochondrial dysfunction: A vicious circle in neurodegenerative disorders? Neurosci. Lett. 2019, 710, 132931. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Balestri, W.; Sharma, R.; da Silva, V.A.; Bobotis, B.C.; Curle, A.J.; Kothakota, V.; Kalantarnia, F.; Hangad, M.V.; Hoorfar, M.; Jones, J.L.; et al. Modeling the neuroimmune system in Alzheimer’s and Parkinson’s diseases. J. Neuroinflamm. 2024, 21, 32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Qin, X.Y.; Zhang, S.P.; Cao, C.; Loh, Y.P.; Cheng, Y. Aberrations in Peripheral Inflammatory Cytokine Levels in Parkinson Disease: A Systematic Review and Meta-analysis. JAMA Neurol. 2016, 73, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Santaella, A.; Kuiperij, H.B.; van Rumund, A.; Esselink, R.A.J.; van Gool, A.J.; Bloem, B.R.; Verbeek, M.M. Cerebrospinal fluid monocyte chemoattractant protein 1 correlates with progression of Parkinson’s disease. NPJ Park. Dis. 2020, 6, 21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kwon, E.H.; Tennagels, S.; Gold, R.; Gerwert, K.; Beyer, L.; Tönges, L. Update on CSF Biomarkers in Parkinson’s Disease. Biomolecules 2022, 12, 329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Griseta, C.; Battista, P.; Castellana, F.; Colonna, I.; Sciarra, S.; Zupo, R.; Bortone, I.; Lampignano, L.; Tirelli, S.; Bernardino, G.; et al. Serum levels of IL-6 are associated with cognitive impairment in the salus in apulia population-based study. Heliyon 2023, 9, e13972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yacoubian, T.A.; Fang, Y.D.; Gerstenecker, A.; Amara, A.; Stover, N.; Ruffrage, L.; Collette, C.; Kennedy, R.; Zhang, Y.; Hong, H.; et al. Brain and Systemic Inflammation in De Novo Parkinson’s Disease. Mov. Disord. 2023, 38, 743–754. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Muñoz-Delgado, L.; Labrador-Espinosa, M.Á.; Macías-García, D.; Jesús, S.; Benítez Zamora, B.; Fernández-Rodríguez, P.; Adarmes-Gómez, A.D.; Reina Castillo, M.I.; Castro-Labrador, S.; Silva-Rodríguez, J.; et al. Peripheral Inflammation Is Associated with Dopaminergic Degeneration in Parkinson’s Disease. Mov. Disord. 2023, 38, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Chong, L.; Li, X.; Liu, Y.; Liu, P.; Hou, C.; Li, R. Correlation between serum RANTES levels and the severity of Parkinson’s disease. Oxid. Med. Cell Longev. 2014, 2014, 208408. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Magnusen, A.F.; Hatton, S.L.; Rani, R.; Pandey, M.K. Genetic Defects and Pro-inflammatory Cytokines in Parkinson’s Disease. Front Neurol. 2021, 12, 636139. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Su, H.; Lei, C.T.; Zhang, C. Interleukin-6 Signaling Pathway and Its Role in Kidney Disease: An Update. Front. Immunol. 2017, 8, 405. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Santaella, A.; Kuiperij, H.B.; van Rumund, A.; Esselink, R.A.J.; van Gool, A.J.; Bloem, B.R.; Verbeek, M.M. Inflammation biomarker discovery in Parkinson’s disease and atypical parkinsonisms. BMC Neurol. 2020, 20, 26. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, Y.; Xu, K.; Xiang, Y.; Ma, B.; Li, H.; Li, Y.; Shi, Y.; Li, S.; Bai, Y. Role of MCP-1 as an inflammatory biomarker in nephropathy. Front. Immunol. 2024, 14, 1303076. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Mahajan, V.; Deygen, I.; Klyachko, N.L.; Inskoe, E.; Piroyan, A.; Sokolsky, M.; Okolie, O.; et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine 2016, 12, 655–664. [Google Scholar] [CrossRef]
- Kim, M.S.; Haney, M.J.; Zhao, Y.; Yuan, D.; Deygen, I.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: In vitro and in vivo evaluations. Nanomedicine 2018, 14, 195–204. [Google Scholar] [CrossRef]
- Haney, M.J.; Zhao, Y.; Jin, Y.S.; Li, S.M.; Bago, J.R.; Klyachko, N.L.; Kabanov, A.V.; Batrakova, E.V. Macrophage-Derived Extracellular Vesicles as Drug Delivery Systems for Triple Negative Breast Cancer (TNBC) Therapy. J. Neuroimmune Pharmacol. 2020, 15, 487–500. [Google Scholar] [CrossRef]
- Haney, M.J.; Suresh, P.; Zhao, Y.; Kanmogne, G.D.; Kadiu, I.; Sokolsky-Papkov, M.; Klyachko, N.L.; Mosley, R.L.; Kabanov, A.V.; Gendelman, H.E.; et al. Blood-borne macrophage-neural cell interactions hitchhike on endosome networks for cell-based nanozyme brain delivery. Nanomedicine 2012, 7, 815–833. [Google Scholar] [CrossRef]
- Yuan, D.; Zhao, Y.; Banks, W.A.; Bullock, K.M.; Haney, M.; Batrakova, E.; Kabanov, A.V. Macrophage exosomes as natural nanocarriers for protein delivery to inflamed brain. Biomaterials 2017, 142, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Klyachko, N.L.; Arzt, C.J.; Li, S.M.; Gololobova, O.A.; Batrakova, E.V. Extracellular Vesicle-Based Therapeutics: Preclinical and Clinical Investigations. Pharmaceutics 2020, 12, 1171. [Google Scholar] [CrossRef]
- Batrakova, E.V.; Kim, M.S. Using exosomes, naturally-equipped nanocarriers, for drug delivery. J. Control Release 2015, 219, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Harrison, E.; Zhao, Y.; Kabanov, A.; Batrakova, E.V. TPP1 Delivery to Lysosomes with Extracellular Vesicles and their Enhanced Brain Distribution in the Animal Model of Batten Disease. Adv. Healthc. Mater. 2019, 8, e1801271. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Y.; Zhou, J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl. Neurodegener. 2015, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.M.; Burgess, T.; Lee, J.; Blackburn, D.J.; Allen, S.P.; Mortiboys, H. Peripheral Glycolysis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 8924. [Google Scholar] [CrossRef] [PubMed]
- Cai, R.; Zhang, Y.; Simmering, J.E.; Schultz, J.L.; Li, Y.; Fernandez-Carasa, I.; Consiglio, A.; Raya, A.; Polgreen, P.M.; Narayanan, N.S.; et al. Enhancing glycolysis attenuates Parkinson’s disease progression in models and clinical databases. J. Clin. Investig. 2019, 129, 4539–4549. [Google Scholar] [CrossRef] [PubMed]
- El-Hage, N.; Podhaizer, E.M.; Sturgill, J.; Hauser, K.F. Toll-like receptor expression and activation in astroglia: Differential regulation by HIV-1 Tat, gp120, and morphine. Immunol. Investig. 2011, 40, 498–522. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbajal, C.; Rodriguez, M.; Owens, F.; Stone, N.; Veeragoni, D.; Fan, R.Z.; Tieu, K.; El-Hage, N. Therapeutic Efficacy of Small Extracellular Vesicles Loaded with ROCK Inhibitor in Parkinson’s Disease. Pharmaceutics 2025, 17, 365. https://doi.org/10.3390/pharmaceutics17030365
Carbajal C, Rodriguez M, Owens F, Stone N, Veeragoni D, Fan RZ, Tieu K, El-Hage N. Therapeutic Efficacy of Small Extracellular Vesicles Loaded with ROCK Inhibitor in Parkinson’s Disease. Pharmaceutics. 2025; 17(3):365. https://doi.org/10.3390/pharmaceutics17030365
Chicago/Turabian StyleCarbajal, Candy, Myosotys Rodriguez, Florida Owens, Nicole Stone, Dileepkumar Veeragoni, Rebecca Z. Fan, Kim Tieu, and Nazira El-Hage. 2025. "Therapeutic Efficacy of Small Extracellular Vesicles Loaded with ROCK Inhibitor in Parkinson’s Disease" Pharmaceutics 17, no. 3: 365. https://doi.org/10.3390/pharmaceutics17030365
APA StyleCarbajal, C., Rodriguez, M., Owens, F., Stone, N., Veeragoni, D., Fan, R. Z., Tieu, K., & El-Hage, N. (2025). Therapeutic Efficacy of Small Extracellular Vesicles Loaded with ROCK Inhibitor in Parkinson’s Disease. Pharmaceutics, 17(3), 365. https://doi.org/10.3390/pharmaceutics17030365