
BRCT Domains: Structure, Functions, and Implications in Disease—New Therapeutic Targets for Innovative Drug Discovery against Infections



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
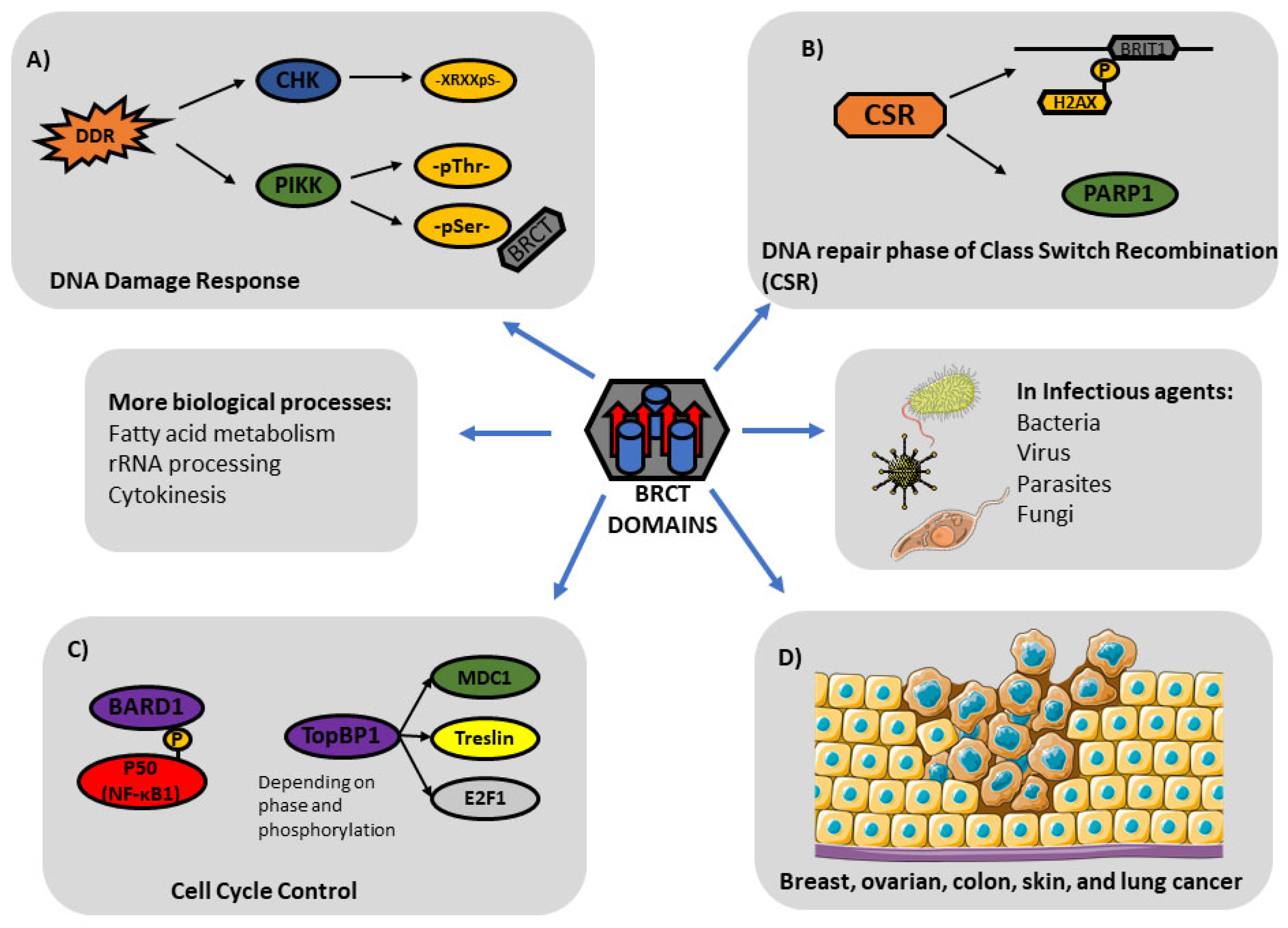
1. Introduction
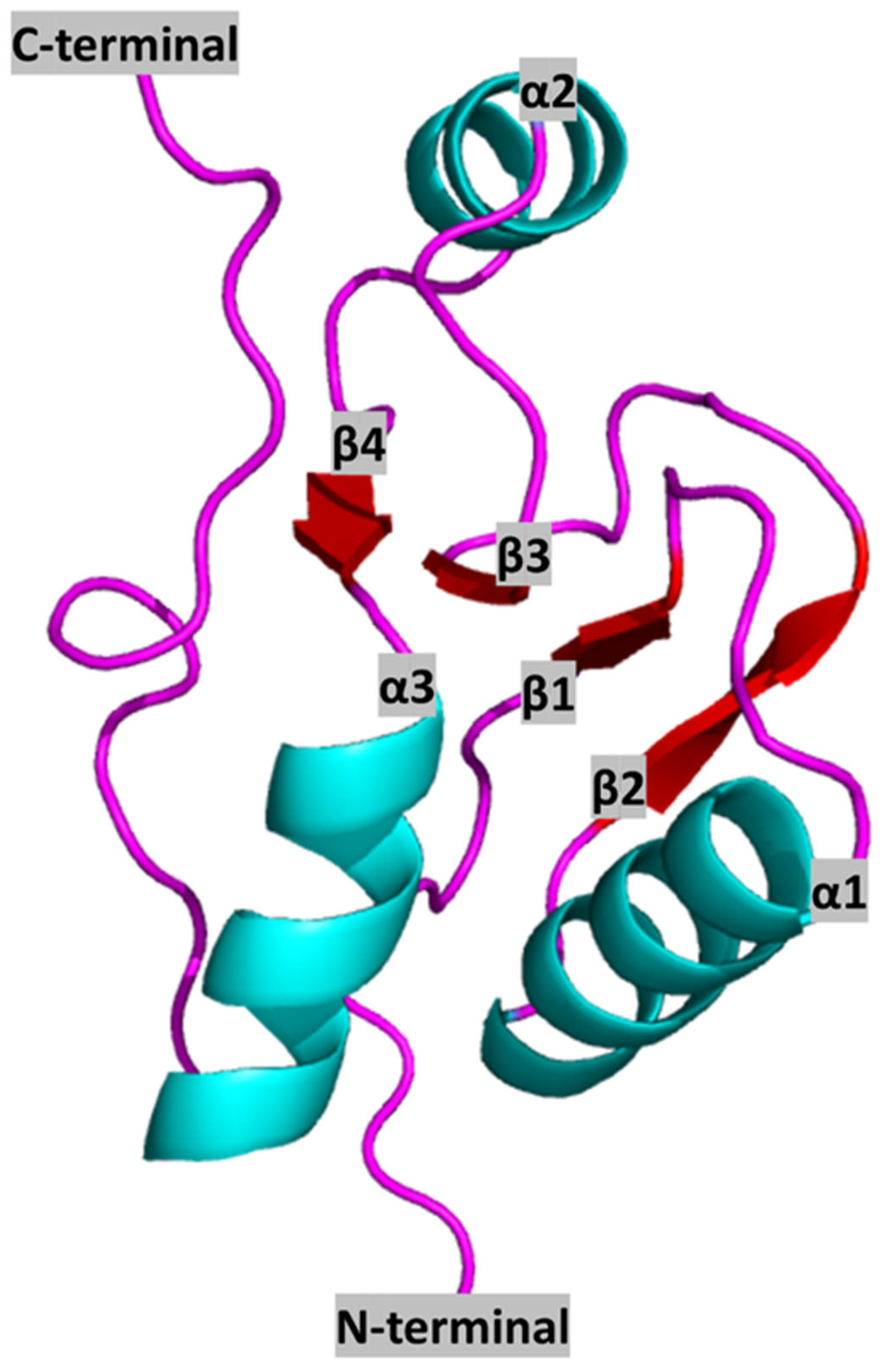
2. BRCT Domains: Structure and Architecture
3. Interactions Mediated by BRCT Domains
3.1. Phosphate-Dependent Interactions
3.2. Phosphate-Independent Interactions
4. The Role of BRCT in Cell Processes
4.1. BRCT in DNA Damage Response
4.2. BRCT in Cell Cycle Control
4.3. Other Biological Roles of BCRT
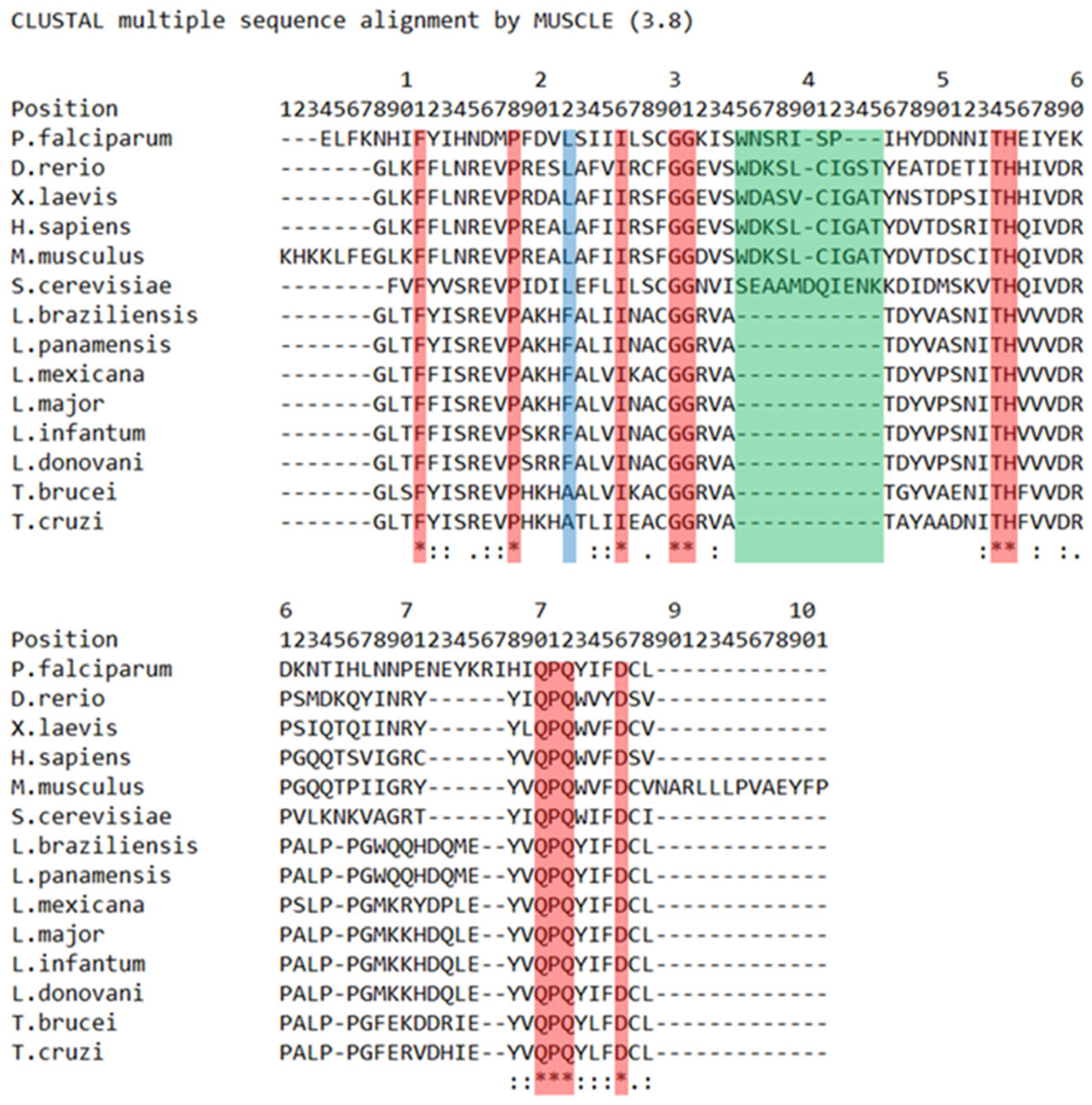
5. BRCT Domains in Infectious Agents
5.1. In Bacteria and Viruses
5.2. In Parasites
5.3. In Fungi
6. Implication of BRCT Domains in Cancer and Other Pathologic Processes
6.1. BRCT Domains and Cancer
6.2. Implication of BRCT in Other Pathologic Processes
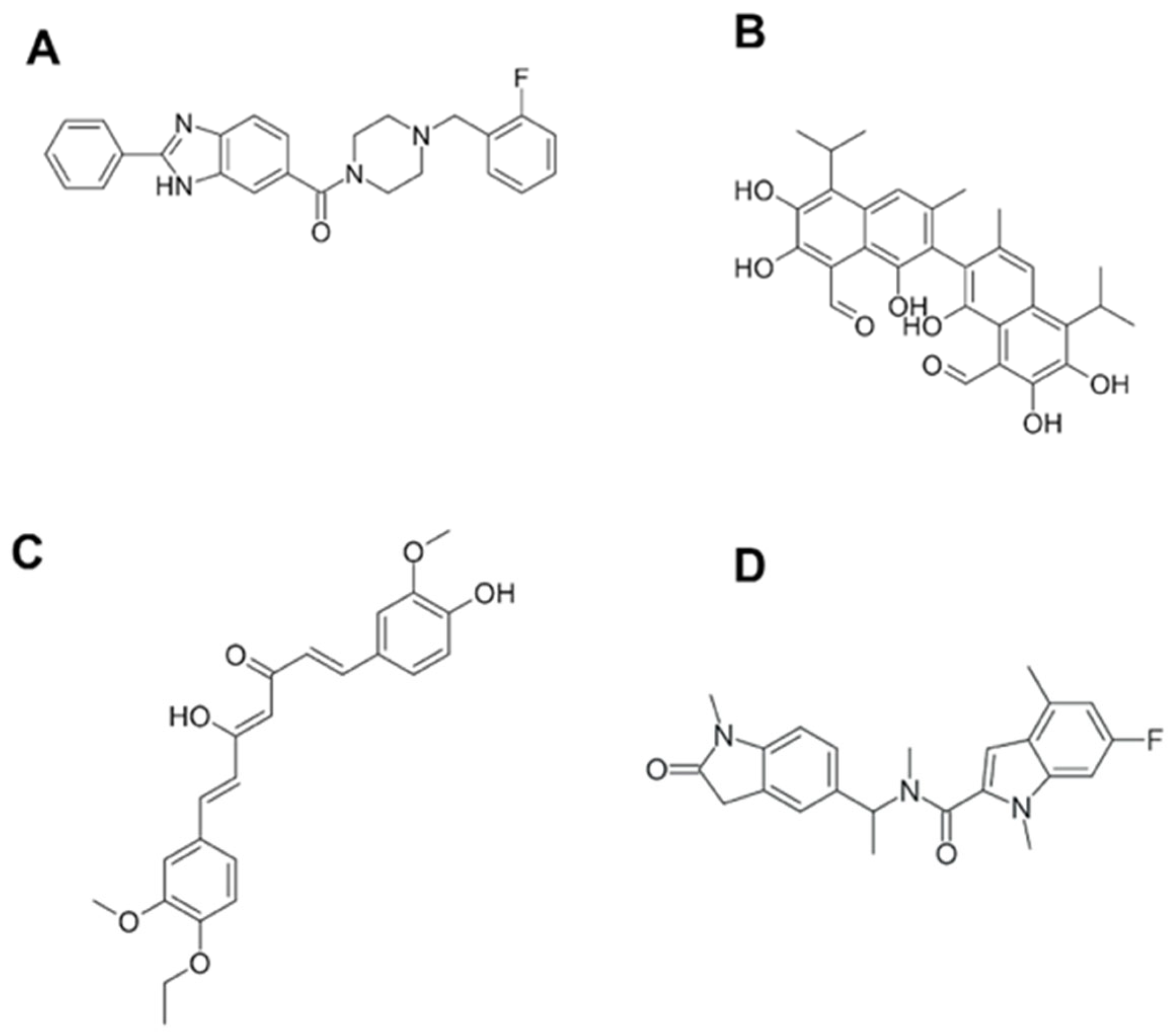
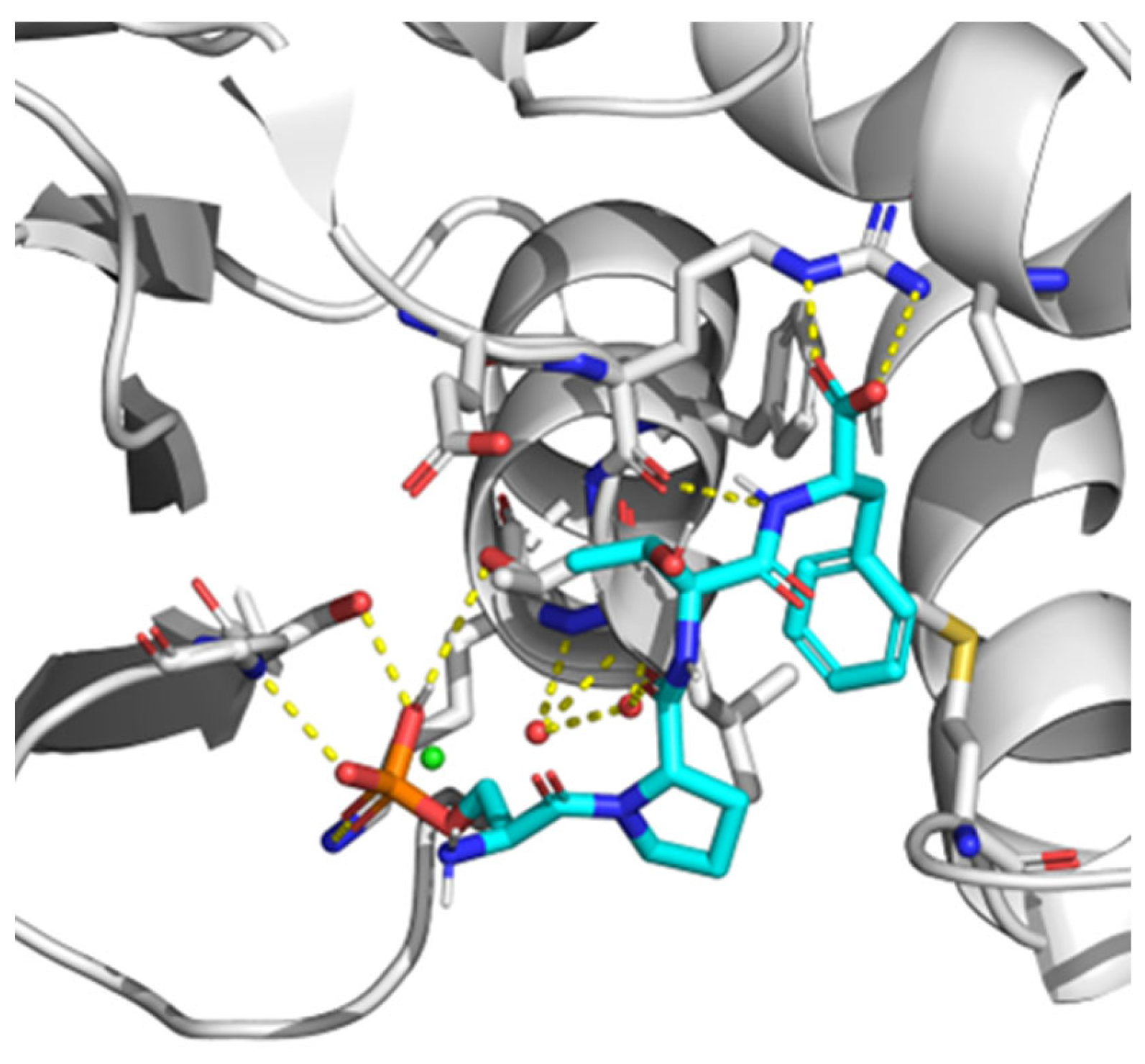
7. BRCT as a Therapeutic Target. Novel Techniques for BRCT Domain Research
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ghadermarzi, S.; Li, X.; Li, M.; Kurgan, L. Sequence-Derived Markers of Drug Targets and Potentially Druggable Human Proteins. Front. Genet. 2019, 10, 1075. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2016, 16, 19–34. [Google Scholar] [CrossRef]
- Lauss, M.; Kriegner, A.; Vierlinger, K.; Neohammer, C. Characterization of the drugged human genome. Pharmacogenomics 2007, 8, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.J.; Han, L.Y.; Yap, C.W.; Ji, Z.L.; Cao, Z.W.; Chen, Y.Z. Therapeutic targets: Progress of their exploration and investigation of their characteristics. Pharmacol. Rev. 2006, 58, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Bakheet, T.M.; Doig, A.J. Properties and identification of human protein drug targets. Bioinformatics 2009, 25, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Bull, S.C.; Doig, A.J. Properties of protein drug target classes. PLoS ONE 2015, 10, e0117955. [Google Scholar] [CrossRef]
- Kim, B.; Jo, J.; Han, J.; Park, C.; Lee, H. In silico re-identification of properties of drug target proteins. BMC Bioinform. 2017, 18, 35–44. [Google Scholar] [CrossRef][Green Version]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R.; et al. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, eaag1166. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef]
- Makley, L.N.; Gestwicki, J.E. Expanding the Number of “Druggable” Targets: Non-Enzymes and Protein-Protein Interactions. Chem. Biol. Drug Des. 2013, 81, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, E.S.; Halakou, F.; Nussinov, R.; Gursoy, A.; Keskin, O. Methods for Discovering and Targeting Druggable Protein-Protein Interfaces and Their Application to Repurposing. Methods Mol. Biol. 2019, 1903, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Bork, P.; Hofmann, K.; Neuwald, P.F.A.; Altschul, S.F.; Koonin, E.V. A Superfamily of Conserved Domains in DNA Damage-Responsive Cell Cycle Checkpoint Proteins. FASEB J. 1997, 11, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.C.Y.; Glover, J.N.M. BRCT domains: Easy as one, two, three. Cell Cycle 2011, 10, 2461–2470. [Google Scholar] [CrossRef] [PubMed]
- Gerloff, D.L.; Woods, N.T.; Farago, A.A.; Monteiro, A.N.A. BRCT domains: A little more than kin, and less than kind. FEBS Lett. 2012, 586, 2711–2716. [Google Scholar] [CrossRef]
- Sheng, Z.-Z.; Zhao, Y.-Q.; Huang, J.-F. Functional Evolution of BRCT Domains from Binding DNA to Protein. Evol. Bioinform. Online 2011, 7, 87–97. [Google Scholar] [CrossRef]
- Zhang, X.; Moréra, S.; Bates, P.A.; Whitehead, P.C.; Coffer, A.I.; Hainbucher, K.; Nash, R.A.; Sternberg, M.J.E.; Lindahl, T.; Freemont, P.S. Structure of an XRCC1 BRCT domain: A new protein-protein interaction module. EMBO J. 1998, 17, 6404–6411. [Google Scholar] [CrossRef]
- Callebaut, I.; Mornon, J.P. From BRCA1 to RAP1: A widespread BRCT module closely associated with DNA repair. FEBS Lett. 1997, 400, 25–30. [Google Scholar] [CrossRef]
- Glover, J.N.M.; Williams, R.S.; Lee, M.S. Interactions between BRCT repeats and phosphoproteins: Tangled up in two. Trends Biochem. Sci. 2004, 29, 579–585. [Google Scholar] [CrossRef]
- Peña-Guerrero, J.; Fernández-Rubio, C.; Burguete-Mikeo, A.; El-Dirany, R.; García-Sosa, A.T.; Nguewa, P. Discovery and Validation of Lmj_04_BRCT Domain, a Novel Therapeutic Target: Identification of Candidate Drugs for Leishmaniasis. Int. J. Mol. Sci. 2021, 22, 10493. [Google Scholar] [CrossRef]
- Haque, J.; Boger, S.; Li, J.; Duncan, S. The murine Pes1 gene encodes a nuclear protein containing a BRCT domain. Genomics 2000, 70, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.; Pethica, R.; Zhou, Y.; Talbot, C.; Vogel, C.; Madera, M.; Chothia, C.; Gough, J. SUPERFAMILY—Sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 2009, 37, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Green, R.; Glover, J.N.M. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat. Struct. Biol. 2001, 8, 838–842. [Google Scholar] [CrossRef]
- Rodriguez, M.C.; Songyang, Z. BRCT domains: Phosphopeptide binding and signaling modules. Front. Biosci. 2008, 13, 5905–5915. [Google Scholar] [CrossRef]
- Liu, X.; Ladias, J.A.A. Structural basis for the brca1 brct interaction with the proteins atrip and baat1. Biochemistry 2013, 52, 7618–7627. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Huang, Y.; Edwards, R.A.; Yang, S.; Blackford, A.N.; Niedzwiedz, W.; Glover, J.N.M. Structural Insight into BLM Recognition by TopBP1. Structure 2017, 25, 1582–1588.e3. [Google Scholar] [CrossRef]
- Mer, G.; Botuyan, M.V. A New BRCT Binding Mode in TopBP1-BLM Helicase Interaction. Structure 2017, 25, 1471–1472. [Google Scholar] [CrossRef]
- Gómez-Cavazos, J.S.; Lee, K.Y.; Lara-González, P.; Li, Y.; Desai, A.; Shiau, A.K.; Oegema, K. A Non-canonical BRCT-Phosphopeptide Recognition Mechanism Underlies RhoA Activation in Cytokinesis. Curr. Biol. 2020, 30, 3101–3115.e11. [Google Scholar] [CrossRef]
- Manke, I.A.; Lowery, D.M.; Nguyen, A.; Yaffe, M.B. BRCT Repeats As Phosphopeptide-Binding Modules Involved in Protein Targeting. Science 2003, 302, 636–639. [Google Scholar] [CrossRef]
- Yu, X.; Chini, C.C.S.; He, M.; Mer, G.; Chen, J. The BRCT Domain Is a Phospho-Protein Binding Domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef]
- Wardlaw, C.P.; Carr, A.M.; Oliver, A.W. TopBP1: A BRCT-scaffold protein functioning in multiple cellular pathways. DNA Repair 2014, 22, 165–174. [Google Scholar] [CrossRef]
- Leung, C.C.Y.; Kellogg, E.; Kuhnert, A.; Hänel, F.; Baker, D.; Glover, J.N.M. Insights from the crystal structure of the sixth BRCT domain of topoisomerase IIβ binding protein 1. Protein Sci. 2010, 19, 162–167. [Google Scholar] [PubMed]
- Leung, C.C.Y.; Sun, L.; Gong, Z.; Burkat, M.; Edwards, R.; Assmus, M.; Chen, J.; Glover, J.N.M. Structural insights into recognition of MDC1 by TopBP1 in DNA replication checkpoint control. Structure 2013, 21, 1450–1459. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rappas, M.; Oliver, A.W.; Pearl, L.H. Structure and function of the Rad9-binding region of the DNA-damage checkpoint adaptor TopBP1. Nucleic Acids Res. 2011, 39, 313–324. [Google Scholar] [CrossRef]
- Yamane, K.; Tsuruo, T. Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene 1999, 18, 5194–5203. [Google Scholar] [CrossRef]
- Matsumoto, T.; Go, K.; Hyodo, M.; Koiwai, K.; Maezawa, S.; Hayano, T.; Suzuki, M.; Koiwai, O. BRCT domain of DNA polymerase μ has DNA-binding activity and promotes the DNA polymerization activity. Genes Cells 2012, 17, 790–806. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ab, E.; Bonvin, A.M.J.J.; Siegal, G. Structure of the DNA-bound BRCA1 C-terminal region from human replication factor C p140 and model of the protein-DNA complex. J. Biol. Chem. 2010, 285, 10087–10097. [Google Scholar] [CrossRef] [PubMed]
- Dulic, A.; Bates, P.A.; Zhang, X.; Martin, S.R.; Freemont, P.S.; Lindahl, T.; Barnes, D.E. BRCT domain interactions in the heterodimeric DNA repair protein XRCC1-DNA ligase III. Biochemistry 2001, 40, 5906–5913. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Rashid, I.; Sverzhinsky, A.; Pourfarjam, Y.; Tsai, M.S.; Ellenberger, T.; Pascal, J.M.; Kim, I.K.; Tainer, J.A.; Tomkinson, A.E. An atypical BRCT-BRCT interaction with the XRCC1 scaffold protein compacts human DNA Ligase IIIα within a flexible DNA repair complex. Nucleic Acids Res. 2021, 49, 306–321. [Google Scholar] [CrossRef]
- Masson, M.; Niedergang, C.; Schreiber, V.; Muller, S.; Murcia, J.M.-D.; de Murcia, G. XRCC1 Is Specifically Associated with Poly(ADP-Ribose) Polymerase and Negatively Regulates Its Activity following DNA Damage. Mol. Cell. Biol. 1998, 18, 3563–3571. [Google Scholar] [CrossRef]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar] [CrossRef]
- Wollmann, Y.; Schmidt, U.; Wieland, G.D.; Zipfel, P.F.; Saluz, H.P.; Hänel, F. The DNA Topoisomerase IIβ Binding Protein 1 (TopBP1) interacts with poly (ADP-ribose) polymerase (PARP-1). J. Cell. Biochem. 2007, 102, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, D.J.; Basu, B.P.; Serpell, L.C.; Joo, W.S.; Date, T.; Iwabuchi, K.; Doherty, A.J. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 2002, 21, 3863–3872. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, D.H.; Yaffe, M.B. 14-3-3 proteins, FHA domains and BRCT domains in the DNA damage response. DNA Repair 2009, 8, 1009–1017. [Google Scholar] [CrossRef]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Huen, M.S.Y.; Grant, R.; Manke, I.; Minn, K.; Yu, X.; Yaffe, M.B.; Chen, J. The E3 ubiquitin ligase RNF8 transduces the DNA damage signal via an ubiquitin-dependent signaling pathway. Cell 2008, 131, 901–914. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Jeggo, P.A. Roles for 53BP1 in the repair of radiation-induced DNA double strand breaks. DNA Repair 2020, 93, 102915. [Google Scholar] [CrossRef]
- Nguewa, P.A.; Fuertes, M.A.; Alonso, C.; Peréz, J.M. Pharmacological modulation of Poly(ADP-ribose) polymerase-mediated cell death: Exploitation in cancer chemotherapy. Mol. Pharmacol. 2003, 64, 1007–1014. [Google Scholar] [CrossRef]
- Nguewa, P.A.; Fuertes, M.A.; Valladares, B.; Alonso, C.; Pérez, J.M. Poly(ADP-ribose) polymerases: Homology, structural domains and functions. Novel therapeutical applications. Prog. Biophys. Mol. Biol. 2005, 88, 143–172. [Google Scholar] [CrossRef]
- Paddock, M.N.; Buelow, B.D.; Takeda, S.; Scharenberg, A.M. The BRCT domain of PARP-1 is required for immunoglobulin gene conversion. PLoS Biol. 2010, 8, e1000428. [Google Scholar] [CrossRef]
- Yen, W.F.; Chaudhry, A.; Vaidyanathan, B.; Yewdell, W.T.; Pucella, J.N.; Sharma, R.; Liang, Y.; Li, K.; Rudensky, A.Y.; Chaudhuri, J. BRCT-domain protein BRIT1 influences class switch recombination. Proc. Natl. Acad. Sci. USA 2017, 114, 8354–8359. [Google Scholar] [CrossRef]
- Wu, L.; Crawley, C.D.; Garofalo, A.; Nichols, J.W.; Campbell, P.A.; Khramtsova, G.F.; Olopade, O.I.; Weichselbaum, R.R.; Yamini, B. p50 mono-ubiquitination and interaction with BARD1 regulates cell cycle progression and maintains genome stability. Nat. Commun. 2020, 11, 5007. [Google Scholar] [CrossRef] [PubMed]
- Groothuizen, F.S.; Sixma, T.K. The conserved molecular machinery in DNA mismatch repair enzyme structures. DNA Repair 2016, 38, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [PubMed]
- Caligo, M.A.; Bonatti, F.; Guidugli, L.; Aretini, P.; Galli, A. A yeast recombination assay to characterize human BRCA1 missense variants of unknown pathological significance. Hum. Mutat. 2009, 30, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Lodovichi, S.; Bellè, F.; Cervelli, T.; Lorenzoni, A.; Maresca, L.; Cozzani, C.; Caligo, M.A.; Galli, A. Effect of BRCA1 missense variants on gene reversion in DNA double-strand break repair mutants and cell cycle-arrested cells of Saccharomyces cerevisiae. Mutagenesis 2020, 35, 189–195. [Google Scholar] [CrossRef]
- Basant, A.; Glotzer, M. Spatiotemporal Regulation of RhoA during Cytokinesis. Curr. Biol. 2018, 28, R570–R580. [Google Scholar] [CrossRef]
- Schneid, S.; Wolff, F.; Buchner, K.; Bertram, N.; Baygün, S.; Barbosa, P.; Mangal, S.; Zanin, E. The BRCT domains of ECT2 have distinct functions during cytokinesis. Cell Rep. 2021, 34, 108805. [Google Scholar] [CrossRef]
- Muhseena, N.K.; Mathukkada, S.; Das, S.P.; Laha, S. The repair gene BACH1—A potential oncogene. Oncol. Rev. 2021, 15, 519. [Google Scholar] [CrossRef]
- Bagge, J.; Oestergaard, V.H.; Lisby, M. Functions of TopBP1 in preserving genome integrity during mitosis. Semin. Cell Dev. Biol. 2021, 113, 57–64. [Google Scholar] [CrossRef]
- Bigot, N.; Day, M.; Baldock, R.A.; Watts, F.Z.; Oliver, A.W.; Pearl, L.H. Phosphorylation-mediated interactions with TOPBP1 couple 53BP1 and 9-1-1 to control the G1 DNA damage checkpoint. Elife 2019, 8, 108805. [Google Scholar] [CrossRef] [PubMed]
- Reubens, M.C.; Rozenzhak, S.; Russell, P. Multi-BRCT domain protein Brc1 links Rhp18/Rad18 and γH2A to maintain genome stability during S phase. Mol. Cell. Biol. 2017, 37, e00260-17. [Google Scholar] [CrossRef]
- Hölzel, M.; Grimm, T.; Rohrmoser, M.; Malamoussi, A.; Harasim, T.; Gruber-Eber, A.; Kremmer, E.; Eick, D. The BRCT domain of mammalian Pes1 is crucial for nucleolar localization and rRNA processing. Nucleic Acids Res. 2007, 35, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Grimm, T.; Hölzel, M.; Rohrmoser, M.; Harasim, T.; Malamoussi, A.; Gruber-Eber, A.; Kremmer, E.; Eick, D. Dominant-negative Pes1 mutants inhibit ribosomal RNA processing and cell proliferation via incorporation into the PeBoW-complex. Nucleic Acids Res. 2006, 34, 3030–3043. [Google Scholar] [CrossRef]
- Ghosh, A.; Shuman, S.; Lima, C.D. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol. Cell 2008, 32, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Perrino, F.; Harvey, S.; McNeill, S. Two functional domains of the epsilon subunit of DNA polymerase III. Biochemistry 1999, 38, 16001–16009. [Google Scholar] [CrossRef] [PubMed]
- Pohlhaus, J.R.; Long, D.T.; O’Reilly, E.; Kreuzer, K.N. The epsilon subunit of DNA polymerase III Is involved in the nalidixic acid-induced SOS response in Escherichia coli. J. Bacteriol. 2008, 190, 5239–5247. [Google Scholar] [CrossRef]
- Hamdan, S.; Bulloch, E.M.; Thompson, P.R.; Beck, J.L.; Yang, J.Y.; Crowther, J.A.; Lilley, P.E.; Carr, P.D.; Ollis, D.L.; Brown, S.E.; et al. Hydrolysis of the 5′-p-nitrophenyl ester of TMP by the proofreading exonuclease (epsilon) subunit of Escherichia coli DNA polymerase III. Biochemistry 2002, 41, 5266–5275. [Google Scholar] [CrossRef]
- Santos, J.A.; Lamers, M.H. Novel Antibiotics Targeting Bacterial Replicative DNA Polymerases. Antibiotics 2020, 9, 776. [Google Scholar] [CrossRef]
- Wilkinson, A.; Day, J.; Bowater, R. Bacterial DNA ligases. Mol. Microbiol. 2001, 40, 1241–1248. [Google Scholar] [CrossRef]
- Martin, I.V.; MacNeill, S.A. ATP-dependent DNA ligases. Genome Biol. 2002, 3, REVIEWS3005. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.; Smith, A.; Bullard, D.; Lavesa-Curto, M.; Sayer, H.; Bonner, A.; Hemmings, A.; Bowater, R. Analysis of ligation and DNA binding by Escherichia coli DNA ligase (LigA). Biochim. Biophys. Acta Proteins Proteom. 2005, 1749, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Li, K.W.; Nair, P.A.; Shuman, S. Structure-guided mutational analysis of the OB, HhH, and BRCT domains of Escherichia coli DNA ligase. J. Biol. Chem. 2008, 283, 23343–23352. [Google Scholar]
- Benarroch, D.; Shuman, S. Characterization of mimivirus NAD+-dependent DNA ligase. Virology 2006, 353, 133–143. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Srivastava, S.K.; Dube, D.; Kukshal, V.; Jha, A.K.; Hajela, K.; Ramachandran, R. NAD+-dependent DNA ligase (Rv3014c) from Mycobacterium tuberculosis: Novel structure-function relationship and identification of a specific inhibitor. Proteins Struct. Funct. Genet. 2007, 69, 97–111. [Google Scholar] [CrossRef]
- Gilson, T.; Greer, A.E.; Vindigni, A.; Ketner, G.; Hanakahi, L.A. The α2 helix in the DNA ligase IV BRCT-1 domain is required for targeted degradation of ligase IV during adenovirus infection. Virology 2012, 428, 128–135. [Google Scholar] [CrossRef][Green Version]
- Fenoy, I.M.; Bogado, S.S.; Contreras, S.M.; Gottifredi, V.; Angel, S.O. The knowns unknowns: Exploring the homologous recombination repair pathway in Toxoplasma gondii. Front. Microbiol. 2016, 7, 627. [Google Scholar] [CrossRef]
- Akiyoshi, B.; Gull, K. Discovery of unconventional kinetochores in kinetoplastids. Cell 2014, 156, 1247–1258. [Google Scholar] [CrossRef]
- Ludzia, P.; Lowe, E.D.; Marcianò, G.; Mohammed, S.; Redfield, C.; Akiyoshi, B. Structural characterization of KKT4, an unconventional microtubule-binding kinetochore protein. Structure 2021, 29, 1014–1028.e8. [Google Scholar] [CrossRef]
- Afrin, M.; Kishmiri, H.; Sandhu, R.; Rabbani, M.A.G.; Li, B. Trypanosoma brucei RAP1 Has Essential Functional Domains That Are Required for Different Protein Interactions. mSphere 2020, 5, e00027-20. [Google Scholar] [CrossRef]
- Yang, X.; Figueiredo, L.M.; Espinal, A.; Okubo, E.; Li, B. RAP1 is essential for silencing telomeric variant surface glycoprotein genes in Trypanosoma brucei. Cell 2009, 137, 99–109. [Google Scholar] [CrossRef]
- Algarabel, M.; Fernández-Rubio, C.; Musilova, K.; Peña-Guerrero, J.; Vacas, A.; Larrea, E.; Nguewa, P.A. In Leishmania major, the Homolog of the Oncogene PES1 May Play a Critical Role in Parasite Infectivity. Int. J. Mol. Sci. 2021, 22, 12592. [Google Scholar] [CrossRef] [PubMed]
- Larrea, E.; Fernández-Rubio, C.; Peña-Guerrero, J.; Guruceaga, E.; Nguewa, P.A. The BRCT Domain from the Homologue of the Oncogene PES1 in Leishmania major (LmjPES) Promotes Malignancy and Drug Resistance in Mammalian Cells. Int. J. Mol. Sci. 2022, 23, 13203. [Google Scholar] [CrossRef]
- Graham, I.R.; Haw, R.A.; Spink, K.G.; Halden, K.A.; Chambers, A. In Vivo Analysis of Functional Regions within Yeast Rap1p. Mol. Cell. Biol. 1999, 19, 7481–7490. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mizuno, T.; Kishimoto, T.; Shinzato, T.; Haw, R.; Chambers, A.; Wood, J.; Sinclair, D.; Uemura, H. Role of the N-terminal region of Rap1p in the transcriptional activation of glycolytic genes in Saccharomyces cerevisiae. Yeast 2004, 21, 851–866. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Singh, V.; Baranwal, S.; Thakare, M.J.; Tomar, R.S. The transcription factor Rap1p is required for tolerance to cell-wall perturbing agents and for cell-wall maintenance in Saccharomyces cerevisiae. FEBS Lett. 2015, 589, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Backhaus, K.; Rippert, D.; Heilmann, C.J.; Sorgo, A.G.; de Koster, C.G.; Klis, F.M.; Rodicio, R.; Heinisch, J.J. Mutations in SNF1 complex genes affect yeast cell wall strength. Eur. J. Cell Biol. 2013, 92, 383–395. [Google Scholar] [CrossRef]
- Li, B.; Oestreich, S.; De Lange, T. Identification of human Rap1: Implications for telomere evolution. Cell 2000, 101, 471–483. [Google Scholar] [CrossRef]
- Uemura, H.; Watanabe-Yoshida, M.; Ishii, N.; Shinzato, T.; Haw, R. Isolation and characterization of Candida albicans homologue of RAP1, a repressor and activator protein gene in Saccharomyces cerevisiae. Yeast 2004, 21, 1–10. [Google Scholar] [CrossRef]
- Wilhelmsen, K.C. The tangled biology of tau. Proc. Natl. Acad. Sci. USA 1999, 96, 7120–7121. [Google Scholar] [CrossRef]
- Wang, B. BRCA1 tumor suppressor network: Focusing on its tail. Cell Biosci. 2012, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Black, D.M.; Solomon, E. The search for the familial breast/ovarian cancer gene. Trends Genet. 1993, 9, 22–26. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.P.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, Å.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef]
- Gaboriau, D.C.A.; Rowling, P.J.E.; Morrison, C.G.; Itzhaki, L.S. Protein stability versus function: Effects of destabilizing missense mutations on BRCA1 DNA repair activity. Biochem. J. 2015, 466, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Ahlborn, L.B.; Dandanell, M.; Steffensen, A.Y.; Jønson, L.; Nielsen, F.C.; Hansen, T.V.O. Splicing analysis of 14 BRCA1 missense variants classifies nine variants as pathogenic. Breast Cancer Res. Treat. 2015, 150, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Bellè, F.; Mercatanti, A.; Lodovichi, S.; Congregati, C.; Guglielmi, C.; Tancredi, M.; Caligo, M.A.; Cervelli, T.; Galli, A. validation and data-integration of yeast-based assays for functional classification of BRCA1 missense variants. Int. J. Mol. Sci. 2022, 23, 4049. [Google Scholar] [CrossRef]
- Iversen, E.S.; Couch, F.J.; Goldgar, D.E.; Tavtigian, S.V.; Monteiro, A.N.A. A computational method to classify variants of uncertain significance using functional assay data with application to BRCA1. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1078–1088. [Google Scholar] [CrossRef]
- Woods, N.T.; Baskin, R.; Golubeva, V.; Jhuraney, A.; De-Gregoriis, G.; Vaclova, T.; Goldgar, D.E.; Couch, F.J.; Carvalho, M.A.; Iversen, E.S.; et al. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom. Med. 2016, 1, 16001. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Wang, Y.; Yin, C.C.; Xie, Y.; Liu, X.; Ding, H.; Tian, Z.; Shen, J.; He, L.; et al. Functional analysis of BRCT missense mutations in BRCA1-mutated chinese han familial breast cancer. Oncol. Lett. 2017, 14, 5839–5844. [Google Scholar] [CrossRef][Green Version]
- De Gregoriis, G.; Ramos, J.A.; Fernandes, P.V.; Vignal, G.M.; Brianese, R.C.; Carraro, D.M.; Monteiro, A.N.; Struchiner, C.J.; Suarez-Kurtz, G.; Vianna-Jorge, R.; et al. DNA repair genes PAXIP1 and TP53BP1 expression is associated with breast cancer prognosis. Cancer Biol. Ther. 2017, 18, 439–449. [Google Scholar] [CrossRef]
- Alsiary, R.; Brownhill, S.C.; Brüning-Richardson, A.; Hutson, R.; Griffin, N.; Morrison, E.E.; Bond, J.; Burchill, S.A.; Bell, S.M. Expression analysis of the MCPH1/BRIT1 and BRCA1 tumor suppressor genes and telomerase splice variants in epithelial ovarian cancer. Gene 2018, 672, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Jhuraney, A.; Woods, N.T.; Wright, G.; Rix, L.; Kinose, F.; Kroeger, J.L.; Remily-Wood, E.; Cress, W.D.; Koomen, J.M.; Brantley, S.G.; et al. PAXIP1 Potentiates the Combination of WEE1 Inhibitor AZD1775 and Platinum Agents in Lung Cancer. Mol Cancer Ther. 2016, 15, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, A.I.; Banerjee, T.; Wingo, M.; Duncan, K.; Ning, J.; Rodrigues, F.M.; Huang, K.L.; Lee, C.; Chen, F.; Ding, L.; et al. Functional analysis of BARD1 missense variants in homology-directed repair and damage sensitivity. PLoS Genet. 2019, 15, e1008049. [Google Scholar] [CrossRef]
- Mok, M.C.Y.; Campalans, A.; Pillon, M.C.; Guarné, A.; Radicella, J.P.; Junop, M.S. Identification of an XRCC1 DNA binding activity essential for retention at sites of DNA damage. Sci. Rep. 2019, 9, 3095. [Google Scholar] [CrossRef]
- Polo, L.M.; Xu, Y.; Hornyak, P.; Garces, F.; Zeng, Z.; Hailstone, R.; Matthews, S.J.; Caldecott, K.W.; Oliver, A.W.; Pearl, L.H. Efficient Single-Strand Break Repair Requires Binding to Both Poly(ADP-Ribose) and DNA by the Central BRCT Domain of XRCC1. Cell Rep. 2019, 26, 573–581.e5. [Google Scholar] [CrossRef]
- Xu, P.; Liu, Q.; Xie, Y.; Shi, X.; Li, Y.; Peng, M.; Quo, H.; Sun, R.; Li, J.; Hong, Y.; et al. Breast cancer susceptibility protein 1 (BRCA1) rescues neurons from cerebral ischemia/reperfusion injury through NRF2-mediated antioxidant pathway. Redox Biol. 2018, 18, 158–172. [Google Scholar] [CrossRef]
- Xu, P.; Shi, X.; Zhang, X.; Liu, Q.; Xie, Y.; Hong, Y.; Li, J.; Peng, M.; Liu, X.; Xu, G. Overexpression of BRCA1 in neural stem cells enhances cell survival and functional recovery after transplantation into experimental ischemic stroke. Oxid. Med. Cell. Longev. 2019, 2019, 8739730. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Yu, X. Human DNA ligase IV is able to use NAD+ as an alternative adenylation donor for DNA ends ligation. Nucleic Acids Res. 2019, 47, 1321–1334. [Google Scholar] [CrossRef]
- Richards, M.W.; Leung, J.W.; Roe, S.M.; Li, K.; Chen, J.; Bayliss, R. A pocket on the surface of the N-terminal BRCT domain of Mcph1 is required to prevent abnormal chromosome condensation. J. Mol. Biol. 2010, 395, 908–915. [Google Scholar] [CrossRef]
- Ghani-Kakhki, M.; Robinson, P.N.; Morlot, S.; Mitter, D.; Trimborn, M.; Albrecht, B.; Varon, R.; Sperling, K.; Neitzel, H. Two missense mutations in the primary autosomal recessive microcephaly gene MCPH1 disrupt the function of the highly conserved n-terminal BRCT domain of microcephalin. Mol. Syndromol. 2012, 3, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Schneble-Löhnert, N.; Kristofova, M.; Qing, X.; Labisch, J.; Hofmann, S.; Ehrenberg, S.; Sannai, M.; Jörß, T.; Ori, A.; et al. The N-terminal BRCT domain determines MCPH1 function in brain development and fertility. Cell Death Dis. 2021, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Ke, Y.; Wang, C.; Zhang, J.; Zhong, X.; Wang, R.; Zeng, X.; Ba, X. The Role of PARPs in Inflammation-and Metabolic-Related Diseases: Molecular Mechanisms and Beyond. Cells 2019, 8, 1047. [Google Scholar] [CrossRef] [PubMed]
- Morrone, M.D.S.; Somensi, N.; Franz, L.; Ramos, V.M.; Gasparotto, J.; da Rosa, H.T.; Sartori, M.; Figueiró, F.; Gelain, D.P.; Zanotto-Filho, A.; et al. BRCA-1 depletion impairs pro-inflammatory polarization and activation of RAW 264.7 macrophages in a NF-κB-dependent mechanism. Mol. Cell. Biochem. 2019, 462, 11–23. [Google Scholar] [CrossRef]
- Takeuchi, T.; Ishidoh, T.; Iijima, H.; Kuriyama, I.; Shimazaki, N.; Koiwai, O.; Kuramochi, K.; Kobayashi, S.; Sugawara, F.; Sakaguchi, K.; et al. Structural relationship of curcumin derivatives binding to the BRCT domain of human DNA polymerase λ. Genes Cells 2006, 11, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Periasamy, J.; Kurdekar, V.; Jasti, S.; Nijaguna, M.B.; Boggaram, S.; Hurakadli, M.A.; Raina, D.; Kurup, L.M.; Chintha, C.; Manjunath, K.; et al. Targeting Phosphopeptide Recognition by the Human BRCA1 Tandem BRCT Domain to Interrupt BRCA1-Dependent Signaling. Cell. Chem. Biol. 2018, 25, 677–690.e12. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Randal, M.; DeLano, W.L.; Hyde, J.; Luong, T.N.; Oslob, J.D.; Raphael, D.R.; Taylor, L.; Wang, J.; McDowell, R.S.; et al. Binding of small molecules to an adaptive protein-protein interface. Proc. Natl. Acad. Sci. USA 2003, 100, 1603–1608. [Google Scholar] [CrossRef]
- Simeonov, A.; Yasgar, A.; Jadhav, A.; Lokesh, G.L.; Klumpp, C.; Michael, S.; Austin, C.P.; Natarajan, A.; Inglese, J. Dual-fluorophore quantitative high-throughput screen for inhibitors of BRCT-phosphoprotein interaction. Anal. Biochem. 2008, 375, 60–70. [Google Scholar] [CrossRef][Green Version]
- Lokesh, G.L.; Rachamallu, A.; Kumar, G.D.K.; Natarajan, A. High-throughput fluorescence polarization assay to identify small molecule inhibitors of BRCT domains of breast cancer gene 1. Anal. Biochem. 2006, 352, 135–141. [Google Scholar] [CrossRef]
- Lokesh, G.L.; Muralidhara, B.K.; Negi, S.S.; Natarajan, A. Thermodynamics of phosphopeptide tethering to BRCT: The structural minima for inhibitor design. J. Am. Chem. Soc. 2007, 129, 10658–10659. [Google Scholar] [CrossRef]
- Joseph, P.R.B.; Yuan, Z.; Kumar, E.A.; Lokesh, G.L.; Kizhake, S.; Rajarathnam, K.; Natarajan, A. Structural characterization of BRCT-tetrapeptide binding interactions. Biochem. Biophys. Res. Commun. 2010, 393, 207–210. [Google Scholar] [CrossRef][Green Version]
- Yuan, Z.; Kumar, E.A.; Campbell, S.J.; Palermo, N.Y.; Kizhake, S.; Glover, J.N.M.; Natarajan, A. Exploiting the P-1 pocket of BRCT domains toward a structure guided inhibitor design. ACS Med. Chem. Lett. 2011, 2, 764–767. [Google Scholar] [CrossRef]
- Pessetto, Z.Y.; Yan, Y.; Bessho, T.; Natarajan, A. Inhibition of BRCT(BRCA1)-phosphoprotein interaction enhances the cytotoxic effect of olaparib in breast cancer cells: A proof of concept study for synthetic lethal therapeutic option. Breast Cancer Res. Treat. 2012, 134, 511–517. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Na, Z.; Pan, S.; Uttamchandani, M.; Yao, S.Q. Discovery of cell-permeable inhibitors that target the BRCT domain of BRCA1 protein by using a small-molecule microarray. Angew. Chem. Int. Ed. 2014, 53, 8421–8426. [Google Scholar] [CrossRef] [PubMed]
- Na, Z.; Peng, B.; Ng, S.; Pan, S.; Lee, J.-S.; Shen, H.-M.; Yao, S.Q. A Small-Molecule Protein-Protein Interaction Inhibitor of PARP1 That Targets Its BRCT Domain. Angew. Chem. Int. Ed. 2015, 54, 2515–2519. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.A. β-hairpin peptidomimetics: Design, structures and biological activities. Acc. Chem. Res. 2008, 41, 1278–1288. [Google Scholar] [CrossRef]
- Dias, A.M.G.C.; Roque, A.C.A. The future of protein scaffolds as affinity reagents for purification. Biotechnol. Bioeng. 2017, 114, 481–491. [Google Scholar] [CrossRef]
- Batalha, I.L.; Lychko, I.; Branco, R.J.F.; Iranzo, O.; Roque, A.C.A. β-Hairpins as peptidomimetics of human phosphoprotein-binding domains. Org. Biomol. Chem. 2019, 17, 3996–4004. [Google Scholar] [CrossRef]
- Choulli, S.; Legrand, A.J.; Vicogne, D.; Villeret, V.; Aumercier, M. Ets-1 interacts through a similar binding interface with Ku70 and poly (ADP-Ribose) polymerase-1. Biosci. Biotechnol. Biochem. 2018, 82, 1753–1759. [Google Scholar] [CrossRef]
- Brysbaert, G.; de Ruyck, J.; Aumercier, M.; Lensink, M.F. Identification of novel interaction partners of ets-1: Focus on DNA repair. Genes 2019, 10, 206. [Google Scholar] [CrossRef]
- Arshad, S.; Ishaque, I.; Mumtaz, S.; Rashid, M.U.; Malkani, N. In-Silico Analyses of Nonsynonymous Variants in the BRCA1 Gene. Biochem. Genet. 2021, 59, 1506–1526. [Google Scholar] [CrossRef]
- Kumar, C.; Lakshmi, P.T.V.; Arunachalam, A. Computational Investigation of FDA Approved Drugs as Selective PARP-1 Inhibitors by Targeting BRCT domain for Cancer Therapy. J. Mol. Graph. Model. 2021, 108, 107919. [Google Scholar] [CrossRef] [PubMed]
- You, W.; Huang, Y.-M.M.; Kizhake, S.; Natarajan, A.; Chang, C.-E.A. Characterization of Promiscuous Binding of Phosphor Ligands to Breast-Cancer-Gene 1 (BRCA1) C-Terminal (BRCT): Molecular Dynamics, Free Energy, Entropy and Inhibitor Design. PLoS Comput. Biol. 2016, 12, e1005057. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peña-Guerrero, J.; Fernández-Rubio, C.; García-Sosa, A.T.; Nguewa, P.A. BRCT Domains: Structure, Functions, and Implications in Disease—New Therapeutic Targets for Innovative Drug Discovery against Infections. Pharmaceutics 2023, 15, 1839. https://doi.org/10.3390/pharmaceutics15071839
Peña-Guerrero J, Fernández-Rubio C, García-Sosa AT, Nguewa PA. BRCT Domains: Structure, Functions, and Implications in Disease—New Therapeutic Targets for Innovative Drug Discovery against Infections. Pharmaceutics. 2023; 15(7):1839. https://doi.org/10.3390/pharmaceutics15071839
Chicago/Turabian StylePeña-Guerrero, José, Celia Fernández-Rubio, Alfonso T. García-Sosa, and Paul A. Nguewa. 2023. "BRCT Domains: Structure, Functions, and Implications in Disease—New Therapeutic Targets for Innovative Drug Discovery against Infections" Pharmaceutics 15, no. 7: 1839. https://doi.org/10.3390/pharmaceutics15071839
APA StylePeña-Guerrero, J., Fernández-Rubio, C., García-Sosa, A. T., & Nguewa, P. A. (2023). BRCT Domains: Structure, Functions, and Implications in Disease—New Therapeutic Targets for Innovative Drug Discovery against Infections. Pharmaceutics, 15(7), 1839. https://doi.org/10.3390/pharmaceutics15071839