
Using ChEMBL to Complement Schistosome Drug Discovery
 , and
, and
Abstract
1. Introduction
2. Materials and Methods
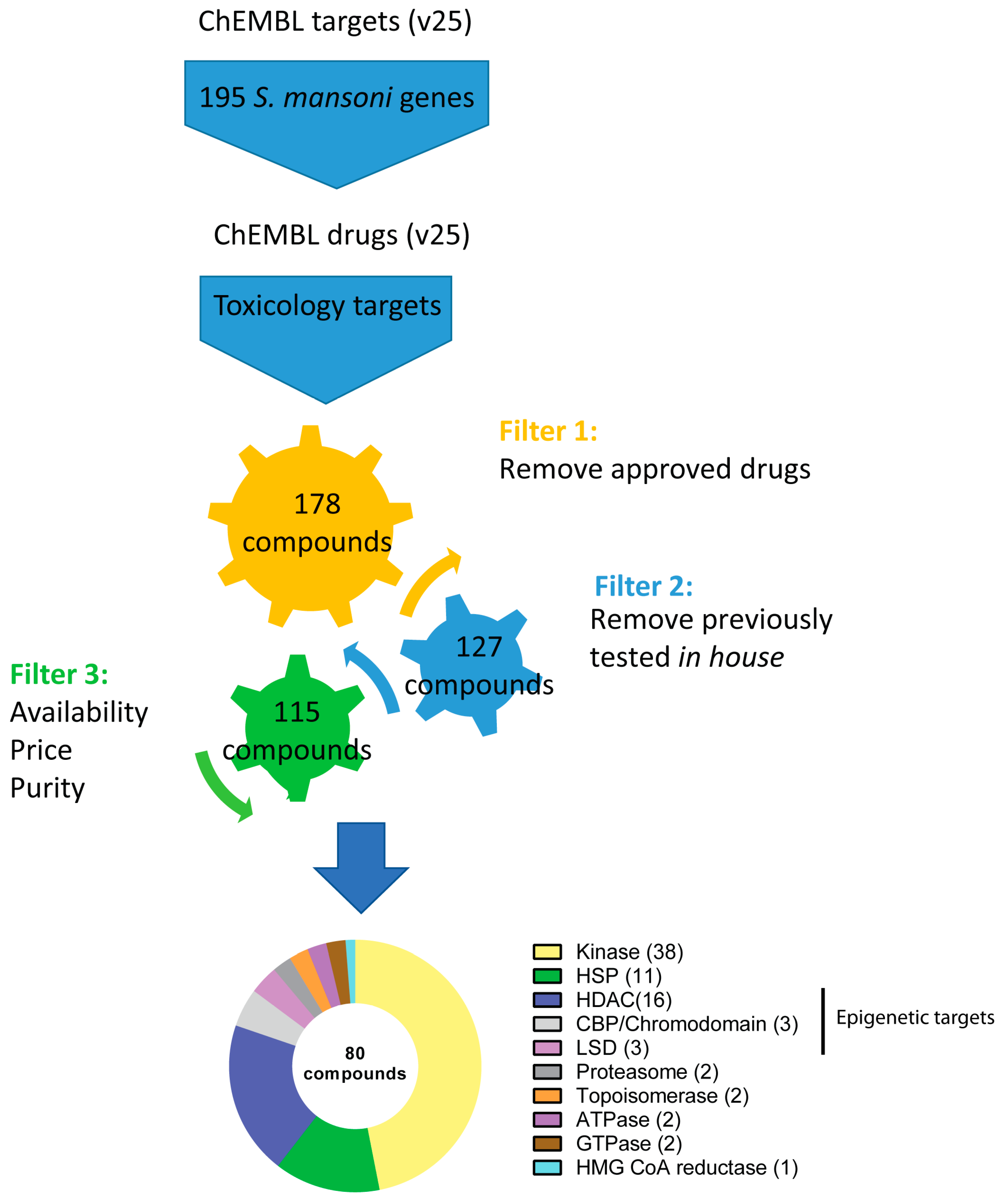
2.1. Bioinformatic Pipeline and Compound Selection
2.2. Literature Research
2.3. Target Prediction in S. mansoni and RNA-Seq Meta Data Analysis
2.4. Parasite Maintenance and Preparation
2.5. Ex Vivo Schistosomula Screening
2.6. Ex Vivo Adult Worm Screening
2.7. Statistics
2.8. Physiochemical Properties Analysis
2.9. Collection of Literature-Based Cytotoxicity Data from ChemBL
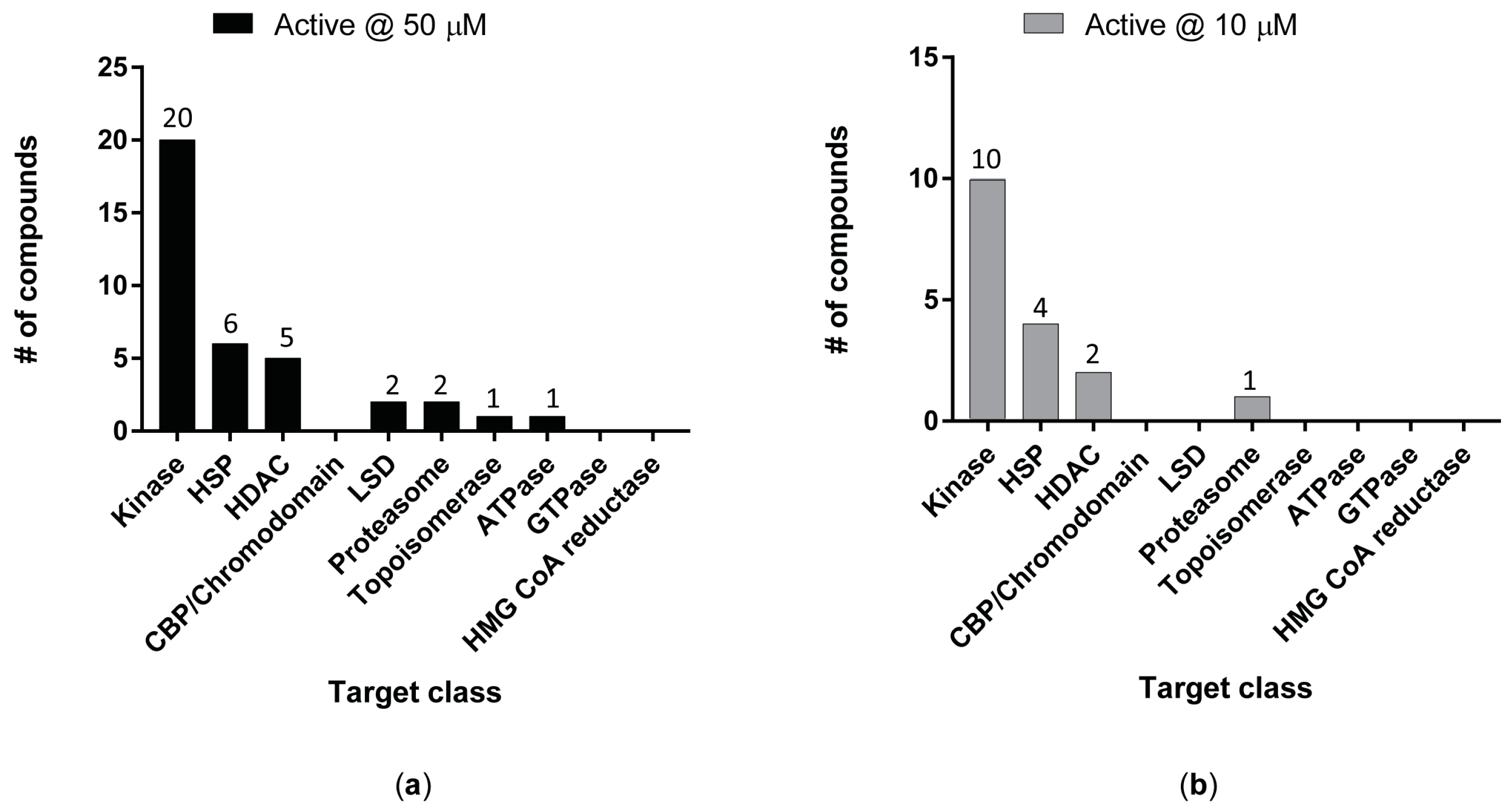
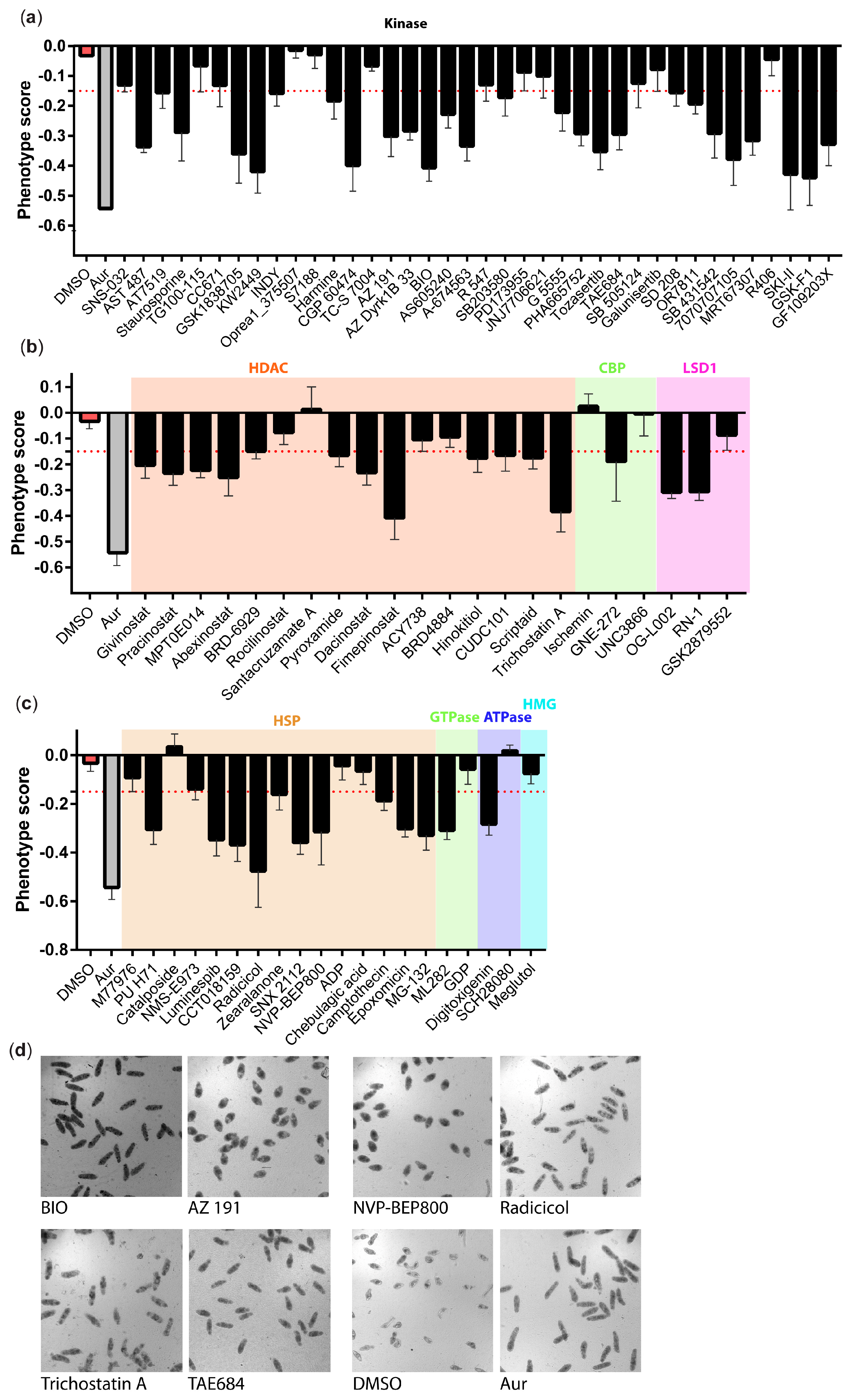
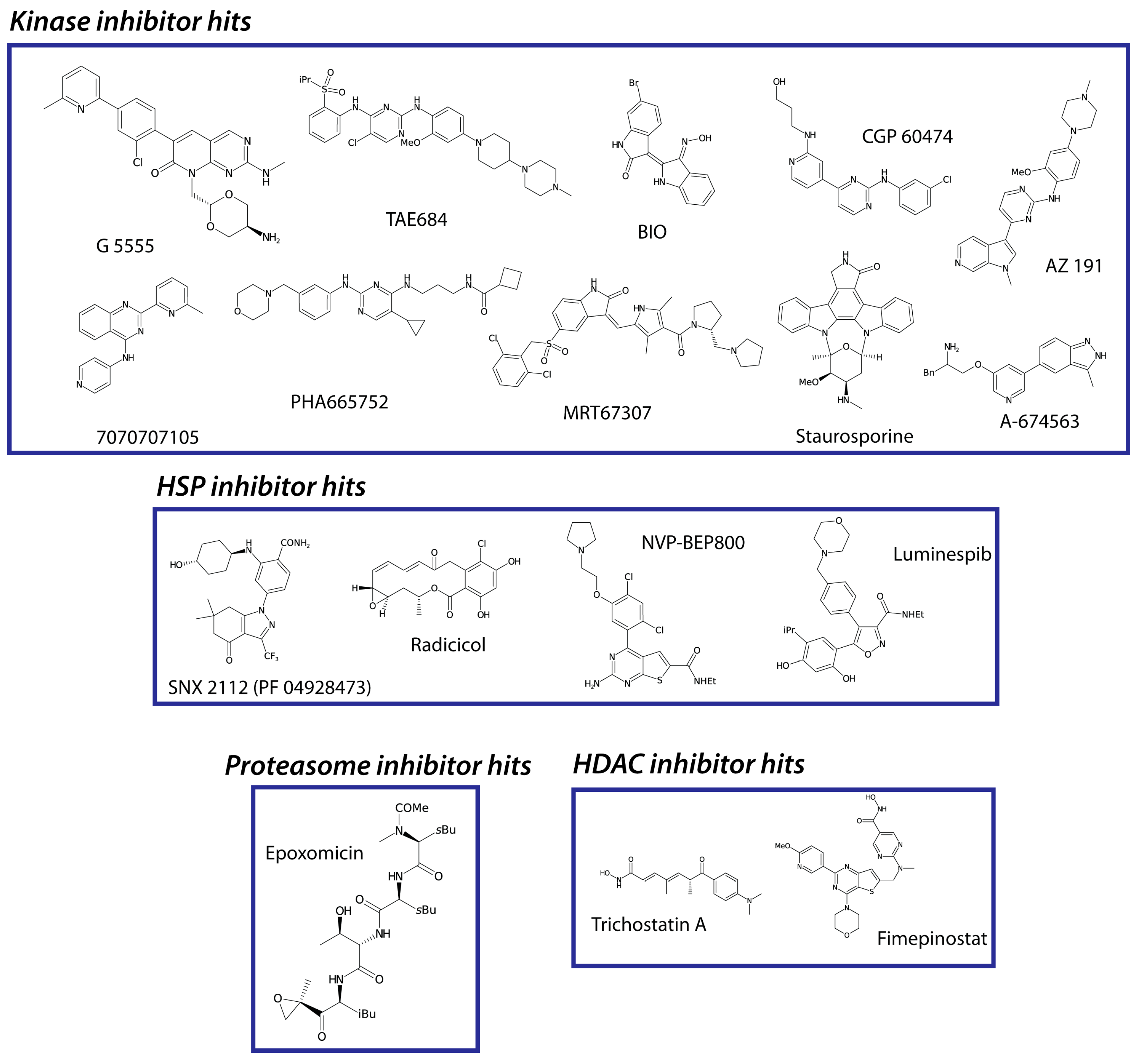
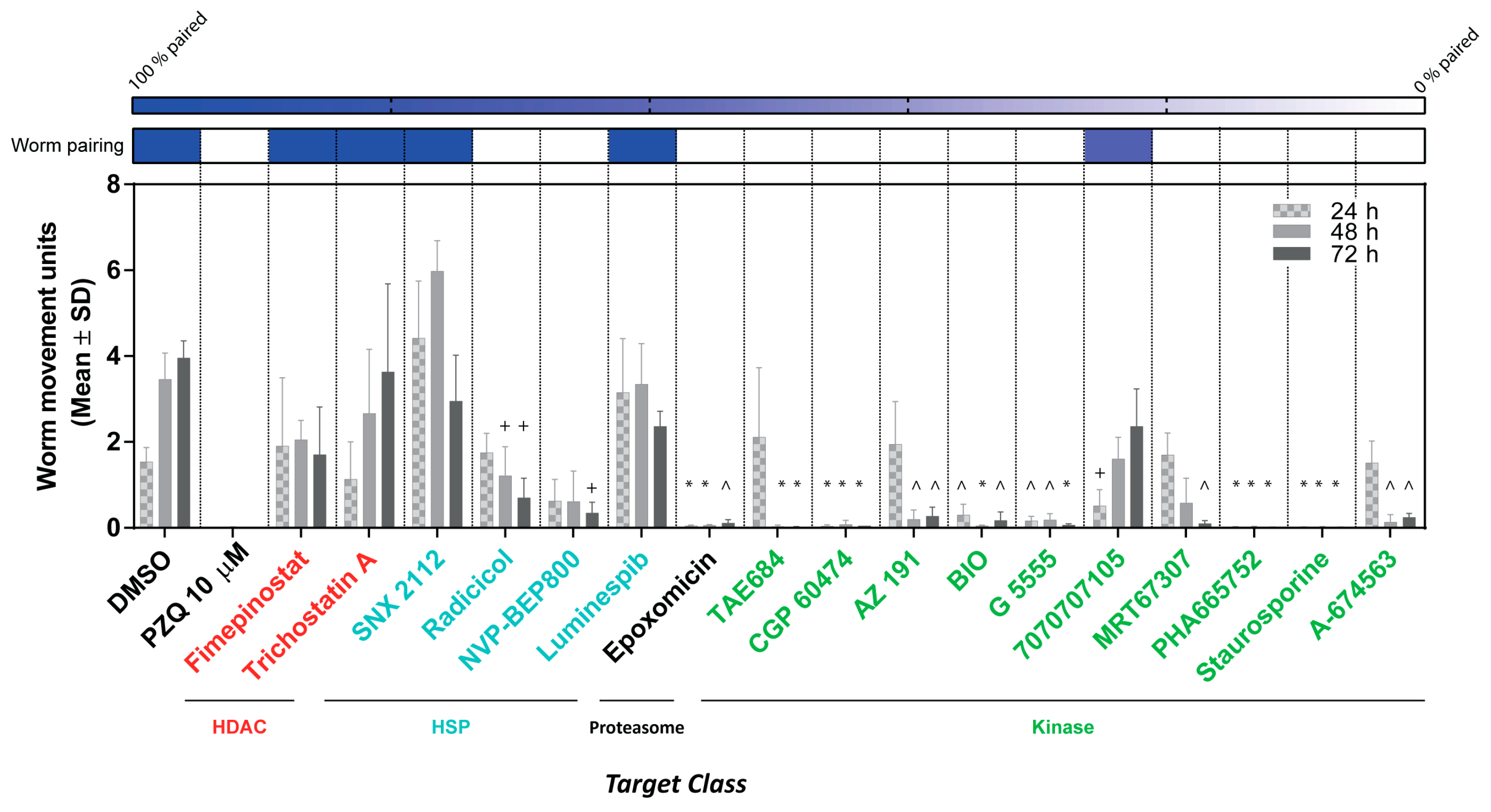
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Health Estimates: Life Expectancy and Leading Causes of Death and Disability. 2023. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 10 January 2023).
- McManus, D.P.; Dunne, D.W.; Sacko, M.; Utzinger, J.; Vennervald, B.J.; Zhou, X.N. Schistosomiasis. Nat. Rev. Dis. Prim. 2018, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Vale, N.; Gouveia, M.J.; Rinaldi, G.; Brindley, P.J.; Gartner, F.; Correia da Costa, J.M. Praziquantel for Schistosomiasis: Single-Drug Metabolism Revisited, Mode of Action, and Resistance. Antimicrob. Agents Chemother. 2017, 61, e02582-16. [Google Scholar] [CrossRef] [PubMed]
- Katz, N.; Coelho, P.M. Clinical therapy of Schistosomiasis mansoni: The Brazilian contribution. Acta Trop. 2008, 108, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Friedrich, L.; Yahya, N.A.; Rohr, C.M.; Chulkov, E.G.; Maillard, D.; Rippmann, F.; Spangenberg, T.; Marchant, J.S. Mechanism of praziquantel action at a parasitic flatworm ion channel. Sci. Transl. Med. 2021, 13, eabj5832. [Google Scholar] [CrossRef] [PubMed]
- Le Clec’h, W.; Chevalier, F.D.; Mattos, A.C.A.; Strickland, A.; Diaz, R.; McDew-White, M.; Rohr, C.M.; Kinung’hi, S.; Allan, F.; Webster, B.L.; et al. Genetic analysis of praziquantel response in schistosome parasites implicates a transient receptor potential channel. Sci. Transl. Med. 2021, 13, eabj9114. [Google Scholar] [CrossRef]
- Valentim, C.L.; Cioli, D.; Chevalier, F.D.; Cao, X.; Taylor, A.B.; Holloway, S.P.; Pica-Mattoccia, L.; Guidi, A.; Basso, A.; Tsai, I.J.; et al. Genetic and molecular basis of drug resistance and species-specific drug action in schistosome parasites. Science 2013, 342, 1385–1389. [Google Scholar] [CrossRef]
- Wu, W.; Wang, W.; Huang, Y.X. New insight into praziquantel against various developmental stages of schistosomes. Parasitol. Res. 2011, 109, 1501–1507. [Google Scholar] [CrossRef]
- Rugel, A.R.; Guzman, M.A.; Taylor, A.B.; Chevalier, F.D.; Tarpley, R.S.; McHardy, S.F.; Cao, X.; Holloway, S.P.; Anderson, T.J.C.; Hart, P.J.; et al. Why does oxamniquine kill Schistosoma mansoni and not S. haematobium and S. japonicum? Int. J. Parasitol. Drugs Drug Resist. 2020, 13, 8–15. [Google Scholar] [CrossRef]
- Striebel, H.P. 4-Isothiocyanato-4′-Nitrodiphenylamine (C 9333-Go/CGP 4540), an Antischistosomal Compound with an Unusual Spectrum of Anthelminthic Activity Against Intestinal Nematodes, Filariae and Trematodes. In Chemotherapy; Adolphe, M., Ed.; Pergamon: Oxford, UK, 1979; pp. 17–26. [Google Scholar] [CrossRef]
- Barreau, M.; Cotrel, C.; Jeanmart, C. Derivatives of 1,2-Dithiole and Anti-Bilharzia Compositions Thereof. U.S. Patent 4,104,386, 1 August 1978. [Google Scholar]
- Baard, A.P.; Sommers, D.K.; Honiball, P.J.; Fourie, E.D.; Du Toit, L.E. Ro 11-3128, a novel benzodiazepine schistosomicide: Results in human schistosomiasis. Curr. Chemother. Infect. Dis. 1980, 2, 1112–1113. [Google Scholar]
- Saeed, M.E.M.; Krishna, S.; Greten, H.J.; Kremsner, P.G.; Efferth, T. Antischistosomal activity of artemisinin derivatives in vivo and in patients. Pharmacol. Res. 2016, 110, 216–226. [Google Scholar] [CrossRef]
- Adenowo, A.F.; Oyinloye, B.E.; Ogunyinka, B.I.; Kappo, A.P. Impact of human schistosomiasis in sub-Saharan Africa. Braz. J. Infect. Dis. 2015, 19, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Bout, D.; Deslèe, D.; Capron, A. Antischistosomal effect of cyclosporin A: Cure and prevention of mouse and rat schistosomiasis mansoni. Infect. Immun. 1986, 52, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.H.; Coelho, P.M.; Costa, J.O.; de Mello, R.T. Activity of 9-acridanone-hydrazone drugs detected at the pre-postural phase, in the experimental schistosomiasis mansoni. Mem. Inst. Oswaldo Cruz 1995, 90, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, F.D.; Le Clec’h, W.; McDew-White, M.; Menon, V.; Guzman, M.A.; Holloway, S.P.; Cao, X.; Taylor, A.B.; Kinung’hi, S.; Gouvras, A.N.; et al. Oxamniquine resistance alleles are widespread in Old World Schistosoma mansoni and predate drug deployment. PLoS Pathog. 2019, 15, e1007881. [Google Scholar] [CrossRef]
- Moreira-Filho, J.T.; Silva, A.C.; Dantas, R.F.; Gomes, B.F.; Souza Neto, L.R.; Brandao-Neto, J.; Owens, R.J.; Furnham, N.; Neves, B.J.; Silva-Junior, F.P.; et al. Schistosomiasis Drug Discovery in the Era of Automation and Artificial Intelligence. Front. Immunol. 2021, 12, 642383. [Google Scholar] [CrossRef]
- Cheuka, P.M. Drug Discovery and Target Identification against Schistosomiasis: A Reality Check on Progress and Future Prospects. Curr. Top. Med. Chem. 2022, 22, 1595–1610. [Google Scholar] [CrossRef]
- Mansour, N.R.; Paveley, R.; Gardner, J.M.F.; Bell, A.S.; Parkinson, T.; Bickle, Q. High Throughput Screening Identifies Novel Lead Compounds with Activity against Larval, Juvenile and Adult Schistosoma mansoni. PLoS Negl. Trop. Dis. 2016, 10, e0004659. [Google Scholar] [CrossRef]
- Gardner, J.M.F.; Mansour, N.R.; Bell, A.S.; Helmby, H.; Bickle, Q. The discovery of a novel series of compounds with single-dose efficacy against juvenile and adult Schistosoma species. PLoS Negl. Trop. Dis. 2021, 15, e0009490. [Google Scholar] [CrossRef]
- Hoffmann, K.F.; Brindley, P.J.; Berriman, M. Halting harmful helminths. Science 2014, 346, 168–169. [Google Scholar] [CrossRef]
- Naidoo, P.; Mkhize-Kwitshana, Z.L. Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9-mediated editing of Schistosoma mansoni genes: Identifying genes for immunologically potent drug and vaccine development. Rev. Soc. Bras. Med. Trop. 2022, 55, e0131. [Google Scholar] [CrossRef]
- Wang, J.; Paz, C.; Padalino, G.; Coghlan, A.; Lu, Z.; Gradinaru, I.; Collins, J.N.R.; Berriman, M.; Hoffmann, K.F.; Collins, J.J. Large-scale RNAi screening uncovers therapeutic targets in the parasite Schistosoma mansoni. Science 2020, 369, 1649–1653. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Kruger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [PubMed]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [PubMed]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards direct deposition of bioassay data. Nucleic Acids Res. 2019, 47, D930–D940. [Google Scholar] [CrossRef]
- International Helminth Genomes Consortium. Comparative genomics of the major parasitic worms. Nat. Genet. 2019, 51, 163–174. [Google Scholar] [CrossRef]
- Lamore, S.D.; Ahlberg, E.; Boyer, S.; Lamb, M.L.; Hortigon-Vinagre, M.P.; Rodriguez, V.; Smith, G.L.; Sagemark, J.; Carlsson, L.; Bates, S.M.; et al. Deconvoluting Kinase Inhibitor Induced Cardiotoxicity. Toxicol. Sci. 2017, 158, 213–226. [Google Scholar] [CrossRef]
- Lynch, J.J., III; Van Vleet, T.R.; Mittelstadt, S.W.; Blomme, E.A.G. Potential functional and pathological side effects related to off-target pharmacological activity. J. Pharmacol. Toxicol. Methods 2017, 87, 108–126. [Google Scholar] [CrossRef]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef]
- Janes, J.; Young, M.E.; Chen, E.; Rogers, N.H.; Burgstaller-Muehlbacher, S.; Hughes, L.D.; Love, M.S.; Hull, M.V.; Kuhen, K.L.; Woods, A.K.; et al. The ReFRAME library as a comprehensive drug repurposing library and its application to the treatment of cryptosporidiosis. Proc. Natl. Acad. Sci. USA 2018, 115, 10750–10755. [Google Scholar] [CrossRef]
- Whatley, K.C.L.; Padalino, G.; Whiteland, H.; Geyer, K.K.; Hulme, B.J.; Chalmers, I.W.; Forde-Thomas, J.; Ferla, S.; Brancale, A.; Hoffmann, K.F. The repositioning of epigenetic probes/inhibitors identifies new anti-schistosomal lead compounds and chemotherapeutic targets. PLoS Negl. Trop. Dis. 2019, 13, e0007693. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, A.; Padalino, G.; O’Boyle, N.M.; Hoffmann, K.F.; Berriman, M. Identification of anti-schistosomal, anthelmintic and anti-parasitic compounds curated and text-mined from the scientific literature. Wellcome Open Res. 2022, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Sander, T.; Freyss, J.; von Korff, M.; Rufener, C. DataWarrior: An open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015, 55, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, Y.; Berriman, M. A web portal for gene expression across all life stages of Schistosoma mansoni. bioRxiv 2018. [Google Scholar] [CrossRef]
- Anderson, L.; Amaral, M.S.; Beckedorff, F.; Silva, L.F.; Dazzani, B.; Oliveira, K.C.; Almeida, G.T.; Gomes, M.R.; Pires, D.S.; Setubal, J.C. Schistosoma mansoni egg, adult male and female comparative gene expression analysis and identification of novel genes by RNA-Seq. PLoS Negl. Trop. Dis. 2015, 9, e0004334. [Google Scholar] [CrossRef]
- Wang, B.; Collins, J.J., III; Newmark, P.A. Functional genomic characterization of neoblast-like stem cells in larval Schistosoma mansoni. eLife 2013, 2, e00768. [Google Scholar] [CrossRef]
- Protasio, A.V.; Tsai, I.J.; Babbage, A.; Nichol, S.; Hunt, M.; Aslett, M.A.; De Silva, N.; Velarde, G.S.; Anderson, T.J.C.; Clark, R.C.; et al. A Systematically Improved High Quality Genome and Transcriptome of the Human Blood Fluke Schistosoma mansoni. PLoS Negl. Trop. Dis. 2012, 6, e1455. [Google Scholar] [CrossRef]
- Protasio, A.V.; van Dongen, S.; Collins, J.; Quintais, L.; Ribeiro, D.M.; Sessler, F.; Hunt, M.; Rinaldi, G.; Collins, J.J.; Enright, A.J.; et al. MiR-277/4989 regulate transcriptional landscape during juvenile to adult transition in the parasitic helminth Schistosoma mansoni. PLoS Negl. Trop. Dis. 2017, 11, e0005559. [Google Scholar] [CrossRef]
- Lu, Z.; Sessler, F.; Holroyd, N.; Hahnel, S.; Quack, T.; Berriman, M.; Grevelding, C.G. Schistosome sex matters: A deep view into gonad-specific and pairing-dependent transcriptomes reveals a complex gender interplay. Sci. Rep. 2016, 6, 31150. [Google Scholar] [CrossRef]
- Lu, Z.; Sessler, F.; Holroyd, N.; Hahnel, S.; Quack, T.; Berriman, M.; Grevelding, C.G. A gene expression atlas of adult Schistosoma mansoni and their gonads. Sci. Data 2017, 4, 170118. [Google Scholar] [CrossRef] [PubMed]
- Geyer, K.K.; Niazi, U.H.; Duval, D.; Cosseau, C.; Tomlinson, C.; Chalmers, I.W.; Swain, M.T.; Cutress, D.J.; Bickham-Wright, U.; Munshi, S.E.; et al. The Biomphalaria glabrata DNA methylation machinery displays spatial tissue expression, is differentially active in distinct snail populations and is modulated by interactions with Schistosoma mansoni. PLOS Negl. Trop. Dis. 2017, 11, e0005246. [Google Scholar] [CrossRef] [PubMed]
- Colley, D.G.; Wikel, S.K. Schistosoma mansoni: Simplified method for the production of schistosomules. Exp. Parasitol. 1974, 35, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Smithers, S.; Terry, R. The infection of laboratory hosts with cercariae of Schistosoma mansoni and the recovery of the adult worms. Parasitology 1965, 55, 695–700. [Google Scholar] [CrossRef]
- Craven, H.M.; Bonsignore, R.; Lenis, V.; Santi, N.; Berrar, D.; Swain, M.; Whiteland, H.; Casini, A.; Hoffmann, K.F. Identifying and validating the presence of Guanine-Quadruplexes (G4) within the blood fluke parasite Schistosoma mansoni. PLoS Negl. Trop. Dis. 2021, 15, e0008770. [Google Scholar] [CrossRef] [PubMed]
- Padalino, G.; El-Sakkary, N.; Liu, L.J.; Liu, C.; Harte, D.S.G.; Barnes, R.E.; Sayers, E.; Forde-Thomas, J.; Whiteland, H.; Bassetto, M.; et al. Anti-schistosomal activities of quinoxaline-containing compounds: From hit identification to lead optimisation. Eur. J. Med. Chem. 2021, 226, 113823. [Google Scholar] [CrossRef] [PubMed]
- Whiteland, H.; Crusco, A.; Bloemberg, L.W.; Tibble-Howlings, J.; Forde-Thomas, J.; Coghlan, A.; Murphy, P.J.; Hoffmann, K.F. Quorum sensing N-Acyl homoserine lactones are a new class of anti-schistosomal. PLoS Negl. Trop. Dis. 2020, 14, e0008630. [Google Scholar] [CrossRef]
- Crusco, A.; Whiteland, H.; Baptista, R.; Forde-Thomas, J.E.; Beckmann, M.; Mur, L.A.J.; Nash, R.J.; Westwell, A.D.; Hoffmann, K.F. Antischistosomal Properties of Sclareol and Its Heck-Coupled Derivatives: Design, Synthesis, Biological Evaluation, and Untargeted Metabolomics. ACS Infect. Dis. 2019, 5, 1188–1199. [Google Scholar] [CrossRef]
- Paveley, R.; Mansour, N.; Hallyburton, I.; Bleicher, L.; Alex, E.; Mikic, I.; Guidi, A.; Gilbert, I.H.; Hopkins, A.L.; Bickle, Q.D. Whole Organism High-Content Screening by Label-Free, Image-Based Bayesian Classification for Parasitic Diseases. PLoS Negl. Trop. Dis. 2012, 6, e1762. [Google Scholar] [CrossRef]
- Padalino, G. WormassayGP2. Zenodo 2020. [Google Scholar] [CrossRef]
- Marcellino, C.; Gut, J.; Lim, K.C.; Singh, R.; McKerrow, J.; Sakanari, J. WormAssay: A Novel Computer Application for Whole-Plate Motion-based Screening of Macroscopic Parasites. PLoS Negl. Trop. Dis. 2012, 6, e1494. [Google Scholar] [CrossRef] [PubMed]
- Basch, P.F. Cultivation of Schistosoma mansoni In vitro. II. Production of Infertile Eggs by Worm Pairs Cultured from Cercariae. J. Parasitol. 1981, 67, 186–190. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Collins, J.J., III. Systematically improved in vitro culture conditions reveal new insights into the reproductive biology of the human parasite Schistosoma mansoni. PLoS Biol. 2019, 17, e3000254. [Google Scholar] [CrossRef]
- Waisberg, M.; Lobo, F.P.; Cerqueira, G.C.; Passos, L.K.J.; Carvalho, O.S.; Franco, G.R.; El-Sayed, N.M. Microarray analysis of gene expression induced by sexual contact in Schistosoma mansoni. BMC Genom. 2007, 8, 181. [Google Scholar] [CrossRef]
- Chen, D.; Hou, L.; Wei, J.; Guo, S.; Cui, W.; Yang, P.; Kang, L.; Wang, X. Aggregation pheromone 4-vinylanisole promotes the synchrony of sexual maturation in female locusts. eLife 2022, 11, e74581. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Santos, G.B.; Ganesan, A.; Emery, F.S. Oral Administration of Peptide-Based Drugs: Beyond Lipinski’s Rule. ChemMedChem 2016, 11, 2245–2251. [Google Scholar] [CrossRef]
- Schaftenaar, G.; de Vlieg, J. Quantum mechanical polar surface area. J. Comput.-Aided Mol. Des. 2012, 26, 311–318. [Google Scholar] [CrossRef]
- Mugumbate, G.; Overington, J.P. The relationship between target-class and the physicochemical properties of antibacterial drugs. Bioorg. Med. Chem. 2015, 23, 5218–5224. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Berriman, M.; Coghlan, A.; Mutowo, P.; O’Boyle, N.; Lomax, J.; Leach, A.R. Creating a screening set of potential anthelmintic compounds using ChEMBL. Protoc. Exch. 2018. [Google Scholar] [CrossRef]
- Park, S.; Park, J.A.; Jeon, J.H.; Lee, Y. Traditional and Novel Mechanisms of Heat Shock Protein 90 (HSP90) Inhibition in Cancer Chemotherapy Including HSP90 Cleavage. Biomol. Ther. 2019, 27, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.; Carter, T.R.; Cohen, M.S.; Blagg, B.S.J. Old and New Approaches to Target the Hsp90 Chaperone. Curr. Cancer Drug Targets 2020, 20, 253–270. [Google Scholar] [CrossRef]
- Massey, A.J.; Schoepfer, J.; Brough, P.A.; Brueggen, J.; Chène, P.; Drysdale, M.J.; Pfaar, U.; Radimerski, T.; Ruetz, S.; Schweitzer, A.; et al. Preclinical antitumor activity of the orally available heat shock protein 90 inhibitor NVP-BEP800. Mol. Cancer Ther. 2010, 9, 906–919. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Liu, Q.; Zeng, Q.; Wu, P.; Yu, Q.; Gu, K.; Xue, J.; Wei, X. Radicicol, a Novel Lead Compound against the Migratory-Stage Schistosomula of Schistosoma japonicum. Antimicrob. Agents Chemother. 2021, 65, e01781-20. [Google Scholar] [CrossRef] [PubMed]
- Gillan, V.; O’Neill, K.; Maitland, K.; Sverdrup, F.M.; Devaney, E. A repurposing strategy for Hsp90 inhibitors demonstrates their potency against filarial nematodes. PLoS Negl. Trop. Dis. 2014, 8, e2699. [Google Scholar] [CrossRef]
- Xu, Z.; Ji, M.; Li, C.; Du, X.; Hu, W.; McManus, D.P.; You, H. A Biological and Immunological Characterization of Schistosoma japonicum Heat Shock Proteins 40 and 90α. Int. J. Mol. Sci. 2020, 21, 4034. [Google Scholar] [CrossRef]
- Guerra-Sá, R.; Castro-Borges, W.; Evangelista, E.A.; Kettelhut, I.C.; Rodrigues, V. Schistosoma mansoni: Functional proteasomes are required for development in the vertebrate host. Exp. Parasitol. 2005, 109, 228–236. [Google Scholar] [CrossRef]
- Morais, E.R.; Oliveira, K.C.; Paula, R.G.d.; Ornelas, A.M.M.; Moreira, É.B.C.; Badoco, F.R.; Magalhães, L.G.; Verjovski-Almeida, S.; Rodrigues, V. Effects of proteasome inhibitor MG-132 on the parasite Schistosoma mansoni. PLoS ONE 2017, 12, e0184192. [Google Scholar] [CrossRef]
- Bibo-Verdugo, B.; Wang, S.C.; Almaliti, J.; Ta, A.P.; Jiang, Z.; Wong, D.A.; Lietz, C.B.; Suzuki, B.M.; El-Sakkary, N.; Hook, V.; et al. The Proteasome as a Drug Target in the Metazoan Pathogen, Schistosoma mansoni. ACS Infect. Dis. 2019, 5, 1802–1812. [Google Scholar] [CrossRef]
- Lindsten, K.; Menéndez-Benito, V.; Masucci, M.G.; Dantuma, N.P. A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 2003, 21, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Arimondo, P.; Rots, M.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenet. 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Cosseau, C.; Grunau, C. Schistosoma mansoni: Developmental arrest of miracidia treated with histone deacetylase inhibitors. Exp. Parasitol. 2009, 121, 288–291. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dubois, F.; Caby, S.; Oger, F.; Cosseau, C.; Capron, M.; Grunau, C.; Dissous, C.; Pierce, R.J. Histone deacetylase inhibitors induce apoptosis, histone hyperacetylation and up-regulation of gene transcription in Schistosoma mansoni. Mol. Biochem. Parasitol. 2009, 168, 7–15. [Google Scholar] [CrossRef]
- Zagni, C.; Citarella, A.; Oussama, M.; Rescifina, A.; Maugeri, A.; Navarra, M.; Scala, A.; Piperno, A.; Micale, N. Hydroxamic Acid-Based Histone Deacetylase (HDAC) Inhibitors Bearing a Pyrazole Scaffold and a Cinnamoyl Linker. Int. J. Mol. Sci. 2019, 20, 945. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, H.; Lirussi, F.; Garrido, C.; Ye, X.Y.; Xie, T. Dual inhibitors of histone deacetylases and other cancer-related targets: A pharmacological perspective. Biochem. Pharm. 2020, 182, 114224. [Google Scholar] [CrossRef]
- Neves, B.J.; Braga, R.C.; Bezerra, J.C.; Cravo, P.V.; Andrade, C.H. In silico repositioning-chemogenomics strategy identifies new drugs with potential activity against multiple life stages of Schistosoma mansoni. PLoS Negl. Trop. Dis. 2015, 9, e3435. [Google Scholar] [CrossRef]
- Chua, M.J.; Arnold, M.S.J.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.P.; Skinner-Adams, T.S.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef]
- Citarella, A.; Moi, D.; Pinzi, L.; Bonanni, D.; Rastelli, G. Hydroxamic Acid Derivatives: From Synthetic Strategies to Medicinal Chemistry Applications. ACS Omega 2021, 6, 21843–21849. [Google Scholar] [CrossRef]
- Ononye, S.N.; VanHeyst, M.D.; Oblak, E.Z.; Zhou, W.; Ammar, M.; Anderson, A.C.; Wright, D.L. Tropolones as lead-like natural products: The development of potent and selective histone deacetylase inhibitors. ACS Med. Chem. Lett. 2013, 4, 757–761. [Google Scholar] [CrossRef]
- Chisty, M.M.; Nargis, M.; Inaba, T.; Ishita, K.; Osanai, A.; Kamiya, H. Transmission Electron Microscopy of Schistosoma mansoni Cercariae Treated with Hinokitiol β-thujaplicin), a Compound for Potential Skin Application against Cercarial Penetration. Tohoku J. Exp. Med. 2004, 202, 63–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saccoccia, F.; Brindisi, M.; Gimmelli, R.; Relitti, N.; Guidi, A.; Saraswati, A.P.; Cavella, C.; Brogi, S.; Chemi, G.; Butini, S.; et al. Screening and Phenotypical Characterization of Schistosoma mansoni Histone Deacetylase 8 (SmHDAC8) Inhibitors as Multistage Antischistosomal Agents. ACS Infect. Dis. 2020, 6, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Marek, M.; Oliveira, G.; Pierce, R.J.; Jung, M.; Sippl, W.; Romier, C. Drugging the schistosome zinc-dependent HDACs: Current progress and future perspectives. Future Med. Chem. 2015, 7, 783–800. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2023 update. Pharm. Res 2023, 187, 106552. [Google Scholar] [CrossRef]
- Pereira Moreira, B.; Weber, M.H.W.; Haeberlein, S.; Mokosch, A.S.; Spengler, B.; Grevelding, C.G.; Falcone, F.H. Drug Repurposing and De Novo Drug Discovery of Protein Kinase Inhibitors as New Drugs against Schistosomiasis. Molecules 2022, 27, 1414. [Google Scholar] [CrossRef]
- Moreira, B.P.; Batista, I.C.A.; Tavares, N.C.; Armstrong, T.; Gava, S.G.; Torres, G.P.; Mourão, M.M.; Falcone, F.H. Docking-Based Virtual Screening Enables Prioritizing Protein Kinase Inhibitors with In Vitro Phenotypic Activity Against Schistosoma mansoni. Front. Cell. Infect. Microbiol. 2022, 12, 884. [Google Scholar] [CrossRef]
- Hasan, M.; Yan, N. Therapeutic potential of targeting TBK1 in autoimmune diseases and interferonopathies. Pharm. Res. 2016, 111, 336–342. [Google Scholar] [CrossRef]
- Cowan, N.; Keiser, J. Repurposing of anticancer drugs: In vitro and in vivo activities against Schistosoma mansoni. Parasites Vectors 2015, 8, 417. [Google Scholar] [CrossRef]
- Ashford, A.; Oxley, D.; Kettle, J.; Hudson, K.; Guichard, S.; Cook, S.; Lochhead, P. A novel DYRK1B inhibitor AZ191 demonstrates that DYRK1B acts independently of GSK3β to phosphorylate cyclin D1 at Thr 286, not Thr 288. Biochem. J. 2013, 457, 43–56. [Google Scholar] [CrossRef]
- Knox, J.; Joly, N.; Linossi, E.M.; Carmona-Negrón, J.A.; Jura, N.; Pintard, L.; Zuercher, W.; Roy, P.J. A survey of the Kinome pharmacopeia reveals multiple scaffolds and targets for the development of novel anthelmintics. Sci. Rep. 2021, 11, 9161. [Google Scholar] [CrossRef]
- Ferraris, D.; Yang, Z.; Welsbie, D. Dual leucine zipper kinase as a therapeutic target for neurodegenerative conditions. Future Med. Chem. 2013, 5, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Nawaratna, S.S.K.; McManus, D.P.; Gasser, R.B.; Brindley, P.J.; Boyle, G.M.; Rivera, V.; Ranasinghe, S.L.; Jones, M.K.; You, H.; Gobert, G.N. Use of kinase inhibitors against schistosomes to improve and broaden praziquantel efficacy. Parasitology 2020, 147, 1488–1498. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Suzuki, B.M.; Dohrmann, J.; Singh, R.; Arkin, M.R.; Caffrey, C.R. A multi-dimensional, time-lapse, high content screening platform applied to schistosomiasis drug discovery. Commun. Biol. 2020, 3, 747. [Google Scholar] [CrossRef] [PubMed]
- Ressurreição, M.; De Saram, P.; Kirk, R.S.; Rollinson, D.; Emery, A.M.; Page, N.M.; Davies, A.J.; Walker, A.J. Protein kinase C and extracellular signal-regulated kinase regulate movement, attachment, pairing and egg release in Schistosoma mansoni. PLoS Negl. Trop. Dis. 2014, 8, e2924. [Google Scholar] [CrossRef]
- Ndubaku, C.O.; Crawford, J.J.; Drobnick, J.; Aliagas, I.; Campbell, D.; Dong, P.; Dornan, L.M.; Duron, S.; Epler, J.; Gazzard, L.; et al. Design of Selective PAK1 Inhibitor G-5555: Improving Properties by Employing an Unorthodox Low-pK a Polar Moiety. ACS Med. Chem. Lett. 2015, 6, 1241–1246. [Google Scholar] [CrossRef]
- Chorner, P.M.; Moorehead, R.A. A-674563, a putative AKT1 inhibitor that also suppresses CDK2 activity, inhibits human NSCLC cell growth more effectively than the pan-AKT inhibitor, MK-2206. PLoS ONE 2018, 13, e0193344. [Google Scholar] [CrossRef]
- Ressurreição, M.; Rollinson, D.; Emery, A.M.; Walker, A.J. A role for p38 mitogen-activated protein kinase in early post-embryonic development of Schistosoma mansoni. Mol. Biochem. Parasitol. 2011, 180, 51–55. [Google Scholar] [CrossRef]
- Ressurreição, M.; Rollinson, D.; Emery, A.M.; Walker, A.J. A role for p38 MAPK in the regulation of ciliary motion in a eukaryote. BMC Cell Biol. 2011, 12, 6. [Google Scholar] [CrossRef]
- Knobloch, J.; Beckmann, S.; Burmeister, C.; Quack, T.; Grevelding, C.G. Tyrosine kinase and cooperative TGFβ signaling in the reproductive organs of Schistosoma mansoni. Exp. Parasitol. 2007, 117, 318–336. [Google Scholar] [CrossRef]
- Buro, C.; Oliveira, K.C.; Lu, Z.; Leutner, S.; Beckmann, S.; Dissous, C.; Cailliau, K.; Verjovski-Almeida, S.; Grevelding, C.G. Transcriptome analyses of inhibitor-treated schistosome females provide evidence for cooperating Src-kinase and TGFβ receptor pathways controlling mitosis and eggshell formation. PLoS Pathog. 2013, 9, e1003448. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EC50 (µM) 1 | |||||
|---|---|---|---|---|---|
| Target Class | Compound Name | Phenotype | 95% CI | Motility | 95% CI |
| HDAC | Fimepinostat | 0.447 | (0.350 to 0.543) | 0.766 | (0.256 to 1.275) |
| Trichostatin A | 0.448 | (0.177 to 0.719) | 0.667 | (0.416 to 0.918) | |
| HSP | SNX 2112 | 1.799 | (1.156 to 2.443) | 2.084 | (1.732 to 2.436) |
| Radicicol | 2.167 | (1.057 to 3.277) | 1.489 | (0.871 to 2.106) | |
| NVP-BEP800 | 0.541 | (0.154 to 0.929) | 0.552 | (0.425 to 0.678) | |
| Luminespib | 0.197 | (0.158 to 0.235) | 0.191 | (0.152 to 0.230) | |
| Proteasome | Epoxomicin | 0.239 | (0.223 to 0.255) | 0.212 | (0.176 to 0.248) |
| Kinase | TAE684 | 2.886 | (2.615 to 3.156) | 1.911 | (1.801 to 2.011) |
| CGP 60474 | 0.301 | (0.270 to 0.331) | 0.518 | (0.389 to 0.648) | |
| AZ 191 | 4.628 | (4.242 to 5.014) | 2.177 | (1.858 to 2.496) | |
| BIO | 2.518 | (1.618 to 3.419) | 2.163 | (1.847 to 2.480) | |
| G 5555 | 4.433 | (3.074 to 5.792) | 1.238 | (0.406 to 2.071) | |
| 7070707105 | 2.570 | (1.598 to 3.542) | 2.667 | (2.001 to 3.333) | |
| MRT67307 | 4.736 | (3.973 to 5.499) | 1.532 | (1.224 to 1.840) | |
| PHA665752 | 5.682 | (5.472 to 5.892) | 3.345 | (2.806 to 3.884) | |
| Staurosporine | 0.218 | (0.163 to 0.273) | 0.187 | (0.154 to 0.221) | |
| A-674563 | 4.124 | (3.564 to 4.683) | 1.732 | (1.127 to 2.337) | |
| Cell Line (CC50, nM) | |||||
|---|---|---|---|---|---|
| Compound name | K562 | HCT116 | A549 | MCF7 | Huh-7 |
| BIO | 1300 | 5200 | - | - | 6200 |
| A-674563 | - | - | - | - | - |
| AZ 191 | - | - | - | - | - |
| CGP 60474 | - | - | - | - | - |
| Epoxomicin | - | - | - | - | - |
| Fimepinostat | 280 * | 5 * | - | 41 * | 560 * |
| G 5555 | - | - | >1000 | - | - |
| Luminespib | 8.7 | 121/16 | 39/60 | 6.4/3.35 | - |
| NVP-BEP800 | - | - | - | - | - |
| PHA665752 | - | - | - | - | - |
| Radicicol | - | - | 100 | 30/47.7/23/30 | - |
| SNX 2112 | 6 | - | - | 53 | - |
| Staurosporine | 40/80/100/76.8 | 52/37/28/48 | 7470/9500 (1) 13,220 (2) | 8810/520/64 | 230 |
| TAE684 | - | - | - | - | - |
| Trichostatin A | 410/120/160 | 800/900 | 20/160/80/160/50 | 60/60/100 | 60 |
| CC50 (nM) | |||
|---|---|---|---|
| Cell line | CGP 60474 | PHA665752 | TAE684 |
| COLO-800 | 940.23 | 27,169.99 | 766.95 |
| COLO-684 | 94.89 | - | 11,691.29 |
| COLO-824 | 354.71 | - | 10,866.86 |
| COLO-829 | 4176.22 | - | 28,459.47 |
| COLO-320-HSR | 82.86 | - | 197.76 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padalino, G.; Coghlan, A.; Pagliuca, G.; Forde-Thomas, J.E.; Berriman, M.; Hoffmann, K.F. Using ChEMBL to Complement Schistosome Drug Discovery. Pharmaceutics 2023, 15, 1359. https://doi.org/10.3390/pharmaceutics15051359
Padalino G, Coghlan A, Pagliuca G, Forde-Thomas JE, Berriman M, Hoffmann KF. Using ChEMBL to Complement Schistosome Drug Discovery. Pharmaceutics. 2023; 15(5):1359. https://doi.org/10.3390/pharmaceutics15051359
Chicago/Turabian StylePadalino, Gilda, Avril Coghlan, Giampaolo Pagliuca, Josephine E. Forde-Thomas, Matthew Berriman, and Karl F. Hoffmann. 2023. "Using ChEMBL to Complement Schistosome Drug Discovery" Pharmaceutics 15, no. 5: 1359. https://doi.org/10.3390/pharmaceutics15051359
APA StylePadalino, G., Coghlan, A., Pagliuca, G., Forde-Thomas, J. E., Berriman, M., & Hoffmann, K. F. (2023). Using ChEMBL to Complement Schistosome Drug Discovery. Pharmaceutics, 15(5), 1359. https://doi.org/10.3390/pharmaceutics15051359