
Novel Therapeutics for Malaria

 ,
, 
Abstract
1. Malaria
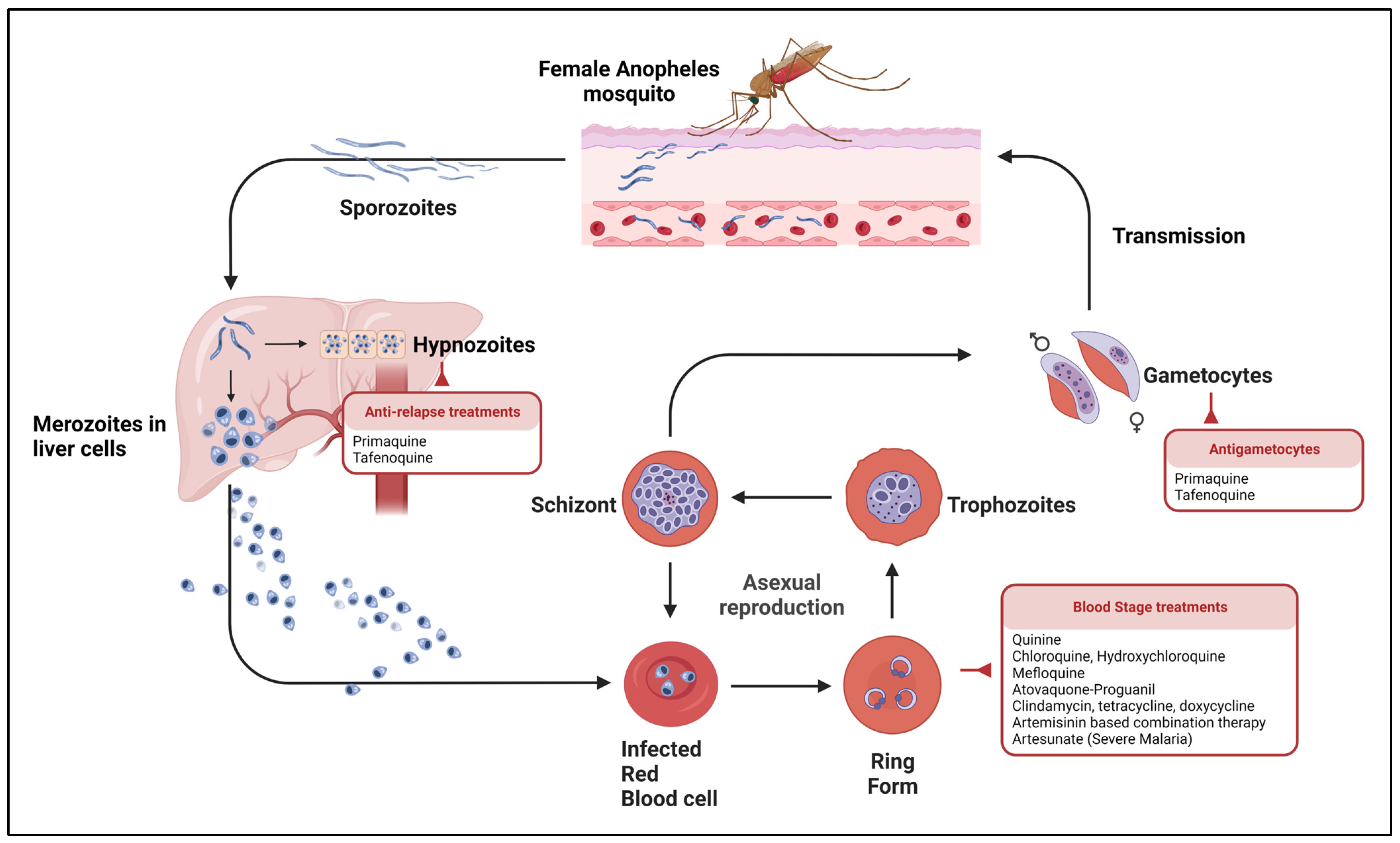
1.1. Parasite Life Cycle and Clinical Manifestations
1.2. Diagnostic Modalities
2. Antimalarial Drugs
2.1. Current Treatments

{kind=link}
| Class | Category | Drugs | Side Effects | Mechanism of Action | Ref. |
|---|---|---|---|---|---|
| Quinoline | Cinchona alkaloid | Quinine, quinidine | Hypoglycemia, cinchonism (tinnitus, hearing loss, visual disturbances, headache, dysphoria, nausea, vomiting), hypotension | Toxic accumulation of heme inside the infected red blood cell and inhibition of glycolytic enzymes | [14,26] |
| 4-AQ | Chloroquine | Pruritis, nausea, vomiting, diarrhea, keratopathy, retinopathy (with prolonged use) | [14,27,28] | ||
| Amodiaquine | Hepatitis, myelotoxicity, agranulocytosis | [29,30] | |||
| Piperaquine | Headache, dizziness, nausea, abdominal pain | [31] | |||
| 8-AQ | Primaquine, tafenoquine | Hemolytic anemia in G6PD deficiency | Inhibit pyrimidine synthesis and disrupt mitochondrial electron transport chain by producing oxidative metabolites | [32] | |
| Quinoline-menthol | Mefloquine | Dizziness, anorexia, vomiting | Toxic accumulation of heme inside the infected red blood cell | [33,34] | |
| Aryl amino-alcohol | Lumefantrine, halofantrine | Nausea, vomiting, diarrhea, pruritis, rash, cardiac toxicity (with halofantrine) | [35] | ||
| Benzonaphthyridine derivative | Pyronaridine | Headache, vomiting, abdominal pain, bradycardia, hypoglycemia | [36] | ||
| Naphthoquinones | Hydroxynaphthoquinone | Atovaquone | Nausea, vomiting, diarrhea, headache, fever, transient liver enzyme elevation | Inhibit the electron transport system at the cytochrome bc1 complex | [17] |
| Antifolate | Diaminopyrimidine | Pyrimethamine | Bone marrow suppression, glossitis, stomatitis, exfoliative dermatitis, hair loss | Interfere with DNA synthesis by inhibiting DHFR, parasite mitochondrial toxicity seen with proguanil | [20,37] |
| Biguanide | Proguanil | ||||
| Sulfonamides | Sulfadoxine | Fever, arthralgia, bone marrow suppression, SJS, hemolytic anemia in G6PD | Interfere with DNA synthesis by inhibiting DHPS enzyme | [38,39] | |
| Artemisinin | Sesquiterpene lactone endoperoxides | Artemether, artesunate, dihydroartemisinin | Nausea, vomiting, diarrhea | Protein alkylation and DNA damage by free radical generation | [21,40] |
| Trioxolanes | Trioxolane peroxide | Arterolane | Headache, nausea, vomiting | Inhibit heme detoxification and PfATP6 | [41] |
| Antimicrobials | Lincosamide | Clindamycin | Rash, DRESS, SJS, diarrhea, C. difficile | Interfere with protein translation | [42] |
| Tetracycline | Doxycycline, tetracycline | Nausea, vomiting, diarrhea, epigastric pain | [43] | ||
| Macrolide | Azithromycin | Nausea, vomiting, diarrhea | [44] |
2.2. Plasmodium Drug Resistance
2.3. Novel Therapies
2.3.1. Triple Combination Therapy
2.3.2. Phosphatidylinositol 4-Kinase (PI4K) Inhibitors
2.3.3. Plasmodium falciparum P-Type Na+ ATPase (PfATP4) Inhibitors
2.3.4. Dihydroorotate Dehydrogenase (DHODH) Inhibitors
2.3.5. Dihydrofolate Reductase (DHFR) and Thymidylate Synthase (TS) Inhibitors
2.3.6. Isoprenoid Biosynthesis Inhibitors
2.3.7. Choline Transport Inhibitors
2.3.8. P. falciparum Translational Elongation Factor 2 Inhibitors
2.3.9. Triaminopyrimidine
2.3.10. Adhesive Polysaccharide Inhibitors
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Conroy, A.L.; Datta, D.; John, C.C. What causes severe malaria and its complications in children? Lessons learned over the past 15 years. BMC Med. 2019, 17, 52. [Google Scholar] [CrossRef] [PubMed]
- Bykersma, A. The New Zoonotic Malaria: Plasmodium cynomolgi. Trop. Med. Infect. Dis. 2021, 6, 46. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report; World Health Organization: Geneva, Switzerland, 2022. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022 (accessed on 6 February 2023).
- Anand, A.; Mace, K.E.; Townsend, R.L.; Madison-Antenucci, S.; Grimm, K.E.; Espina, N.; Losco, P.; Lucchi, N.W.; Rivera, H.; Breen, K.; et al. Investigation of a case of suspected transfusion-transmitted malaria. Transfusion 2018, 58, 2115–2121. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, L.A.; Price, M.; Noland, D.; Fuchs, J.; Filkins, L.; McElvania, E.; Luu, H.S.; Sebert, M.; Waters, A.; Hsiang, M.S. Transfusion-Transmitted Malaria: Two Pediatric Cases From the United States and Their Relevance in an Increasingly Globalized World. J. Pediatr. Infect. Dis. Soc. 2021, 10, 1092–1095. [Google Scholar] [CrossRef]
- World Health Organization. WHO Guidelines for Malaria; World Health Organization: Geneva, Switzerland, 2022; p. 426.
- Goldman-Yassen, A.E.; Mony, V.K.; Arguin, P.M.; Daily, J.P. Higher Rates of Misdiagnosis in Pediatric Patients Versus Adults Hospitalized With Imported Malaria. Pediatr. Emerg. Care 2016, 32, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Mace, K.E.; Lucchi, N.W.; Tan, K.R. Malaria Surveillance—United States, 2017. MMWR Surveill. Summ. 2021, 70, 1. [Google Scholar] [CrossRef]
- Pritt, B.S. Plasmodium and Babesia. In Manual of Clinical Microbiology; Jorgensen, J.H., Carroll, K.C., Funke, G., Pfaller, M.A., Landry, M.L., Richter, S.S., Warnock, D.W., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; Volume 2, pp. 2338–2356. ISBN 978-1-68367-280-7. [Google Scholar]
- Mathison, B.A.; Pritt, B.S. Update on Malaria Diagnostics and Test Utilization. J. Clin. Microbiol. 2017, 55, 2009–2017. [Google Scholar] [CrossRef]
- M15AE: Lab Diagnosis of Blood-Borne Parasitic Diseases. Available online: https://clsi.org/standards/products/microbiology/documents/m15/ (accessed on 18 December 2022).
- CDC–DPDx–Malaria. Available online: https://www.cdc.gov/dpdx/malaria/index.html (accessed on 18 December 2022).
- Gachelin, G.; Garner, P.; Ferroni, E.; Tröhler, U.; Chalmers, I. Evaluating Cinchona bark and quinine for treating and preventing malaria. J. R. Soc. Med. 2017, 110, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.; Tilley, L. Quinoline antimalarials: Mechanisms of action and resistance. Int. J. Parasitol. 1997, 27, 231–240. [Google Scholar] [CrossRef]
- Ippolito, M.M.; Moser, K.A.; Kabuya, J.-B.B.; Cunningham, C.; Juliano, J.J. Antimalarial Drug Resistance and Implications for the WHO Global Technical Strategy. Curr. Epidemiol. Rep. 2021, 8, 46–62. [Google Scholar] [CrossRef]
- Pryce, J.; Hine, P. Pyronaridine-artesunate for treating uncomplicated Plasmodium falciparum malaria. Cochrane Database Syst. Rev. 2019, 2019, CD006404. [Google Scholar] [CrossRef] [PubMed]
- Fry, M.; Pudney, M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4’-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 1992, 43, 1545–1553. [Google Scholar] [CrossRef]
- Goodman, C.D.; Buchanan, H.D.; McFadden, G.I. Is the Mitochondrion a Good Malaria Drug Target? Trends Parasitol. 2017, 33, 185–193. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Ranasinha, C.D.; Kumar, N.; Barditch-Crovo, P. Prophylactic activity of atovaquone against Plasmodium falciparum in humans. Am. J. Trop. Med. Hyg. 1999, 60, 831–836. [Google Scholar] [CrossRef]
- Chulay, J.D.; Watkins, W.M.; Sixsmith, D.G. Synergistic antimalarial activity of pyrimethamine and sulfadoxine against Plasmodium falciparum in vitro. Am. J. Trop. Med. Hyg. 1984, 33, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, A.M.; Kumar, N. Antimalarial action of artesunate involves DNA damage mediated by reactive oxygen species. Antimicrob. Agents Chemother. 2015, 59, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Bridgford, J.L.; Xie, S.C.; Cobbold, S.A.; Pasaje, C.F.A.; Herrmann, S.; Yang, T.; Gillett, D.L.; Dick, L.R.; Ralph, S.A.; Dogovski, C.; et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 2018, 9, 3801. [Google Scholar] [CrossRef]
- Kumar, N.; Zheng, H. Stage-specific gametocytocidal effect in vitro of the antimalaria drug qinghaosu onPlasmodium falciparum. Parasitol. Res. 1990, 76, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Aderibigbe, B.A. Design of Drug Delivery Systems Containing Artemisinin and Its Derivatives. Molecules 2017, 22, 323. [Google Scholar] [CrossRef]
- Liu, C. Discovery and Development of Artemisinin and Related Compounds. Chin. Herb. Med. 2017, 9, 101–114. [Google Scholar] [CrossRef]
- Achan, J.; Talisuna, A.O.; Erhart, A.; Yeka, A.; Tibenderana, J.K.; Baliraine, F.N.; Rosenthal, P.J.; D’Alessandro, U. Quinine, an old anti-malarial drug in a modern world: Role in the treatment of malaria. Malar. J. 2011, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Melles, R.B.; Marmor, M.F. Pericentral retinopathy and racial differences in hydroxychloroquine toxicity. Ophthalmology 2015, 122, 110–116. [Google Scholar] [CrossRef]
- Onyeji, C.O.; Ogunbona, F.A. Pharmacokinetic aspects of chloroquine-induced pruritus: Influence of dose and evidence for varied extent of metabolism of the drug. Eur. J. Pharm. Sci. Off. J. Eur. Fed. Pharm. Sci. 2001, 13, 195–201. [Google Scholar] [CrossRef]
- Hatton, C.S.R.; Bunch, C.; Peto, T.E.A.; Pasvol, G.; Russell, S.J.; Singer, C.R.J.; Edwards, G.; Winstanley, P. Frequency of severe neutropenia associated with amodiaquine prophylaxis against malaria. The Lancet 1986, 327, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Neftel, K.A.; Woodtly, W.; Schmid, M.; Frick, P.G.; Fehr, J. Amodiaquine induced agranulocytosis and liver damage. Br. Med. J. Clin. Res. Ed. 1986, 292, 721–723. [Google Scholar] [CrossRef]
- Tarning, J.; Zongo, I.; Somé, F.; Rouamba, N.; Parikh, S.; Rosenthal, P.; Hanpithakpong, W.; Jongrak, N.; Day, N.; White, N.; et al. Population Pharmacokinetics and Pharmacodynamics of Piperaquine in Children With Uncomplicated Falciparum Malaria. Clin. Pharmacol. Ther. 2012, 91, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Beutler, E. The hemolytic effect of primaquine and related compounds: A review. Blood 1959, 14, 103–139. [Google Scholar] [CrossRef]
- World Health Organization. Mefloquine: An update on safety issues: Reports on individual drugs. WHO Drug Inf. 1996, 102, 58–61. [Google Scholar]
- Zhang, J.; Krugliak, M.; Ginsburg, H. The fate of ferriprotorphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol. Biochem. Parasitol. 1999, 99, 129–141. [Google Scholar] [CrossRef]
- Bakshi, R.; Hermeling-Fritz, I.; Gathmann, I.; Alteri, E. An integrated assessment of the clinical safety of artemether-lumefantrine: A new oral fixed-dose combination antimalarial drug. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 419–424. [Google Scholar] [CrossRef]
- Rueangweerayut, R.; Phyo, A.P.; Uthaisin, C.; Poravuth, Y.; Binh, T.Q.; Tinto, H.; Pénali, L.K.; Valecha, N.; Tien, N.T.; Abdulla, S.; et al. Pyronaridine–Artesunate versus Mefloquine plus Artesunate for Malaria. N. Engl. J. Med. 2012, 366, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Friman, G.; Nyström-Rosander, C.; Jonsell, G.; Björkman, A.; Lekås, G.; Svendsrup, B. Agranulocytosis associated with malaria prophylaxis with Maloprim. Br. Med. J. Clin. Res. Ed. 1983, 286, 1244–1245. [Google Scholar] [CrossRef] [PubMed]
- Wolkenstein, P.; Charue, D.; Laurent, P.; Revuz, J.; Roujeau, J.C.; Bagot, M. Metabolic predisposition to cutaneous adverse drug reactions. Role in toxic epidermal necrolysis caused by sulfonamides and anticonvulsants. Arch. Dermatol. 1995, 131, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Price, E.J.; Venables, P.J. Drug-induced lupus. Drug Saf. 1995, 12, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Price, R.; van Vugt, M.; Phaipun, L.; Luxemburger, C.; Simpson, J.; McGready, R.; ter Kuile, F.; Kham, A.; Chongsuphajaisiddhi, T.; White, N.J.; et al. Adverse effects in patients with acute falciparum malaria treated with artemisinin derivatives. Am. J. Trop. Med. Hyg. 1999, 60, 547–555. [Google Scholar] [CrossRef]
- Patil, C.; Katare, S.; Baig, M.; Doifode, S. Fixed Dose Combination of Arterolane and Piperaquine: A Newer Prospect in Antimalarial Therapy. Ann. Med. Health Sci. Res. 2014, 4, 466–471. [Google Scholar] [CrossRef]
- Yang, Y.; Chen, S.; Yang, F.; Zhang, L.; Alterovitz, G.; Zhu, H.; Xuan, J.; Yang, X.; Luo, H.; Mu, J.; et al. HLA-B*51:01 is strongly associated with clindamycin-related cutaneous adverse drug reactions. Pharm. J. 2017, 17, 501–505. [Google Scholar] [CrossRef]
- Dahl, E.L.; Shock, J.L.; Shenai, B.R.; Gut, J.; DeRisi, J.L.; Rosenthal, P.J. Tetracyclines specifically target the apicoplast of the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 2006, 50, 3124–3131. [Google Scholar] [CrossRef]
- Andersen, S.L.; Ager, A.L.; McGreevy, P.; Schuster, B.G.; Ellis, W.; Berman, J. Efficacy of azithromycin as a causal prophylactic agent against murine malaria. Antimicrob. Agents Chemother. 1994, 38, 1862–1863. [Google Scholar] [CrossRef]
- WHO Scientific Group on Chemotherapy of Malaria; World Health Organization. Chemotherapy of Malaria: Report of a WHO Scientific Group [Meeting Held in Geneva from 25 April to 1 May 1967]; World Health Organization: Geneva, Switzerland, 1967.
- Report on Antimalarial Drug Efficacy, Resistance and Response: 10 Years of Surveillance (2010–2019). Available online: https://www.who.int/publications-detail-redirect/9789240012813 (accessed on 7 March 2023).
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of Artemisinin Resistance in Plasmodium falciparum Malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Smithuis, F.M.; Woodrow, C.; Von Seidlein, L. How to Contain Artemisinin- and Multidrug-Resistant Falciparum Malaria. Trends Parasitol. 2017, 33, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of Artemisinin-Resistant Malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar] [CrossRef]
- Dogovski, C.; Xie, S.C.; Burgio, G.; Bridgford, J.; Mok, S.; McCaw, J.M.; Chotivanich, K.; Kenny, S.; Gnädig, N.; Straimer, J.; et al. Targeting the Cell Stress Response of Plasmodium falciparum to Overcome Artemisinin Resistance. PLoS Biol. 2015, 13, e1002132. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Diagana, T.T. Supporting malaria elimination with 21st century antimalarial agent drug discovery. Drug Discov. Today 2015, 20, 1265–1270. [Google Scholar] [CrossRef]
- Sahu, N.K.; Sahu, S.; Kohli, D.V. Novel Molecular Targets for Antimalarial Drug Development. Chem. Biol. Drug Des. 2008, 71, 287–297. [Google Scholar] [CrossRef]
- Cortopassi, W.A.; Celmar Costa Franca, T.; Krettli, A.U. A systems biology approach to antimalarial drug discovery. Expert Opin. Drug Discov. 2018, 13, 617–626. [Google Scholar] [CrossRef]
- Mathews, E.S.; Odom John, A.R. Tackling resistance: Emerging antimalarials and new parasite targets in the era of elimination. F1000Research 2018, 7, F1000 Faculty Rev-1170. [Google Scholar] [CrossRef]
- Roy, K.K. Targeting the active sites of malarial proteases for antimalarial drug discovery: Approaches, progress and challenges. Int. J. Antimicrob. Agents 2017, 50, 287–302. [Google Scholar] [CrossRef]
- Verma, S.; Dixit, R.; Pandey, K.C. Cysteine Proteases: Modes of Activation and Future Prospects as Pharmacological Targets. Front. Pharmacol. 2016, 7, 107. [Google Scholar] [CrossRef]
- Desai, S.A. Targeting ion channels of Plasmodium falciparum-infected human erythrocytes for antimalarial development. Curr. Drug Targets Infect. Disord. 2004, 4, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Dickerman, B.K.; Elsworth, B.; Cobbold, S.A.; Nie, C.Q.; McConville, M.J.; Crabb, B.S.; Gilson, P.R. Identification of inhibitors that dually target the new permeability pathway and dihydroorotate dehydrogenase in the blood stage of Plasmodium falciparum. Sci. Rep. 2016, 6, 37502. [Google Scholar] [CrossRef] [PubMed]
- Heitmeier, M.R.; Hresko, R.C.; Edwards, R.L.; Prinsen, M.J.; Ilagan, M.X.G.; Odom John, A.R.; Hruz, P.W. Identification of druggable small molecule antagonists of the Plasmodium falciparum hexose transporter PfHT and assessment of ligand access to the glucose permeation pathway via FLAG-mediated protein engineering. PLoS ONE 2019, 14, e0216457. [Google Scholar] [CrossRef] [PubMed]
- Rosling, J.E.O.; Ridgway, M.C.; Summers, R.L.; Kirk, K.; Lehane, A.M. Biochemical characterization and chemical inhibition of PfATP4-associated Na+-ATPase activity in Plasmodium falciparum membranes. J. Biol. Chem. 2018, 293, 13327–13337. [Google Scholar] [CrossRef]
- Walloch, P.; Hansen, C.; Priegann, T.; Schade, D.; Beitz, E. Pentafluoro-3-hydroxy-pent-2-en-1-ones Potently Inhibit FNT-Type Lactate Transporters from all Five Human-Pathogenic Plasmodium Species. Chemmedchem 2021, 16, 1283–1289. [Google Scholar] [CrossRef]
- Yadav, B.S.; Chaturvedi, N.; Marina, N. Recent Advances in System Based Study for Anti-Malarial Drug Development Process. Curr. Pharm. Des. 2019, 25, 3367–3377. [Google Scholar] [CrossRef]
- Bietz, S.; Montilla, I.; Külzer, S.; Przyborski, J.M.; Lingelbach, K. Recruitment of human aquaporin 3 to internal membranes in the Plasmodium falciparum infected erythrocyte. Mol. Biochem. Parasitol. 2009, 167, 48–53. [Google Scholar] [CrossRef]
- Posfai, D.; Sylvester, K.; Reddy, A.; Ganley, J.G.; Wirth, J.; Cullen, Q.E.; Dave, T.; Kato, N.; Dave, S.S.; Derbyshire, E.R. Plasmodium parasite exploits host aquaporin-3 during liver stage malaria infection. PLoS Pathog. 2018, 14, e1007057. [Google Scholar] [CrossRef]
- Wiesner, J.; Kettler, K.; Sakowski, J.; Ortmann, R.; Katzin, A.M.; Kimura, E.A.; Silber, K.; Klebe, G.; Jomaa, H.; Schlitzer, M. Farnesyltransferase inhibitors inhibit the growth of malaria parasites in vitro and in vivo. Angew. Chem. Int. Ed. Engl. 2004, 43, 251–254. [Google Scholar] [CrossRef]
- Ha, Y.R.; Hwang, B.-G.; Hong, Y.; Yang, H.-W.; Lee, S.J. Effect of Farnesyltransferase Inhibitor R115777 on Mitochondria of Plasmodium falciparum. Korean J. Parasitol. 2015, 53, 421–430. [Google Scholar] [CrossRef]
- Tse, E.G.; Korsik, M.; Todd, M.H. The past, present and future of anti-malarial medicines. Malar. J. 2019, 18, 93. [Google Scholar] [CrossRef] [PubMed]
- Worthington, R.J.; Melander, C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31, 177–184. [Google Scholar] [CrossRef] [PubMed]
- van der Pluijm, R.W.; Tripura, R.; Hoglund, R.M.; Pyae Phyo, A.; Lek, D.; Ul Islam, A.; Anvikar, A.R.; Satpathi, P.; Satpathi, S.; Behera, P.K.; et al. Triple artemisinin-based combination therapies versus artemisinin-based combination therapies for uncomplicated Plasmodium falciparum malaria: A multicentre, open-label, randomised clinical trial. Lancet 2020, 395, 1345–1360. [Google Scholar] [CrossRef]
- Chien, H.D.; Pantaleo, A.; Kesely, K.R.; Noomuna, P.; Putt, K.S.; Tuan, T.A.; Low, P.S.; Turrini, F.M. Imatinib augments standard malaria combination therapy without added toxicity. J. Exp. Med. 2021, 218, e20210724. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, A.; Kesely, K.R.; Pau, M.C.; Tsamesidis, I.; Schwarzer, E.; Skorokhod, O.A.; Chien, H.D.; Ponzi, M.; Bertuccini, L.; Low, P.S.; et al. Syk inhibitors interfere with erythrocyte membrane modification during P falciparum growth and suppress parasite egress. Blood 2017, 130, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Kandepedu, N.; Gonzàlez Cabrera, D.; Eedubilli, S.; Taylor, D.; Brunschwig, C.; Gibhard, L.; Njoroge, M.; Lawrence, N.; Paquet, T.; Eyermann, C.J.; et al. Identification, Characterization, and Optimization of 2,8-Disubstituted-1,5-naphthyridines as Novel Plasmodium falciparum Phosphatidylinositol-4-kinase Inhibitors with in Vivo Efficacy in a Humanized Mouse Model of Malaria. J. Med. Chem. 2018, 61, 5692–5703. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.W.; Lee, M.C.S.; Lim, C.S.; Lim, S.H.; Roland, J.; Nagle, A.; Simon, O.; Yeung, B.K.S.; Chatterjee, A.K.; McCormack, S.L.; et al. Targeting Plasmodium PI(4)K to eliminate malaria. Nature 2013, 504, 248–253. [Google Scholar] [CrossRef]
- McCarthy, J.S.; Donini, C.; Chalon, S.; Woodford, J.; Marquart, L.; Collins, K.A.; Rozenberg, F.D.; Fidock, D.A.; Cherkaoui-Rbati, M.H.; Gobeau, N.; et al. A Phase 1, Placebo-controlled, Randomized, Single Ascending Dose Study and a Volunteer Infection Study to Characterize the Safety, Pharmacokinetics, and Antimalarial Activity of the Plasmodium Phosphatidylinositol 4-Kinase Inhibitor MMV390048. Clin. Infect. Dis. 2020, 71, e657–e664. [Google Scholar] [CrossRef]
- White, N.J.; Duong, T.T.; Uthaisin, C.; Nosten, F.; Phyo, A.P.; Hanboonkunupakarn, B.; Pukrittayakamee, S.; Jittamala, P.; Chuthasmit, K.; Cheung, M.S.; et al. Antimalarial Activity of KAF156 in Falciparum and Vivax Malaria. N. Engl. J. Med. 2016, 375, 1152–1160. [Google Scholar] [CrossRef]
- Jiménez-Díaz, M.B.; Ebert, D.; Salinas, Y.; Pradhan, A.; Lehane, A.M.; Myrand-Lapierre, M.-E.; O’Loughlin, K.G.; Shackleford, D.M.; Justino de Almeida, M.; Carrillo, A.K.; et al. (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. USA 2014, 111, E5455–E5462. [Google Scholar] [CrossRef]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- van Pelt-Koops, J.C.; Pett, H.E.; Graumans, W.; van der Vegte-Bolmer, M.; van Gemert, G.J.; Rottmann, M.; Yeung, B.K.S.; Diagana, T.T.; Sauerwein, R.W. The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to anopheles mosquito vector. Antimicrob. Agents Chemother. 2012, 56, 3544–3548. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, E.K.; Ndayisaba, G.; Yeka, A.; Asante, K.P.; Grobusch, M.P.; Karita, E.; Mugerwa, H.; Asiimwe, S.; Oduro, A.; Fofana, B.; et al. Efficacy of Cipargamin (KAE609) in a Randomized, Phase II Dose-Escalation Study in Adults in Sub-Saharan Africa With Uncomplicated Plasmodium falciparum Malaria. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2022, 74, 1831–1839. [Google Scholar] [CrossRef]
- Ndayisaba, G.; Yeka, A.; Asante, K.P.; Grobusch, M.P.; Karita, E.; Mugerwa, H.; Asiimwe, S.; Oduro, A.; Fofana, B.; Doumbia, S.; et al. Hepatic safety and tolerability of cipargamin (KAE609), in adult patients with Plasmodium falciparum malaria: A randomized, phase II, controlled, dose-escalation trial in sub-Saharan Africa. Malar. J. 2021, 20, 478. [Google Scholar] [CrossRef] [PubMed]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; Hooft van Huijsduijnen, R.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 26. [Google Scholar] [CrossRef]
- Gaur, A.H.; McCarthy, J.S.; Panetta, J.C.; Dallas, R.H.; Woodford, J.; Tang, L.; Smith, A.M.; Stewart, T.B.; Branum, K.C.; Freeman, B.B.; et al. Safety, tolerability, pharmacokinetics, and antimalarial efficacy of a novel Plasmodium falciparum ATP4 inhibitor SJ733: A first-in-human and induced blood-stage malaria phase 1a/b trial. Lancet Infect. Dis. 2020, 20, 964–975. [Google Scholar] [CrossRef]
- Xu, M.; Zhu, J.; Diao, Y.; Zhou, H.; Ren, X.; Sun, D.; Huang, J.; Han, D.; Zhao, Z.; Zhu, L.; et al. Novel Selective and Potent Inhibitors of Malaria Parasite Dihydroorotate Dehydrogenase: Discovery and Optimization of Dihydrothiophenone Derivatives. J. Med. Chem. 2013, 56, 7911–7924. [Google Scholar] [CrossRef]
- Llanos-Cuentas, A.; Casapia, M.; Chuquiyauri, R.; Hinojosa, J.-C.; Kerr, N.; Rosario, M.; Toovey, S.; Arch, R.H.; Phillips, M.A.; Rozenberg, F.D.; et al. Antimalarial activity of single-dose DSM265, a novel plasmodium dihydroorotate dehydrogenase inhibitor, in patients with uncomplicated Plasmodium falciparum or Plasmodium vivax malaria infection: A proof-of-concept, open-label, phase 2a study. Lancet Infect. Dis. 2018, 18, 874–883. [Google Scholar] [CrossRef]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef]
- Chughlay, M.F.; Gaaloul, M.E.; Donini, C.; Campo, B.; Berghmans, P.-J.; Lucardie, A.; Marx, M.W.; Cherkaoui-Rbati, M.H.; Langdon, G.; Angulo-Barturen, I.; et al. Chemoprotective Antimalarial Activity of P218 against Plasmodium falciparum: A Randomized, Placebo-Controlled Volunteer Infection Study. Am. J. Trop. Med. Hyg. 2021, 104, 1348. [Google Scholar] [CrossRef]
- Goble, J.L.; Adendorff, M.R.; de Beer, T.A.P.; Stephens, L.L.; Blatch, G.L. The malarial drug target Plasmodium falciparum 1-deoxy-D-xylulose-5-phosphate reductoisomerase (PfDXR): Development of a 3-D model for identification of novel, structural and functional features and for inhibitor screening. Protein Pept. Lett. 2010, 17, 109–120. [Google Scholar] [CrossRef]
- Fernandes, J.F.; Lell, B.; Agnandji, S.T.; Obiang, R.M.; Bassat, Q.; Kremsner, P.G.; Mordmüller, B.; Grobusch, M.P. Fosmidomycin as an antimalarial drug: A meta-analysis of clinical trials. Future Microbiol. 2015, 10, 1375–1390. [Google Scholar] [CrossRef]
- Penarete-Vargas, D.M.; Boisson, A.; Urbach, S.; Chantelauze, H.; Peyrottes, S.; Fraisse, L.; Vial, H.J. A Chemical Proteomics Approach for the Search of Pharmacological Targets of the Antimalarial Clinical Candidate Albitiazolium in Plasmodium falciparum Using Photocrosslinking and Click Chemistry. PLoS ONE 2014, 9, e113918. [Google Scholar] [CrossRef] [PubMed]
- Wengelnik, K.; Vidal, V.; Ancelin, M.L.; Cathiard, A.-M.; Morgat, J.L.; Kocken, C.H.; Calas, M.; Herrera, S.; Thomas, A.W.; Vial, H.J. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science 2002, 295, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.A.; Richier, E.; Bray, P.G.; Calas, M.; Vial, H.; Ward, S.A. Heme binding contributes to antimalarial activity of bis-quaternary ammoniums. Antimicrob. Agents Chemother. 2003, 47, 2584–2589. [Google Scholar] [CrossRef]
- Held, J.; Supan, C.; Salazar, C.L.O.; Tinto, H.; Bonkian, L.N.; Nahum, A.; Sié, A.; Abdulla, S.; Cantalloube, C.; Djeriou, E.; et al. Safety and efficacy of the choline analogue SAR97276 for malaria treatment: Results of two phase 2, open-label, multicenter trials in African patients. Malar. J. 2017, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, R.; Merrill, A.R.; Andersen, G.R. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 2006, 34, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Baragaña, B.; Hallyburton, I.; Lee, M.C.S.; Norcross, N.R.; Grimaldi, R.; Otto, T.D.; Proto, W.R.; Blagborough, A.M.; Meister, S.; Wirjanata, G.; et al. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature 2015, 522, 315–320. [Google Scholar] [CrossRef]
- McCarthy, J.S.; Yalkinoglu, Ö.; Odedra, A.; Webster, R.; Oeuvray, C.; Tappert, A.; Bezuidenhout, D.; Giddins, M.J.; Dhingra, S.K.; Fidock, D.A.; et al. Safety, pharmacokinetics, and antimalarial activity of the novel plasmodium eukaryotic translation elongation factor 2 inhibitor M5717: A first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study and volunteer infection study. Lancet Infect. Dis. 2021, 21, 1713–1724. [Google Scholar] [CrossRef]
- Barber, B.E.; Fernandez, M.; Patel, H.B.; Barcelo, C.; Woolley, S.D.; Patel, H.; Llewellyn, S.; Abd-Rahman, A.N.; Sharma, S.; Jain, M.; et al. Safety, pharmacokinetics, and antimalarial activity of the novel triaminopyrimidine ZY-19489: A first-in-human, randomised, placebo-controlled, double-blind, single ascending dose study, pilot food-effect study, and volunteer infection study. Lancet Infect. Dis. 2022, 22, 879–890. [Google Scholar] [CrossRef]
- Armistead, J.S.; Wilson, I.B.H.; van Kuppevelt, T.H.; Dinglasan, R.R. A role for heparan sulfate proteoglycans in Plasmodium falciparum sporozoite invasion of anopheline mosquito salivary glands. Biochem. J. 2011, 438, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, A.M.; Charunwatthana, P.; Rueangveerayut, R.; Uthaisin, C.; Silamut, K.; Chotivanich, K.; Sila, P.; Moll, K.; Lee, S.J.; Lindgren, M.; et al. Inhibition of merozoite invasion and transient de-sequestration by sevuparin in humans with Plasmodium falciparum malaria. PLoS ONE 2017, 12, e0188754. [Google Scholar] [CrossRef] [PubMed]

| Species | Signs and Symptoms | Laboratory Findings |
|---|---|---|
| P. falciparum | Prostration | Acidosis (lactate > 5 mmol/L or plasma bicarbonate < 15 mEq/L or base deficit of >8 mEq/L) |
| Convulsions (>2 within 24 h) | Anemia: Hgb < 7 g/dL or hematocrit < 20% in adults (≤5 g/dL or hematocrit ≤ 15% in children < 12 years of age) with a parasite count > 10,000/microliter | |
| Coma (Glasgow coma scale < 11) | Hypoglycemia < 40 mg/dL | |
| Abnormal bleeding | Jaundice or bilirubin > 3 mg/dl and parasite count >100,000/microliter | |
| Circulatory shock | Creatinine > 3 mg/dL or blood urea nitrogen > 56 mg/dL) | |
| Pulmonary edema, radiologically confirmed or oxygen saturation < 92% on room air with a respiratory rate > 30/minutes | Hyperparasitemia: P. falciparum parasitemia > 10% | |
| P. vivax, P. ovale, and P. malariae | Defined as per P. falciparum but excluding parasite density thresholds | |
| P. knowlesi | Defined as per P. falciparum except parasite density of >100,000/microliter or jaundice and parasite density > 20,000/microliter | |
| Target | Function | Significance | Drugs in Development | Reference |
|---|---|---|---|---|
| Malaria Proteases | Degradation of hemoglobin and proteins and aid in cell penetration | Development of the parasite, immune evasion, and activation of inflammation | Not available | [56,57] |
| Malaria transporters (PSAC, PVM, PfHT, and lactate transporters) | Diffusion of nutrients to infected erythrocyte | Promote growth of the parasite | Preclinical stage (Pentafluoro-3-hydroxy-pent-2-en-1-ones) | [58,59,60,61,62] |
| V-Type H+ ATPase Channels | Regulate hydrogen ion transportation | Maintain pH for survival of the parasite | Not available | [63] |
| Aquaporin-3 (AQP3) | Facilitate water and glycerol movement in and out of cells | Parasite survival and replication | Pre-clinical stage (auphen) | [64,65] |
| Farnesyltransferase | Catalyzing the transfer of farnesyl group from farnesyl pyrophosphate to the C-terminus of proteins containing the CaaX motif | DNA replication, cell division, binding of intracellular proteins to membranes, and protein to protein interaction | Pre-clinical stage (R115777) | [65,66,67] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alaithan, H.; Kumar, N.; Islam, M.Z.; Liappis, A.P.; Nava, V.E. Novel Therapeutics for Malaria. Pharmaceutics 2023, 15, 1800. https://doi.org/10.3390/pharmaceutics15071800
Alaithan H, Kumar N, Islam MZ, Liappis AP, Nava VE. Novel Therapeutics for Malaria. Pharmaceutics. 2023; 15(7):1800. https://doi.org/10.3390/pharmaceutics15071800
Chicago/Turabian StyleAlaithan, Haitham, Nirbhay Kumar, Mohammad Z. Islam, Angelike P. Liappis, and Victor E. Nava. 2023. "Novel Therapeutics for Malaria" Pharmaceutics 15, no. 7: 1800. https://doi.org/10.3390/pharmaceutics15071800
APA StyleAlaithan, H., Kumar, N., Islam, M. Z., Liappis, A. P., & Nava, V. E. (2023). Novel Therapeutics for Malaria. Pharmaceutics, 15(7), 1800. https://doi.org/10.3390/pharmaceutics15071800