
Challenges in Permeability Assessment for Oral Drug Product Development

 ,
,  ,
,  , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
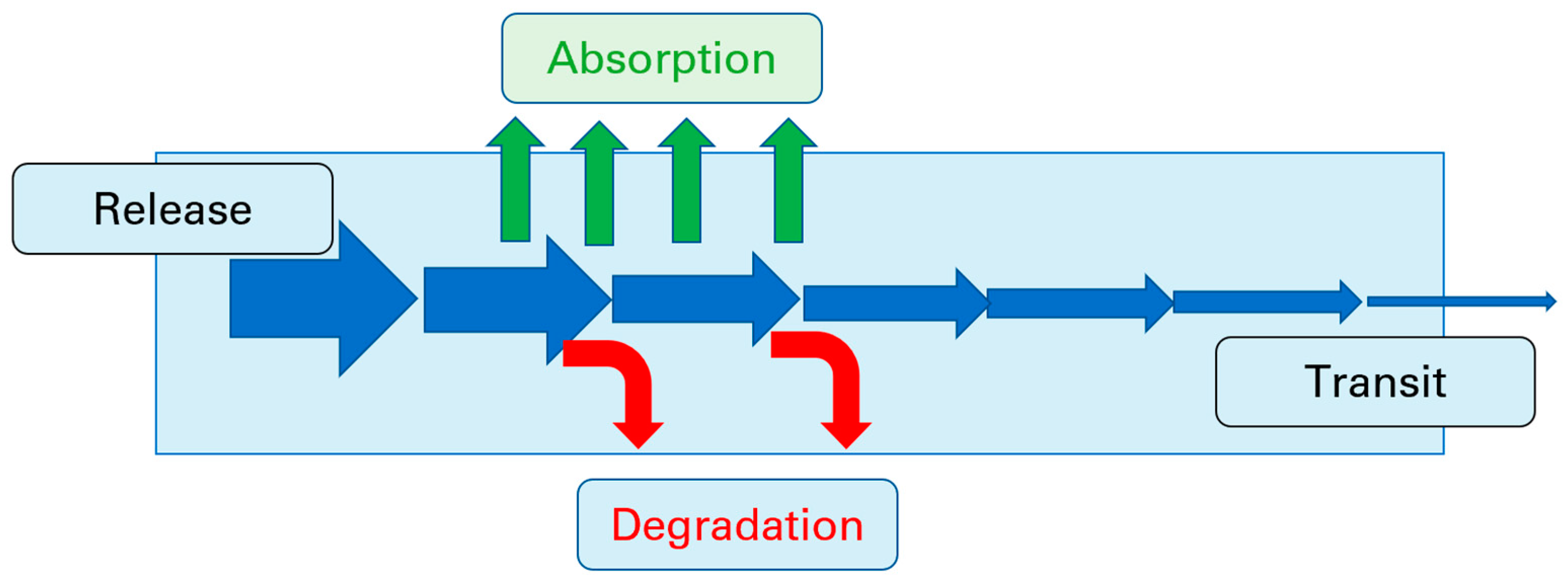
:1. Introduction
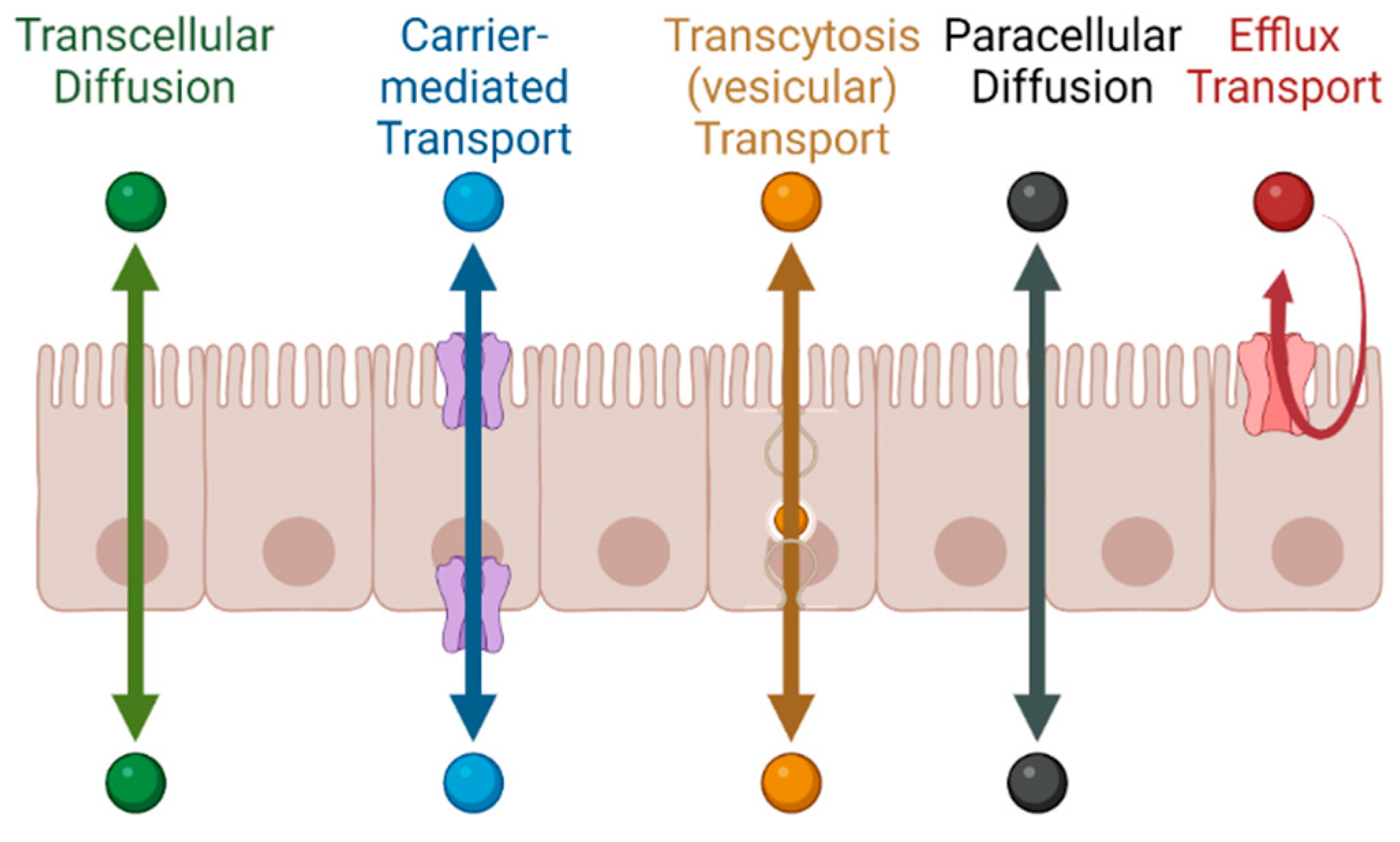
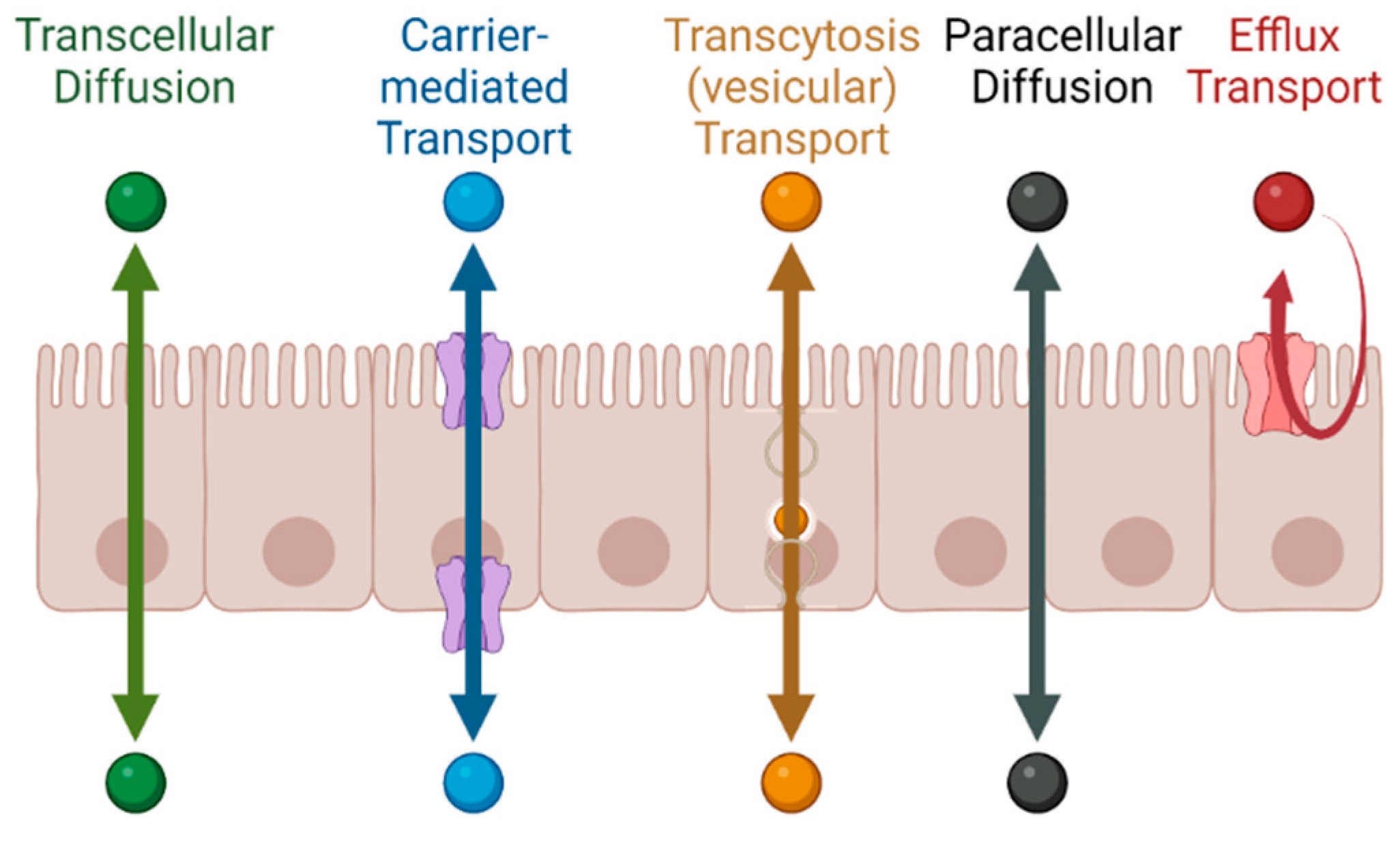
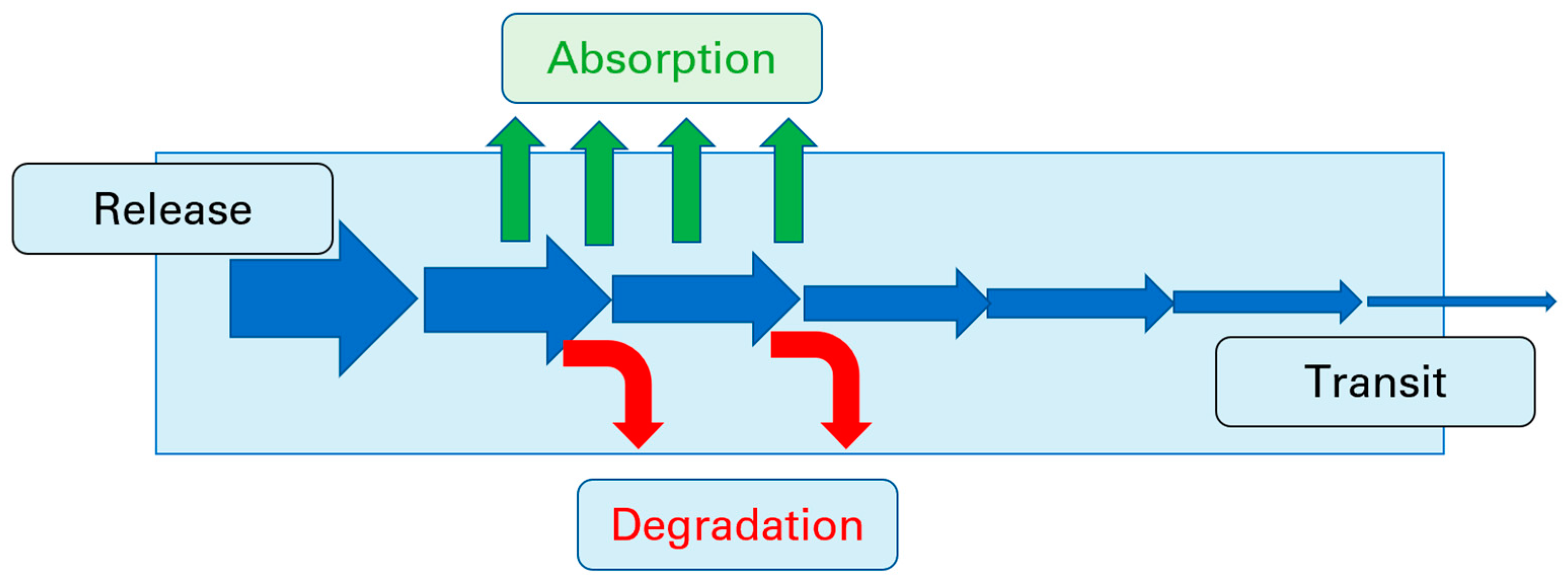
1.1. Permeability across Cellular Barriers
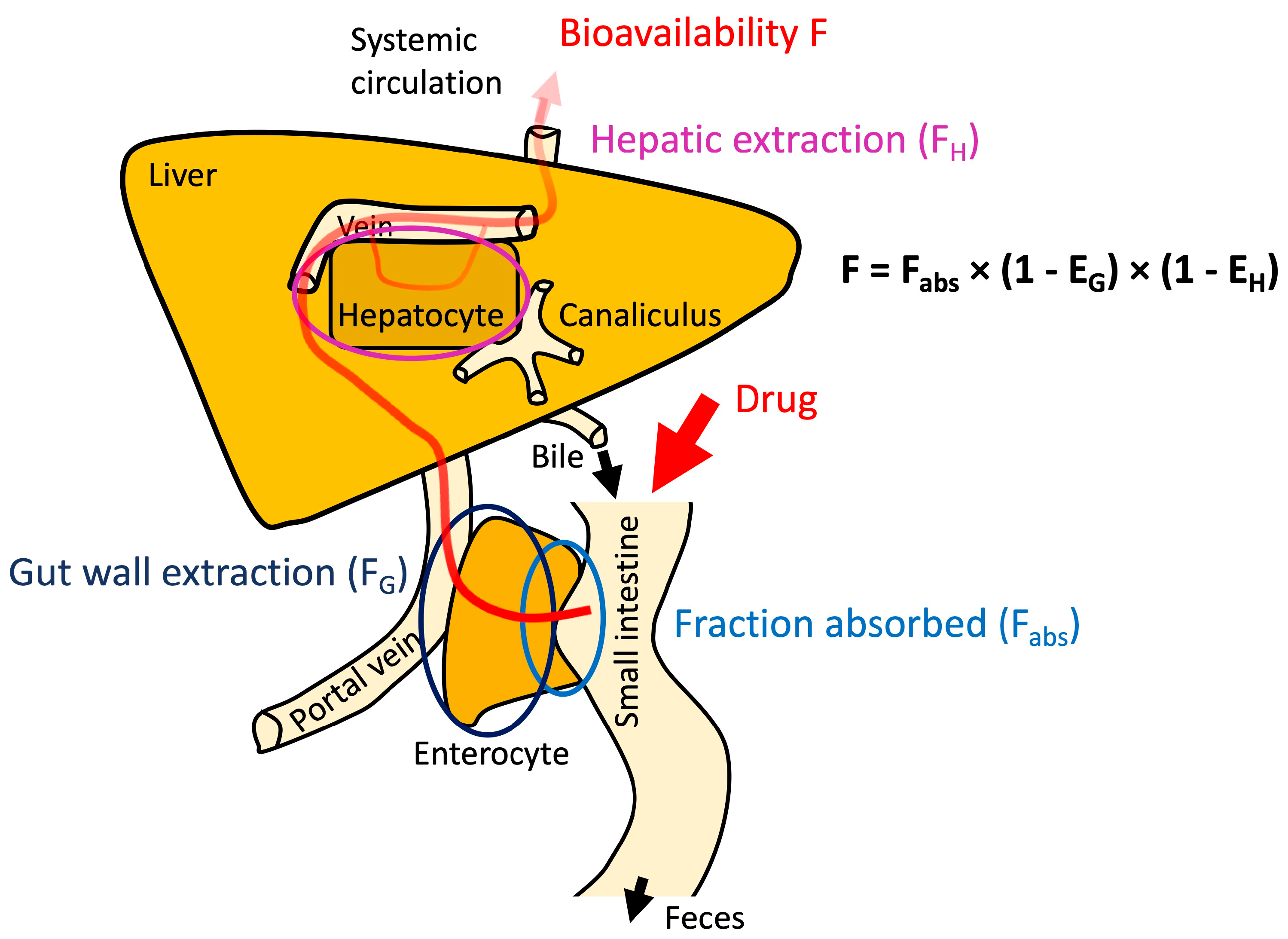
1.2. The Role of Permeability in Pharmaceutical Development
2. Permeability in Drug Discovery and Preclinical Development
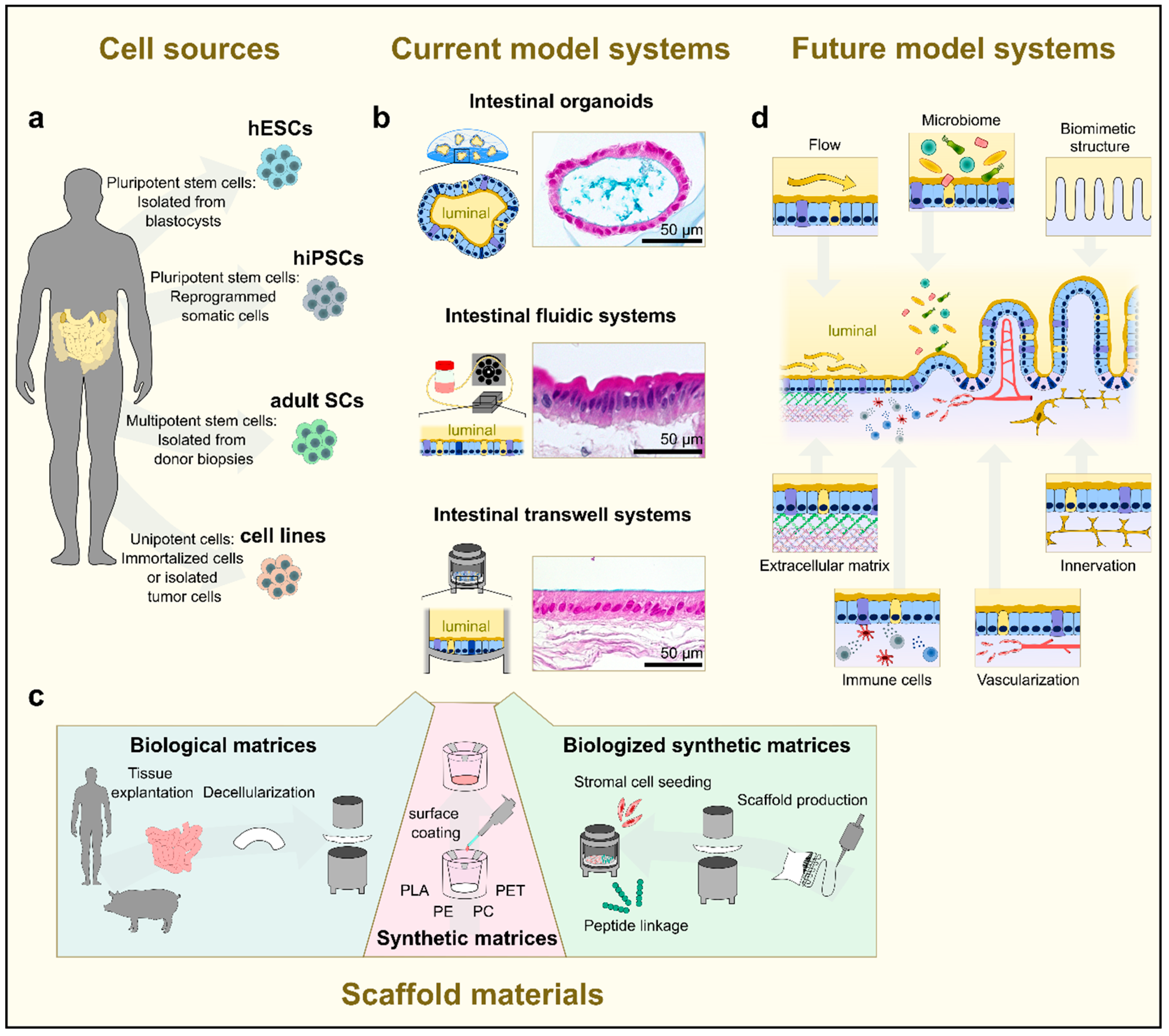
2.1. In Vitro Models
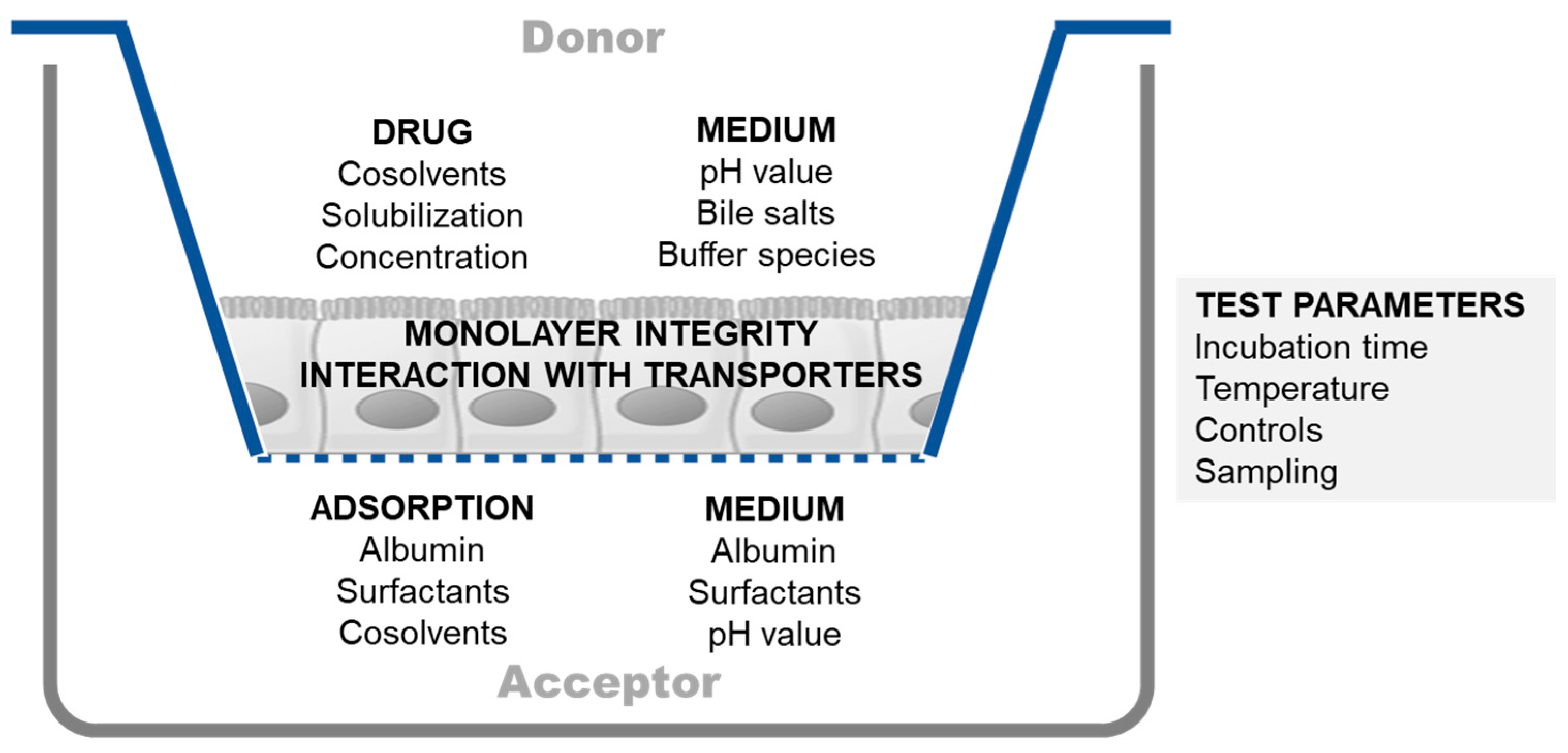
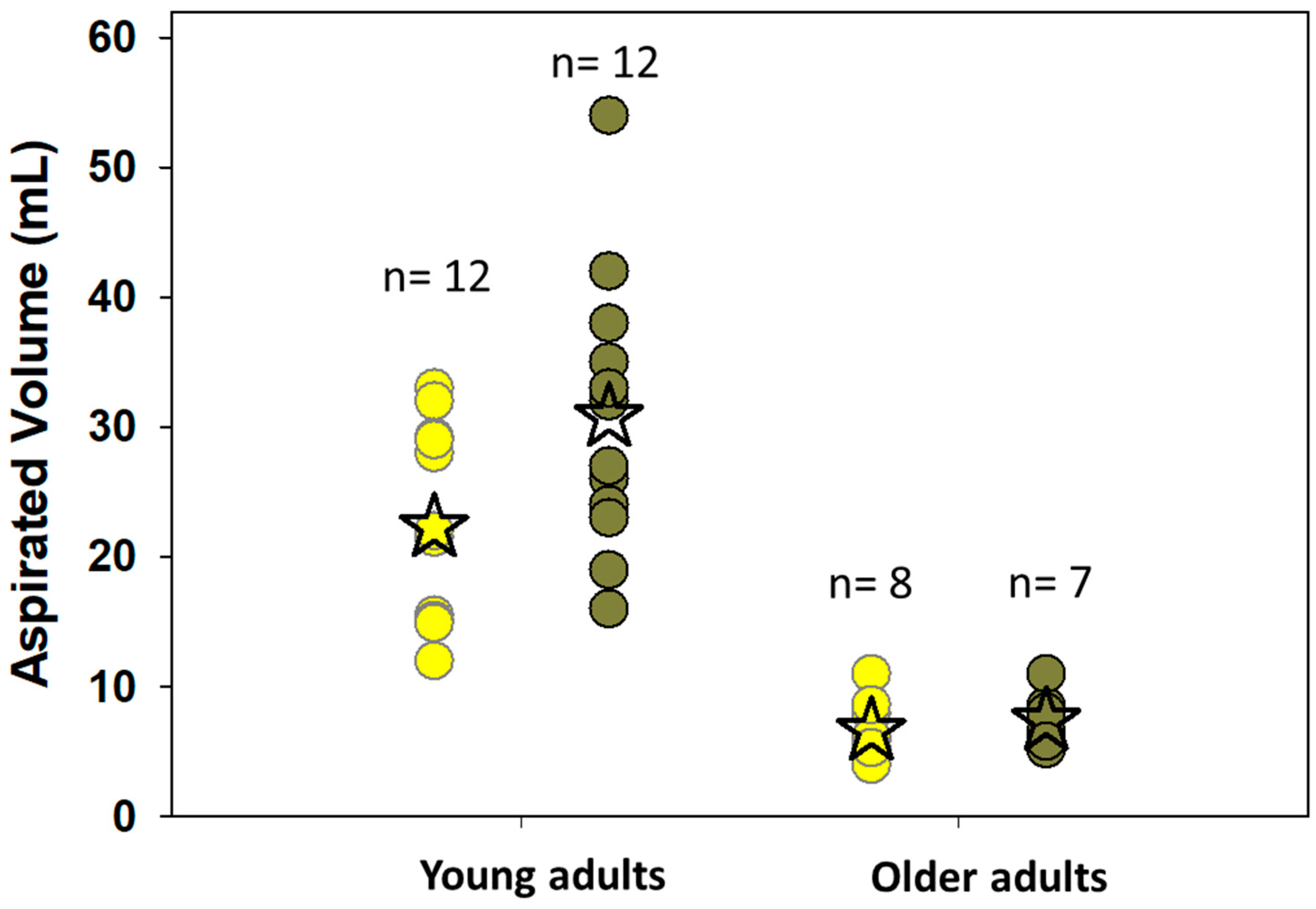
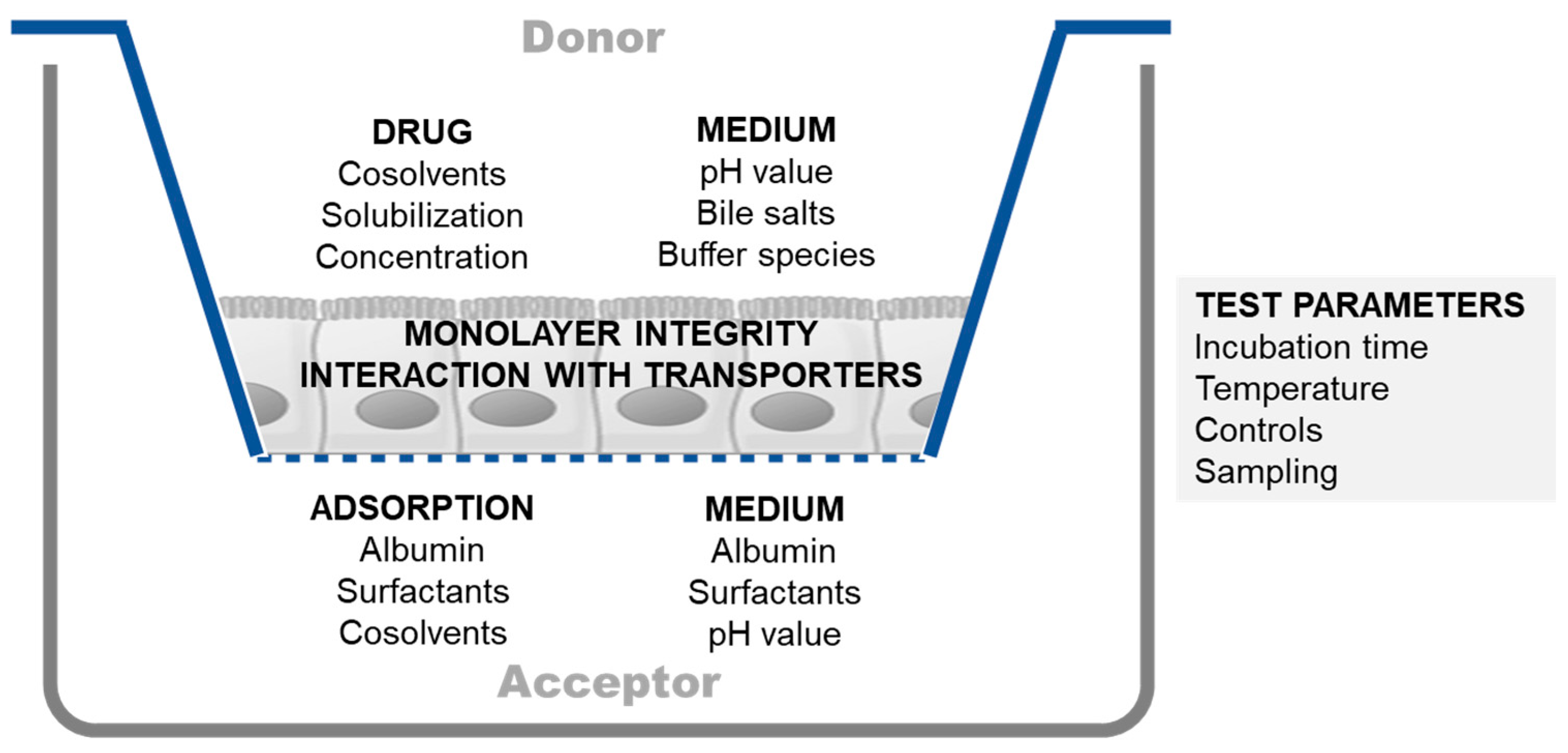
2.1.1. Experimental Challenges in Cell-Based Permeability Assays
2.1.2. Gut-on-Chip Models
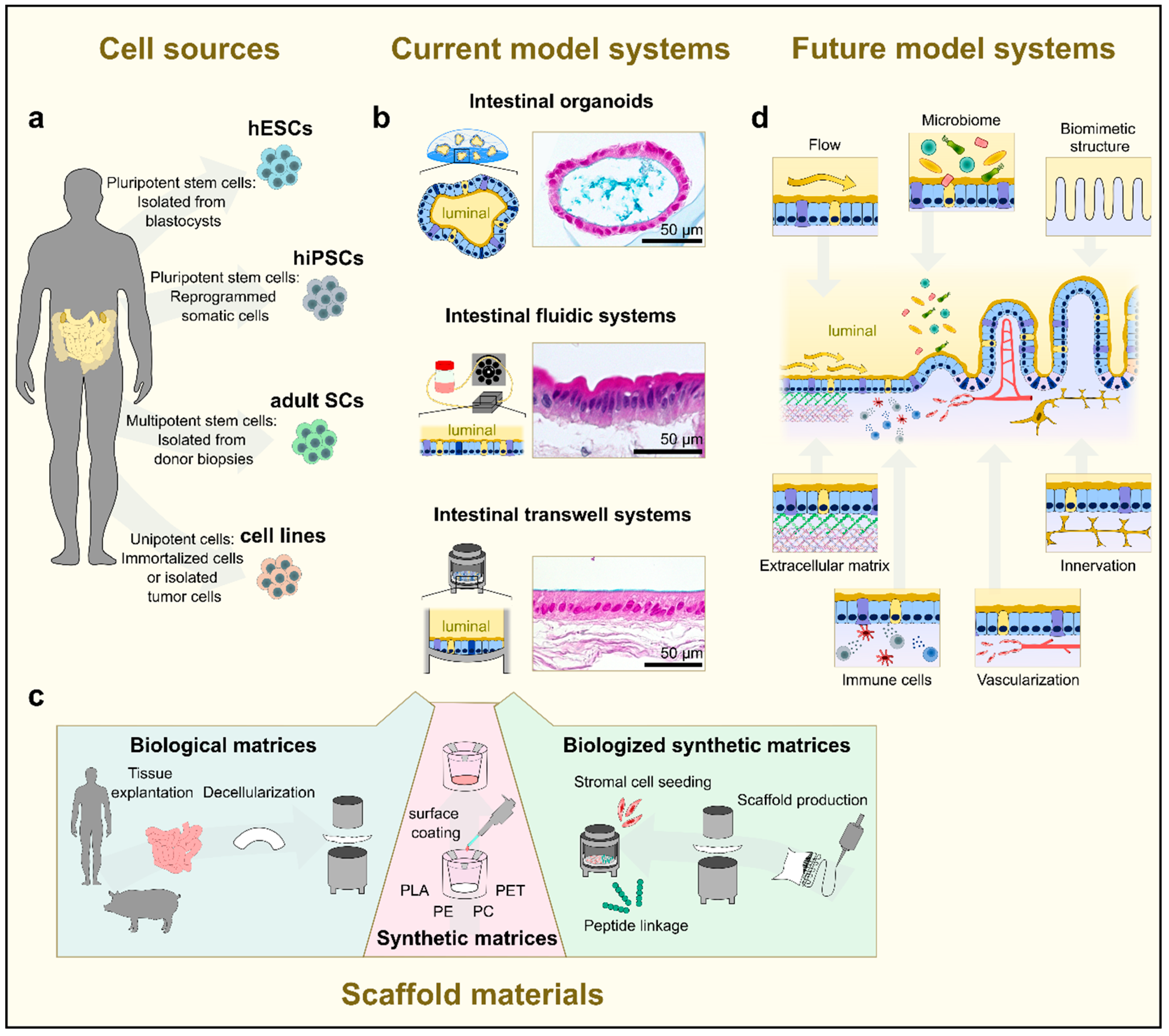
2.1.3. Human Tissue-Based Models
2.2. In Silico Methods
2.2.1. Machine Learning-Based Permeability Modeling
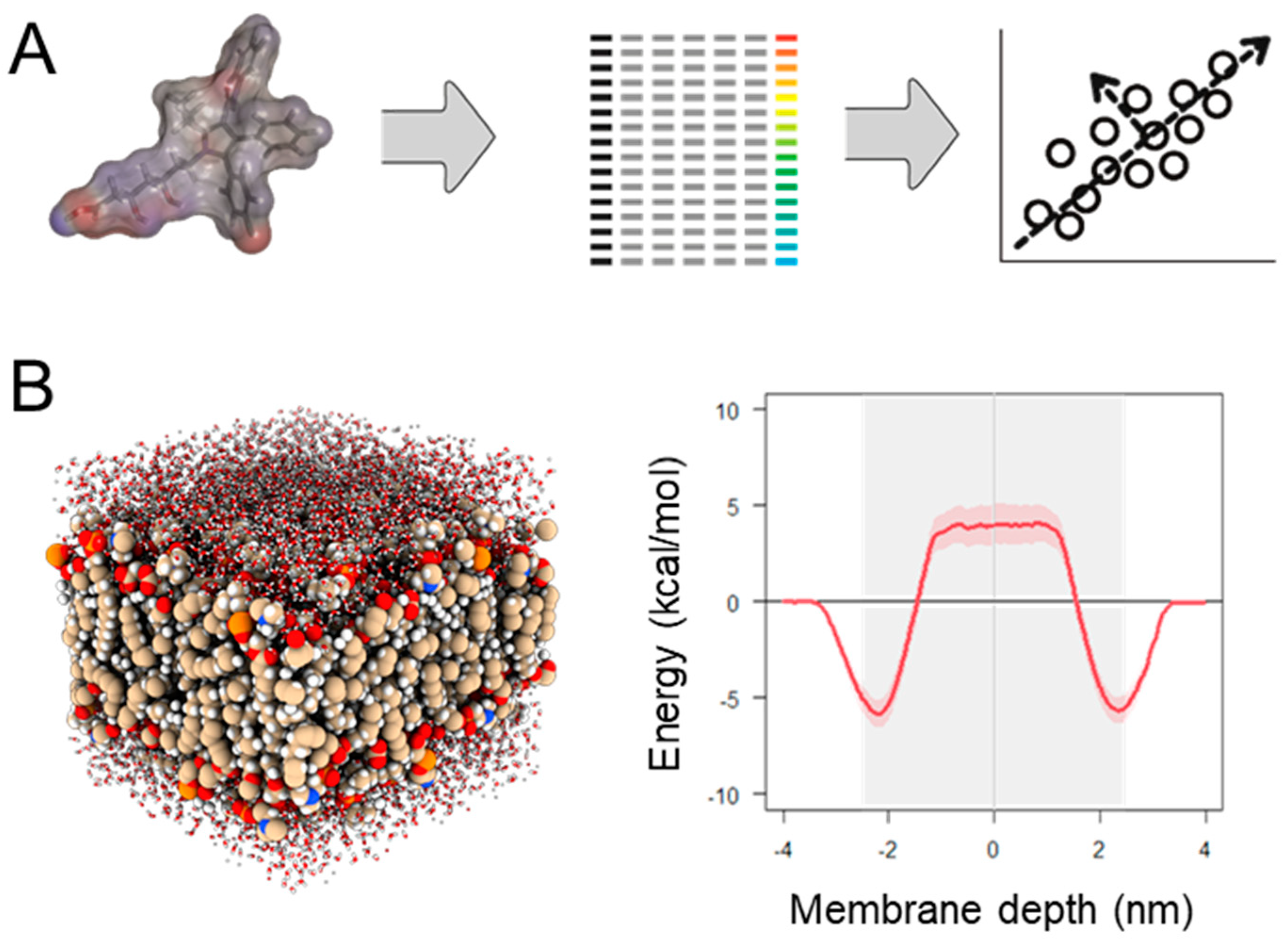
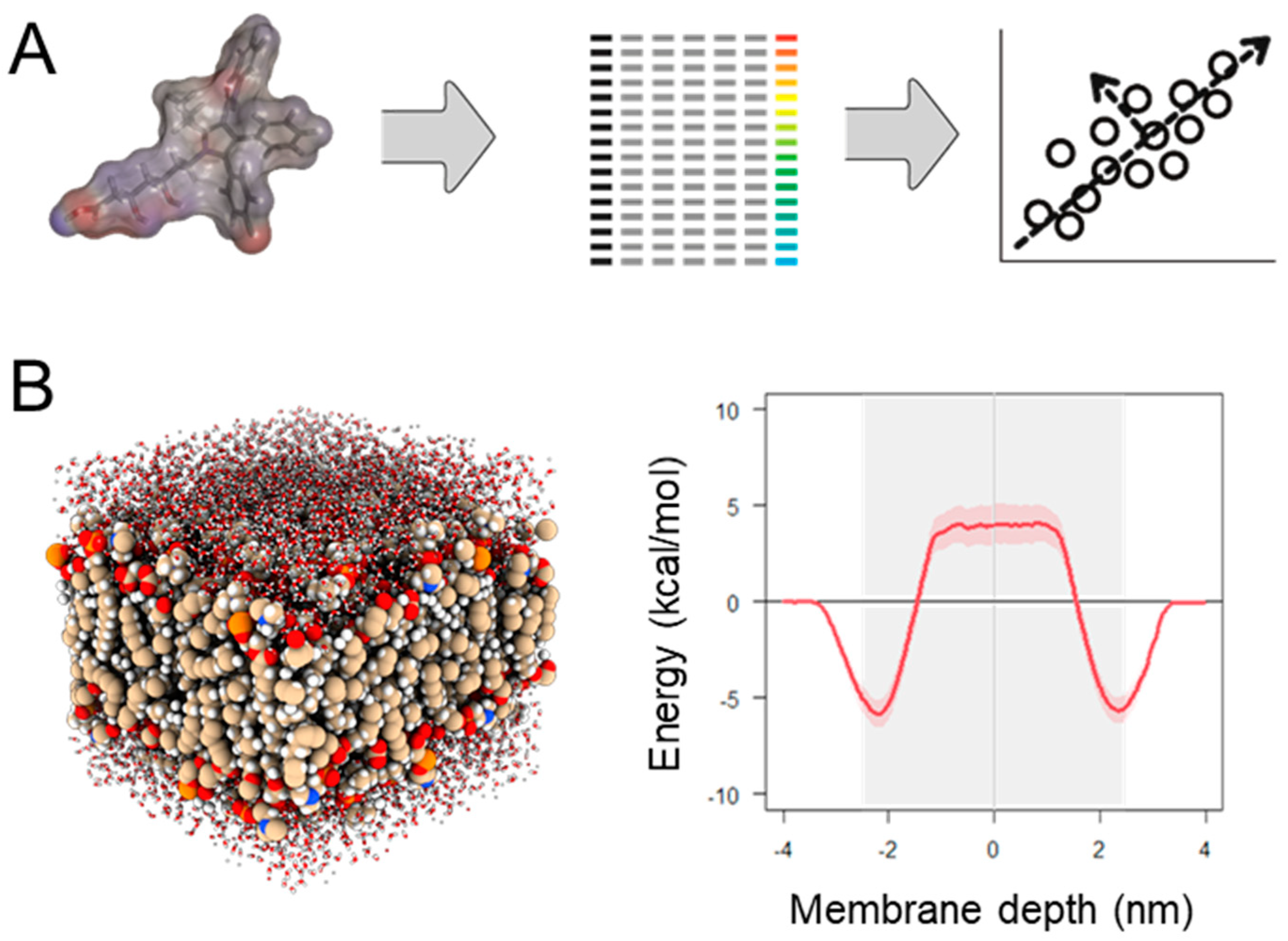
2.2.2. Physics-Based Permeability Simulation
2.2.3. PBPK Modeling
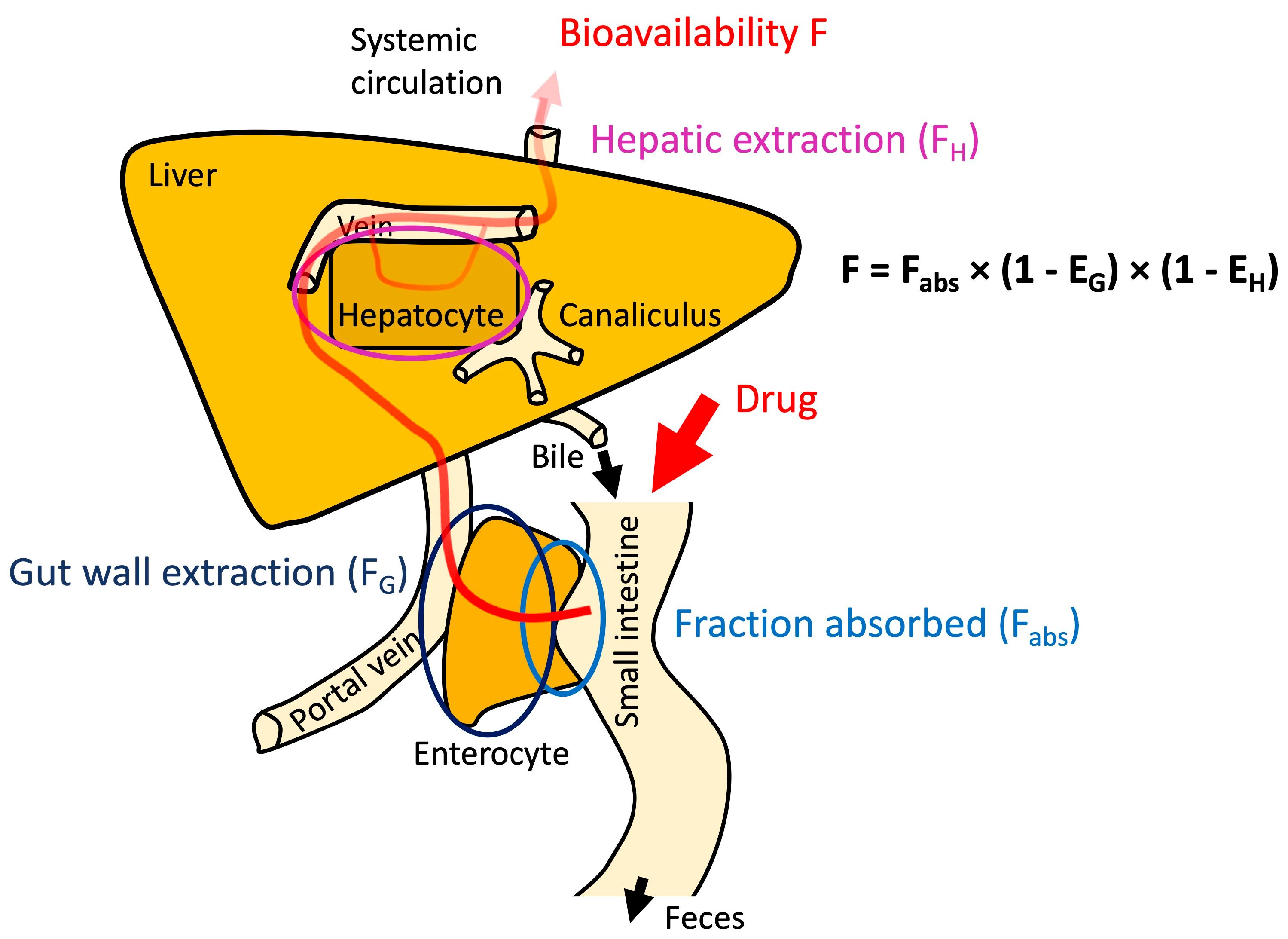
- Handles intrinsic transcellular permeability according to the pH partition hypothesis (but allows the user to select an additional model permitting transcellular ion permeation);
- Considers paracellular permeability separately whereby molecular size in relation to pore size is considered (via a Renkin function), in addition to pore charge–charge interactions (electrolytes can pass through the paracellular pathways);
- Includes consideration of the luminal Unstirred Boundary Layer (UBL), which may be the rate-limiting barrier for otherwise highly permeable drugs.
2.3. In Vivo Models
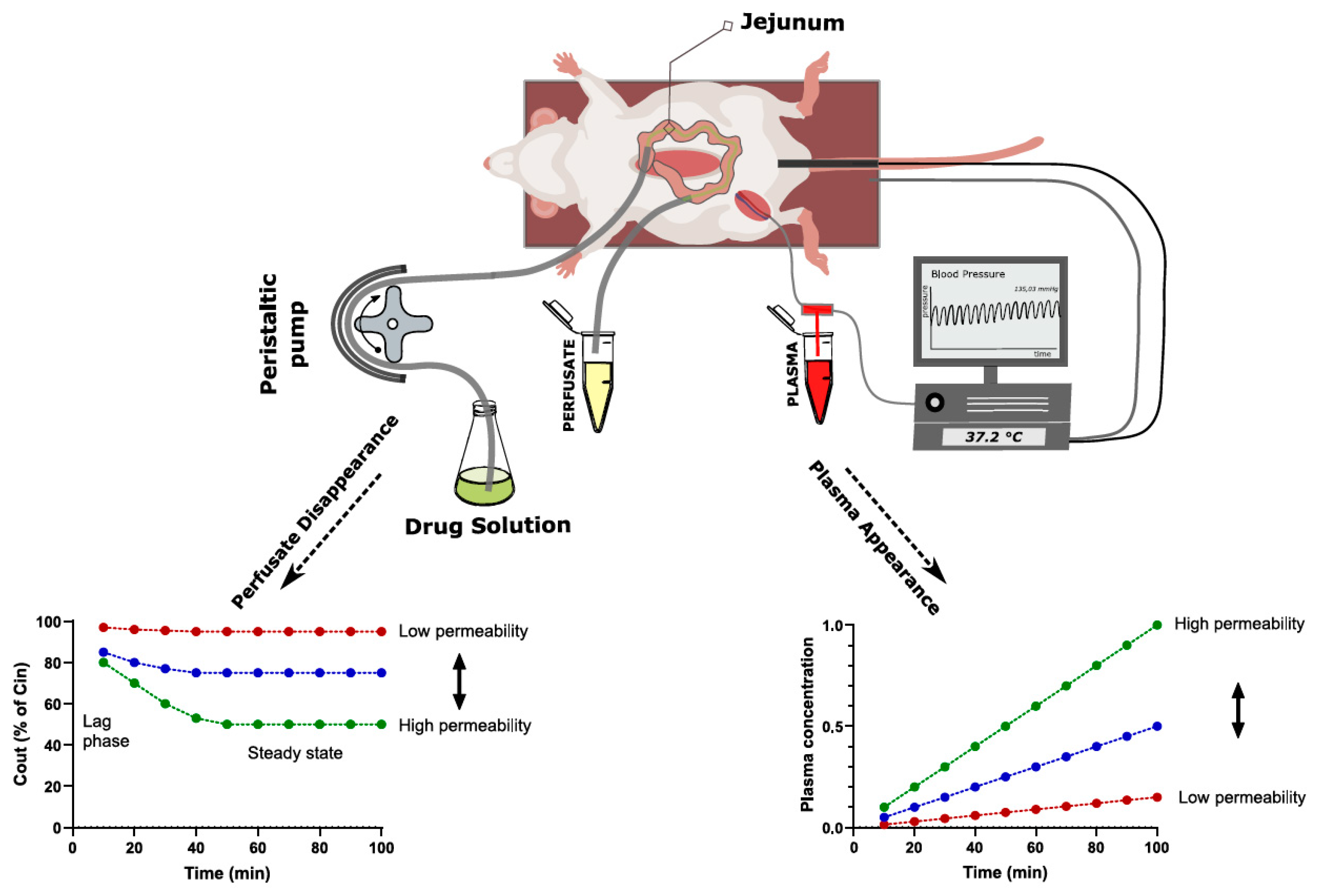
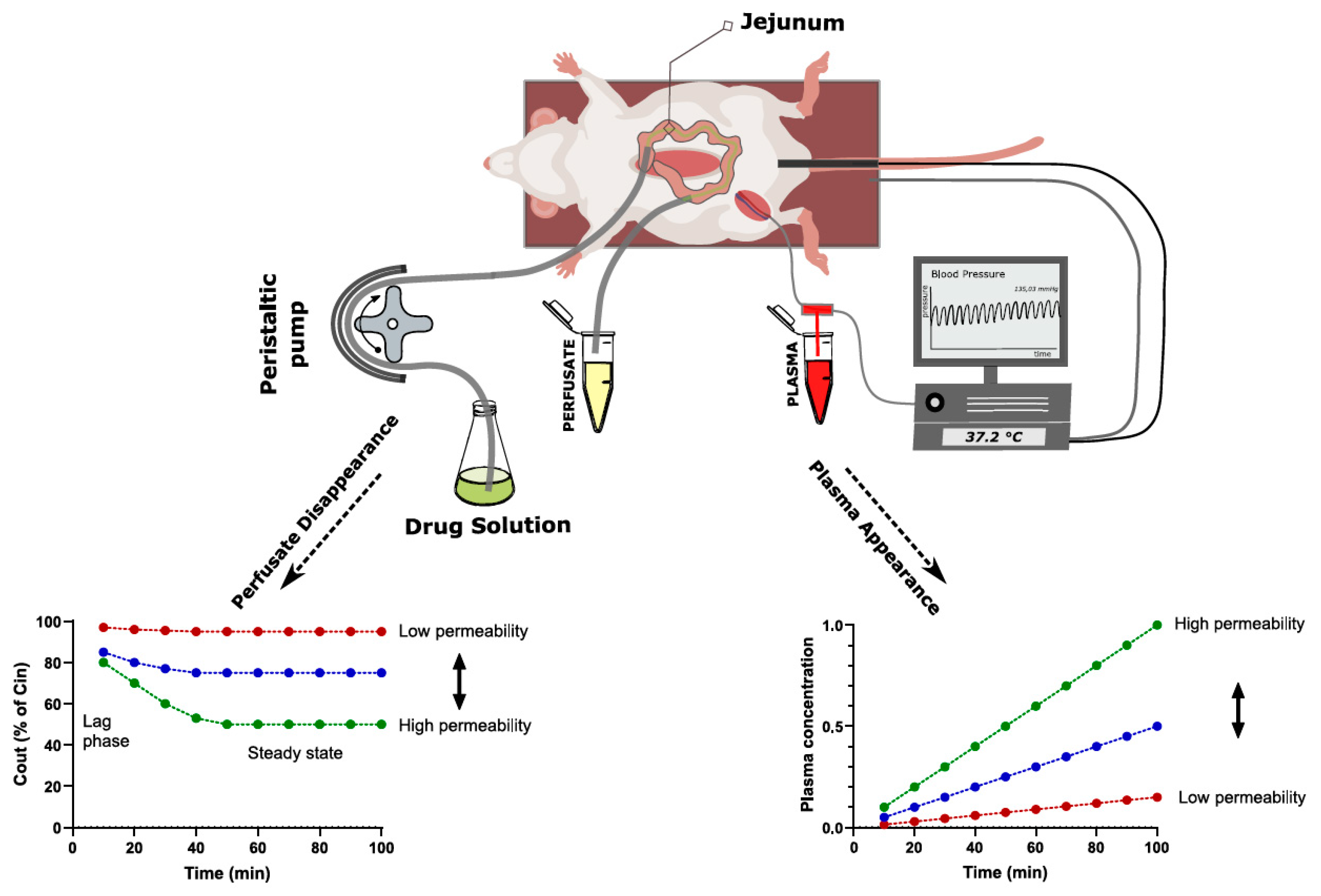
2.3.1. Single-Pass Intestinal Perfusion
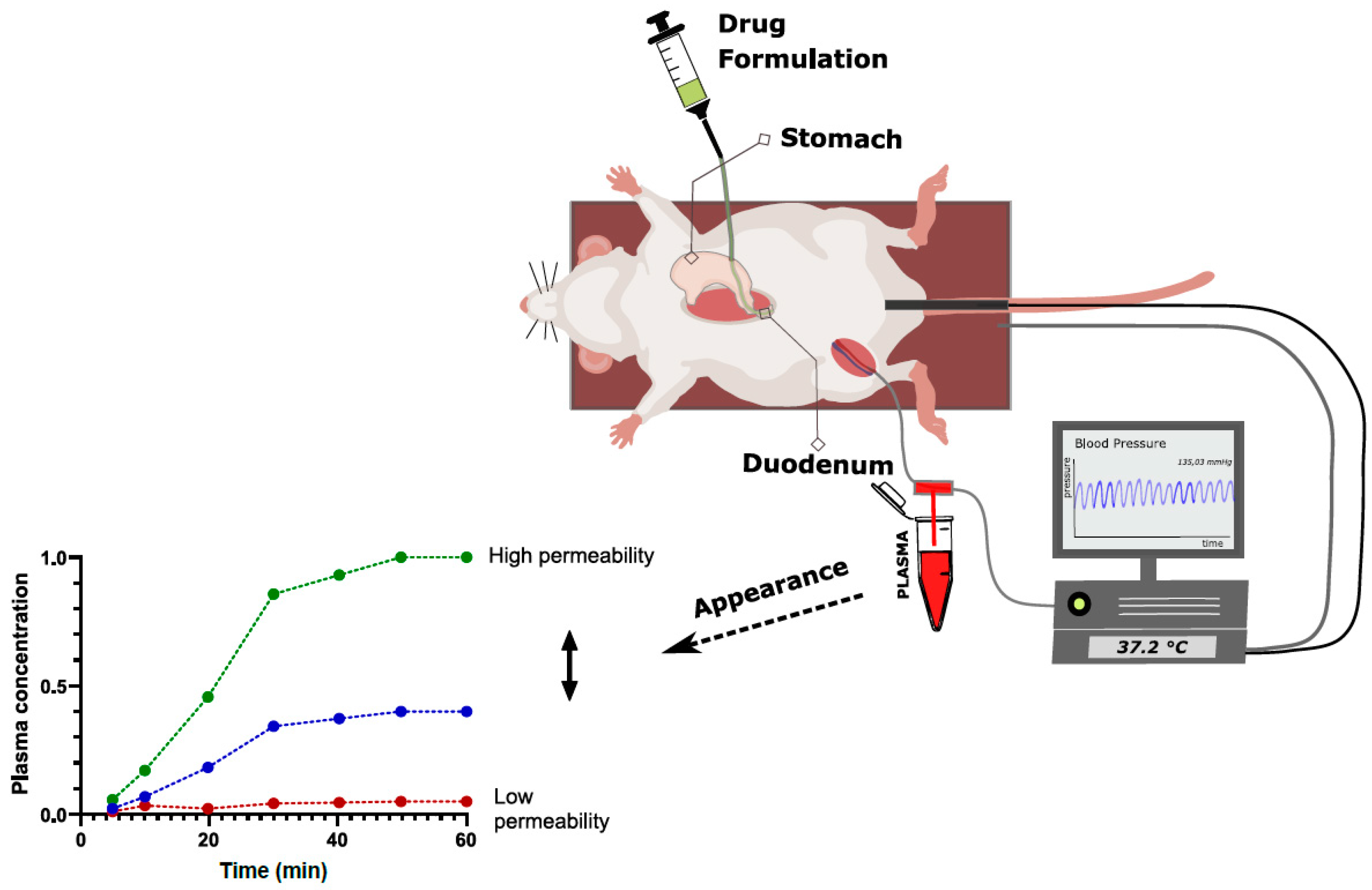
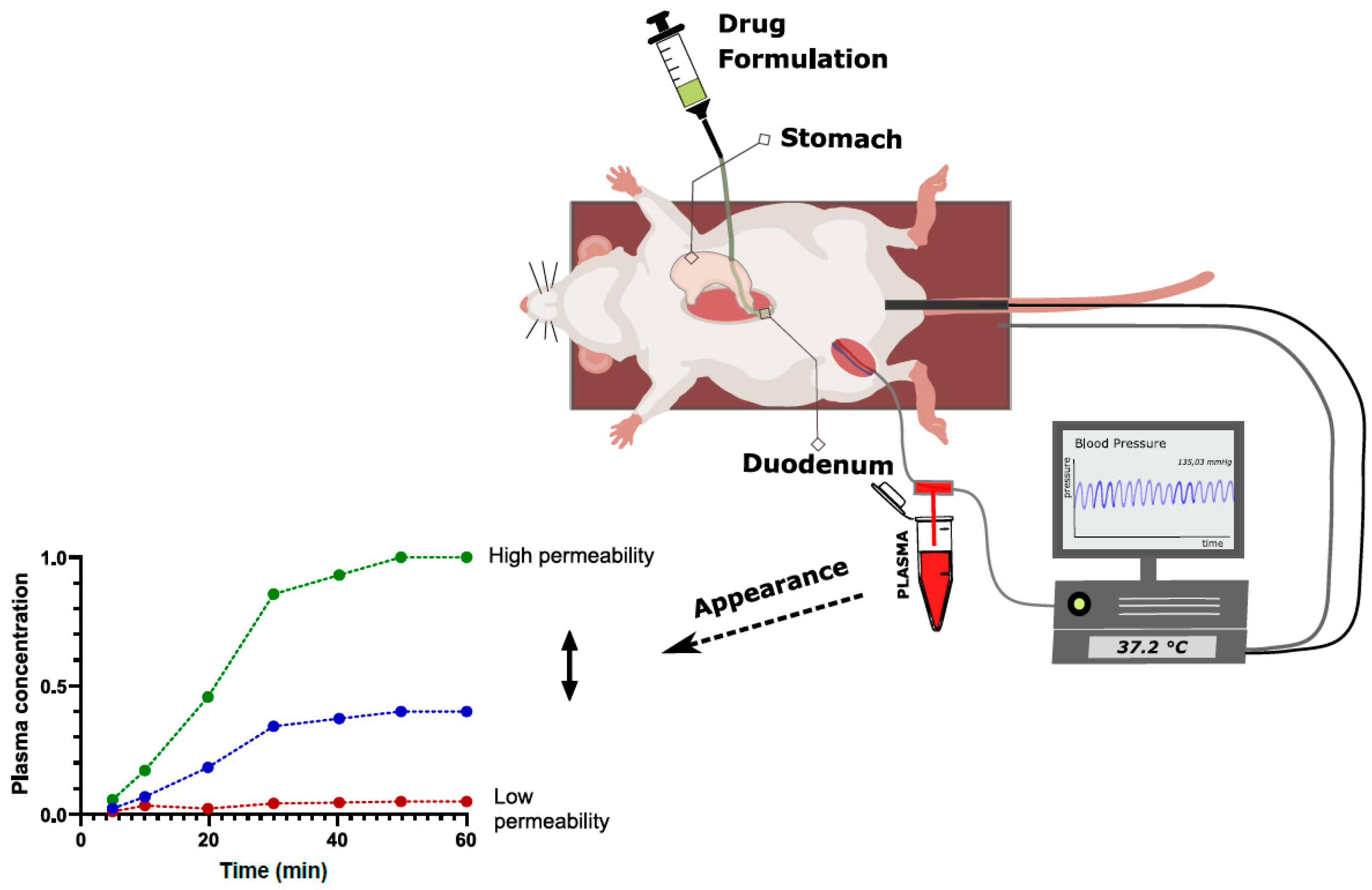
2.3.2. Intraintestinal Dosing
3. Permeability in Clinical Development
3.1. In Vivo Methods Used to Determine Permeability in Clinical Development
3.2. Formulation and Permeability
3.2.1. Excipients and Permeability—Separating Effect from Artefact
Human Studies That Test for Excipient Effects on BCS Class 3 Drug Permeability
Rationale for Discordance between In Vitro and In Vivo Excipient Effects on BCS Class 3 Drug Permeability
3.2.2. Permeation Enhancers—Where Are We?
3.3. Colonic Absorption
4. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohs, R.C.; Greig, N.H. Drug Discovery and Development: Role of Basic Biological Research. Alzheimers Dement. 2017, 3, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, E.H. Drug-Like Properties; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 9780128010761. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jin, J.; Shen, Y.; Zhang, L.; Gong, G.; Bian, H.; Chen, H.; Nagle, D.G.; Wu, Y.; Zhang, W.; et al. Emerging Protein Degradation Strategies: Expanding the Scope to Extracellular and Membrane Proteins. Theranostics 2021, 11, 8337–8349. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q. Site-Specific Antibody Conjugation with Payloads beyond Cytotoxins. Molecules 2023, 28, 917. [Google Scholar] [CrossRef]
- Cole, K.; Al-Kadhimi, Z.; Talmadge, J.E. Highlights into Historical and Current Immune Interventions for Cancer. Int. Immunopharmacol. 2023, 117, 109882. [Google Scholar] [CrossRef]
- Kang, L.; Jin, S.; Wang, J.; Lv, Z.; Xin, C.; Tan, C.; Zhao, M.; Wang, L.; Liu, J. AAV Vectors Applied to the Treatment of CNS Disorders: Clinical Status and Challenges. J. Control. Release 2023, 355, 458–473. [Google Scholar] [CrossRef]
- Matsson, P.; Doak, B.C.; Over, B.; Kihlberg, J. Cell Permeability beyond the Rule of 5. Adv. Drug Deliv. Rev. 2016, 101, 42–61. [Google Scholar] [CrossRef]
- Price, E.; Kalvass, J.C.; DeGoey, D.; Hosmane, B.; Doktor, S.; Desino, K. Global Analysis of Models for Predicting Human Absorption: QSAR, in Vitro, and Preclinical Models. J. Med. Chem. 2021, 64, 9389–9403. [Google Scholar] [CrossRef]
- Dahlgren, D.; Lennernäs, H. Intestinal Permeability and Drug Absorption: Predictive Experimental, Computational and In Vivo Approaches. Pharmaceutics 2019, 11, 411. [Google Scholar] [CrossRef]
- O’Shea, J.P.; Augustijns, P.; Brandl, M.; Brayden, D.J.; Brouwers, J.; Griffin, B.T.; Holm, R.; Jacobsen, A.-C.; Lennernäs, H.; Vinarov, Z.; et al. Best Practices in Current Models Mimicking Drug Permeability in the Gastrointestinal Tract—An UNGAP Review. Eur. J. Pharm. Sci. 2021, 170, 106098. [Google Scholar] [CrossRef]
- Sugano, K.; Kansy, M.; Artursson, P.; Avdeef, A.; Bendels, S.; Di, L.; Ecker, G.F.; Faller, B.; Fischer, H.; Gerebtzoff, G.; et al. Coexistence of Passive and Carrier-Mediated Processes in Drug Transport. Nat. Rev. Drug Discov. 2010, 9, 597–614. [Google Scholar] [CrossRef] [PubMed]
- Schoultz, I.; Keita, Å. V The Intestinal Barrier and Current Techniques for the Assessment of Gut Permeability. Cells 2020, 9, 1909. [Google Scholar] [CrossRef] [PubMed]
- Tuma, P.L.; Hubbard, A.L. Transcytosis: Crossing Cellular Barriers. Physiol. Rev. 2003, 83, 871–932. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Sugiyama, Y. Chapter 5: Membrane Transporters and Drug Response. In Goodman and Gilman’s The Pharmacological Basis of Therapeutics; Brunton, L.L., Hilal-Dandan, R., Knollmann, B.C., Eds.; McGraw-Hill: New York, NY, USA, 2017; ISBN 9781136697142. [Google Scholar]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef]
- Bolhuis, H.; van Veen, H.W.; Poolman, B.; Driessen, A.J.M.; Konings, W.N. Mechanisms of Multidrug Transporters. FEMS Microbiol. Rev. 1997, 21, 55–84. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.-M. ABC Transporters in Multidrug Resistance and Pharmacokinetics, and Strategies for Drug Development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Zhang, L.; Strong, J.M.; Qiu, W.; Lesko, L.J.; Huang, S.-M. Scientific Perspectives on Drug Transporters and Their Role in Drug Interactions. Mol. Pharm. 2006, 3, 62–69. [Google Scholar] [CrossRef]
- Scherrmann, J.-M. Transporters in Absorption, Distribution, and Elimination. Chem. Biodivers. 2009, 6, 1933–1942. [Google Scholar] [CrossRef]
- Avdeef, A. Physicochemical Profiling (Solubility, Permeability and Charge State). Curr. Top. Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef]
- Vinarov, Z.; Abdallah, M.; Agundez, J.A.G.; Allegaert, K.; Basit, A.W.; Braeckmans, M.; Ceulemans, J.; Corsetti, M.; Griffin, B.T.; Grimm, M.; et al. Impact of Gastrointestinal Tract Variability on Oral Drug Absorption and Pharmacokinetics: An UNGAP Review. Eur. J. Pharm. Sci. 2021, 162, 105812. [Google Scholar] [CrossRef]
- Di, L.; Whitney-Pickett, C.; Umland, J.P.; Zhang, H.; Zhang, X.; Gebhard, D.F.; Lai, Y.; Federico, J.J.; Davidson, R.E.; Smith, R.; et al. Development of a New Permeability Assay Using low-Efflux MDCKII Cells. J. Pharm. Sci. 2011, 100, 4974–4985. [Google Scholar] [CrossRef] [PubMed]
- Lennernäs, H. Intestinal Permeability and Its Relevance for Absorption and Elimination. Xenobiotica 2007, 37, 1015–1051. [Google Scholar] [CrossRef]
- Lv, H.; Wang, F.; Reddy, M.V.R.; Zhou, Q.; Zhang, X.; Reddy, E.P.; Gallo, J.M. Screening Candidate Anticancer Drugs for Brain Tumor Chemotherapy: Pharmacokinetic-Driven Approach for a Series of (E)-N-(Substituted Aryl)-3-(Substituted Phenyl)Propenamide Analogues. Investig. New Drugs 2012, 30, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- FDA. M9 Biopharmaceutics Classification SystemBased Biowaivers—Guidance for Industry; FDA: Silver Spring, MD, USA, 2021. [Google Scholar]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theoretical Basis for a Biopharmaceutic Drug Classification: The Correlation of in Vitro Drug Product Dissolution and in Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.Y.; Mattei, A.; Miglani Bhardwaj, R.; Hong, R.S.; Abraham, N.S.; Schneider-Rauber, G.; Engstrom, K.M.; Diwan, M.; Henry, R.F.; Gao, Y.; et al. Implications of the Conformationally Flexible, Macrocyclic Structure of the First-Generation, Direct-Acting Anti-Viral Paritaprevir on Its Solid Form Complexity and Chameleonic Behavior. J. Am. Chem. Soc. 2021, 143, 17479–17491. [Google Scholar] [CrossRef]
- Ye, D.; López Mármol, Á.; Lenz, V.; Muschong, P.; Wilhelm-Alkubaisi, A.; Weinheimer, M.; Koziolek, M.; Sauer, K.A.; Laplanche, L.; Mezler, M. Mucin-Protected Caco-2 Assay to Study Drug Permeation in the Presence of Complex Biorelevant Media. Pharmaceutics 2022, 14, 699. [Google Scholar] [CrossRef]
- Ingels, F.M.; Augustijns, P.F. Biological, Pharmaceutical, and Analytical Considerations with Respect to the Transport Media Used in the Absorption Screening System, Caco-2. J. Pharm. Sci. 2003, 92, 1545–1558. [Google Scholar] [CrossRef]
- Ye, D.; Harder, A.; Fang, Z.; Weinheimer, M.; Laplanche, L.; Mezler, M. Characterization and Validation of Canine P-Glycoprotein-Deficient MDCK II Cell Lines for Efflux Substrate Screening. Pharm. Res. 2020, 37, 194. [Google Scholar] [CrossRef]
- Chen, E.C.; Broccatelli, F.; Plise, E.; Chen, B.; Liu, L.; Cheong, J.; Zhang, S.; Jorski, J.; Gaffney, K.; Umemoto, K.K.; et al. Evaluating the Utility of Canine Mdr1 Knockout Madin-Darby Canine Kidney I Cells in Permeability Screening and Efflux Substrate Determination. Mol. Pharm. 2018, 15, 5103–5113. [Google Scholar] [CrossRef]
- Simoff, I.; Karlgren, M.; Backlund, M.; Lindström, A.-C.; Gaugaz, F.Z.; Matsson, P.; Artursson, P. Complete Knockout of Endogenous Mdr1 (Abcb1) in MDCK Cells by CRISPR-Cas9. J. Pharm. Sci. 2016, 105, 1017–1021. [Google Scholar] [CrossRef]
- Degoey, D.A.; Chen, H.J.; Cox, P.B.; Wendt, M.D. Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J. Med. Chem. 2018, 61, 2636–2651. [Google Scholar] [CrossRef] [PubMed]
- Bransford, P.; Cook, J.; Gupta, M.; Haertter, S.; He, H.; Ju, R.; Kanodia, J.; Lennernäs, H.; Lindley, D.; Polli, J.E.; et al. ICH M9 Guideline in Development on Biopharmaceutics Classification System-Based Biowaivers: An Industrial Perspective from the IQ Consortium. Mol. Pharm. 2020, 17, 361–372. [Google Scholar] [CrossRef] [PubMed]
- EMA. ICH M9 Guideline on Biopharmaceutics Classification System Based Biowaivers; EMA: Amsterdam, The Netherlands, 2018. [Google Scholar]
- FDA. SUPAC-IR: Immediate-Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation; FDA: Silver Spring, MD, USA, 1995. [Google Scholar]
- Hidalgo, I.J.; Raub, T.J.; Borchardt, R.T. Characterization of the Human Colon Carcinoma Cell Line (Caco-2) as a Model System for Intestinal Epithelial Permeability. Gastroenterology 1989, 96, 736–749. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Konishi, K.; Yamazaki, Y.; Taki, Y.; Sakane, T.; Sezaki, H.; Furuyama, Y. New and Better Protocols for a Short-Term Caco-2 Cell Culture System. J. Pharm. Sci. 2002, 91, 669–679. [Google Scholar] [CrossRef]
- Ingels, F.; Ungell, A.-L.; Augustijns, P.F. Selection of Solvent Systems for Membrane-, Cell- and Tissue-Based Permeability Assessment. In Solvent Systems and Their Selection in Pharmaceutics and Biopharmaceutics; Springer: New York, NY, USA, 2007; pp. 179–220. [Google Scholar]
- Miller, J.M.; Beig, A.; Krieg, B.J.; Carr, R.A.; Borchardt, T.B.; Amidon, G.E.; Amidon, G.L.; Dahan, A. The Solubility-Permeability Interplay: Mechanistic Modeling and Predictive Application of the Impact of Micellar Solubilization on Intestinal Permeation. Mol. Pharm. 2011, 8, 1848–1856. [Google Scholar] [CrossRef]
- Augustijns, P.F.; Bradshaw, T.P.; Gan, L.S.L.; Hendren, R.W.; Thakker, D.R. Evidence for a Polarized Efflux System in Caco-2 Cells Capable of Modulating Cyclosporine A Transport. Biochem. Biophys. Res. Commun. 1993, 197, 360–365. [Google Scholar] [CrossRef]
- Ingels, F.; Deferme, S.; Destexhe, E.; Oth, M.; Van den Mooter, G.; Augustijns, P. Simulated Intestinal Fluid as Transport Medium in the Caco-2 Cell Culture Model. Int. J. Pharm. 2002, 232, 183–192. [Google Scholar] [CrossRef]
- Neuhoff, S.; Ungell, A.-L.; Zamora, I.; Artursson, P. PH-Dependent Bidirectional Transport of Weakly Basic Drugs across Caco-2 Monolayers: Implications for Drug-Drug Interactions. Pharm. Res. 2003, 20, 1141–1148. [Google Scholar] [CrossRef]
- Brouwers, J.; Tack, J.; Augustijns, P. In Vitro Behavior of a Phosphate Ester Prodrug of Amprenavir in Human Intestinal Fluids and in the Caco-2 System: Illustration of Intraluminal Supersaturation. Int. J. Pharm. 2007, 336, 302–309. [Google Scholar] [CrossRef]
- Deferme, S.; Tack, J.; Lammert, F.; Augustijns, P. P-Glycoprotein Attenuating Effect of Human Intestinal Fluid. Pharm. Res. 2003, 20, 900–903. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M.; Hoffman, A.; Amidon, G.E.; Amidon, G.L. The Solubility–Permeability Interplay in Using Cyclodextrins as Pharmaceutical Solubilizers: Mechanistic Modeling and Application to Progesterone. J. Pharm. Sci. 2010, 99, 2739–2749. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ingber, D.E. Gut-on-a-Chip Microenvironment Induces Human Intestinal Cells to Undergo Villus Differentiation. Integr. Biol. 2013, 5, 1130. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Ghiboub, M.; Donkers, J.M.; van de Steeg, E.; van Tol, E.A.F.; Hakvoort, T.B.M.; de Jonge, W.J. The Progress of Intestinal Epithelial Models from Cell Lines to Gut-On-Chip. Int. J. Mol. Sci. 2021, 22, 13472. [Google Scholar] [CrossRef] [PubMed]
- Santbergen, M.J.C.; van der Zande, M.; Gerssen, A.; Bouwmeester, H.; Nielen, M.W.F. Dynamic In Vitro Intestinal Barrier Model Coupled to Chip-Based Liquid Chromatography Mass Spectrometry for Oral Bioavailability Studies. Anal. Bioanal. Chem. 2020, 412, 1111–1122. [Google Scholar] [CrossRef]
- Eslami Amirabadi, H.; Donkers, J.M.; Wierenga, E.; Ingenhut, B.; Pieters, L.; Stevens, L.; Donkers, T.; Westerhout, J.; Masereeuw, R.; Bobeldijk-Pastorova, I.; et al. Intestinal Explant Barrier Chip: Long-Term Intestinal Absorption Screening in a Novel Microphysiological System Using Tissue Explants. Lab Chip 2022, 22, 326–342. [Google Scholar] [CrossRef]
- Tsamandouras, N.; Chen, W.L.K.; Edington, C.D.; Stokes, C.L.; Griffith, L.G.; Cirit, M. Integrated Gut and Liver Microphysiological Systems for Quantitative In Vitro Pharmacokinetic Studies. AAPS J. 2017, 19, 1499–1512. [Google Scholar] [CrossRef]
- Trietsch, S.J.; Naumovska, E.; Kurek, D.; Setyawati, M.C.; Vormann, M.K.; Wilschut, K.J.; Lanz, H.L.; Nicolas, A.; Ng, C.P.; Joore, J.; et al. Membrane-Free Culture and Real-Time Barrier Integrity Assessment of Perfused Intestinal Epithelium Tubes. Nat. Commun. 2017, 8, 262. [Google Scholar] [CrossRef]
- Sasaki, Y.; Tatsuoka, H.; Tsuda, M.; Sumi, T.; Eguchi, Y.; So, K.; Higuchi, Y.; Takayama, K.; Torisawa, Y.; Yamashita, F. Intestinal Permeability of Drugs in Caco-2 Cells Cultured in Microfluidic Devices. Biol. Pharm. Bull. 2022, 45, 1246–1253. [Google Scholar] [CrossRef]
- Ashammakhi, N.; Nasiri, R.; de Barros, N.R.; Tebon, P.; Thakor, J.; Goudie, M.; Shamloo, A.; Martin, M.G.; Khademhosseini, A. Gut-on-a-Chip: Current Progress and Future Opportunities. Biomaterials 2020, 255, 120196. [Google Scholar] [CrossRef]
- Kim, H.J.; Huh, D.; Hamilton, G.; Ingber, D.E. Human Gut-on-a-Chip Inhabited by Microbial Flora That Experiences Intestinal Peristalsis-like Motions and Flow. Lab Chip 2012, 12, 2165. [Google Scholar] [CrossRef]
- Kulkarni, G.; Apostolou, A.; Ewart, L.; Lucchesi, C.; Kasendra, M. Combining Human Organoids and Organ-on-a-Chip Technology to Model Intestinal Region-Specific Functionality. J. Vis. Exp. 2022, 183, e63724. [Google Scholar] [CrossRef]
- De Gregorio, V.; Sgambato, C.; Urciuolo, F.; Vecchione, R.; Netti, P.A.; Imparato, G. Immunoresponsive Microbiota-Gut-on-Chip Reproduces Barrier Dysfunction, Stromal Reshaping and Probiotics Translocation under Inflammation. Biomaterials 2022, 286, 121573. [Google Scholar] [CrossRef] [PubMed]
- Maoz, B.M.; Herland, A.; Henry, O.Y.F.; Leineweber, W.D.; Yadid, M.; Doyle, J.; Mannix, R.; Kujala, V.J.; FitzGerald, E.A.; Parker, K.K.; et al. Organs-on-Chips with Combined Multi-Electrode Array and Transepithelial Electrical Resistance Measurement Capabilities. Lab Chip 2017, 17, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.H.; Park, J.-K. Drug Permeability Assay Using Microhole-Trapped Cells in a Microfluidic Device. Anal. Chem. 2009, 81, 1944–1951. [Google Scholar] [CrossRef]
- Milani, N.; Parrott, N.; Ortiz Franyuti, D.; Godoy, P.; Galetin, A.; Gertz, M.; Fowler, S. Application of a Gut–Liver-on-a-Chip Device and Mechanistic Modelling to the Quantitative in Vitro Pharmacokinetic Study of Mycophenolate Mofetil. Lab Chip 2022, 22, 2853–2868. [Google Scholar] [CrossRef] [PubMed]
- Clapp, N.; Amour, A.; Rowan, W.C.; Candarlioglu, P.L. Organ-on-Chip Applications in Drug Discovery: An End User Perspective. Biochem. Soc. Trans. 2021, 49, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Wuyts, B.; Riethorst, D.; Brouwers, J.; Tack, J.; Annaert, P.; Augustijns, P. Evaluation of Fasted State Human Intestinal Fluid as Apical Solvent System in the Caco-2 Absorption Model and Comparison with FaSSIF. Eur. J. Pharm. Sci. 2015, 67, 126–135. [Google Scholar] [CrossRef]
- Fredlund, L.; Winiwarter, S.; Hilgendorf, C. In Vitro Intrinsic Permeability: A Transporter-Independent Measure of Caco-2 Cell Permeability in Drug Design and Development. Mol. Pharm. 2017, 14, 1601–1609. [Google Scholar] [CrossRef]
- Ma, Z.; Li, B.; Peng, J.; Gao, D. Recent Development of Drug Delivery Systems through Microfluidics: From Synthesis to Evaluation. Pharmaceutics 2022, 14, 434. [Google Scholar] [CrossRef]
- Gordon, S. Non-Animal Models of Epithelial Barriers (Skin, Intestine and Lung) in Research, Industrial Applications and Regulatory Toxicology. ALTEX 2015, 32, 327–378. [Google Scholar] [CrossRef]
- Jabaji, Z.; Sears, C.M.; Brinkley, G.J.; Lei, N.Y.; Joshi, V.S.; Wang, J.; Lewis, M.; Stelzner, M.; Martín, M.G.; Dunn, J.C.Y. Use of Collagen Gel as an Alternative Extracellular Matrix for the In Vitro and In Vivo Growth of Murine Small Intestinal Epithelium. Tissue Eng. Part. C Methods 2013, 19, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Dedhia, P.H.; Bertaux-Skeirik, N.; Zavros, Y.; Spence, J.R. Organoid Models of Human Gastrointestinal Development and Disease. Gastroenterology 2016, 150, 1098–1112. [Google Scholar] [CrossRef]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement Membrane Matrix with Biological Activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; van Es, J.H.; van den Brink, S.; van Houdt, W.J.; Pronk, A.; van Gorp, J.; Siersema, P.D.; et al. Long-Term Expansion of Epithelial Organoids From Human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef]
- Cao, L.; Kuratnik, A.; Xu, W.; Gibson, J.D.; Kolling, F.; Falcone, E.R.; Ammar, M.; Van Heyst, M.D.; Wright, D.L.; Nelson, C.E.; et al. Development of Intestinal Organoids as Tissue Surrogates: Cell Composition and the Epigenetic Control of Differentiation. Mol. Carcinog. 2015, 54, 189–202. [Google Scholar] [CrossRef]
- Farin, H.F.; Van Es, J.H.; Clevers, H. Redundant Sources of Wnt Regulate Intestinal Stem Cells and Promote Formation of Paneth Cells. Gastroenterology 2012, 143, 1518–1529.e7. [Google Scholar] [CrossRef]
- Rouch, J.D.; Scott, A.; Lei, N.Y.; Solorzano-Vargas, R.S.; Wang, J.; Hanson, E.M.; Kobayashi, M.; Lewis, M.; Stelzner, M.G.; Dunn, J.C.; et al. 31 Development of Functional Microfold (M) Cells From Intestinal Stem Cells in Primary Human Enteroids. Gastroenterology 2016, 150, S11. [Google Scholar] [CrossRef]
- Basak, O.; Beumer, J.; Wiebrands, K.; Seno, H.; van Oudenaarden, A.; Clevers, H. Induced Quiescence of Lgr5+ Stem Cells in Intestinal Organoids Enables Differentiation of Hormone-Producing Enteroendocrine Cells. Cell Stem Cell 2017, 20, 177–190.e4. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human Intestinal Organoids Maintain Self-Renewal Capacity and Cellular Diversity in Niche-Inspired Culture Condition. Cell Stem Cell 2018, 23, 787–793.e6. [Google Scholar] [CrossRef]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of Notch Processing by γ-Secretase Inhibitors Causes Intestinal Goblet Cell Metaplasia and Induction of Genes Known to Specify Gut Secretory Lineage Differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; van den Brink, S.; Geurts, V.; Beumer, J.; Clevers, H. Establishment and Culture of Human Intestinal Organoids Derived from Adult Stem Cells. Curr. Protoc. Immunol. 2020, 130, e106. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Knoblich, J.A.; Lutolf, M.P.; Martinez-Arias, A. The Hope and the Hype of Organoid Research. Development 2017, 144, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed Differentiation of Human Pluripotent Stem Cells into Intestinal Tissue in Vitro. Nature 2011, 470, 105–109. [Google Scholar] [CrossRef] [PubMed]
- McCracken, K.W.; Howell, J.C.; Wells, J.M.; Spence, J.R. Generating Human Intestinal Tissue from Pluripotent Stem Cells in Vitro. Nat. Prot. 2011, 6, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, S.R.; Hill, D.R.; Altheim, C.H.; Dedhia, P.H.; Taylor, M.J.; Tsai, Y.-H.; Chin, A.M.; Mahe, M.M.; Watson, C.L.; Freeman, J.J.; et al. Transcriptome-Wide Analysis Reveals Hallmarks of Human Intestine Development and Maturation In Vitro and In Vivo. Stem Cell Rep. 2015, 4, 1140–1155. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.B.; Lee, H.; Son, Y.S.; Lee, M.-O.; Kim, Y.-D.; Oh, S.J.; Kwon, O.; Cho, S.; Cho, H.-S.; Kim, D.-S.; et al. Interleukin-2 Induces the in Vitro Maturation of Human Pluripotent Stem Cell-Derived Intestinal Organoids. Nat. Commun. 2018, 9, 3039. [Google Scholar] [CrossRef]
- Mizutani, T.; Nakamura, T.; Morikawa, R.; Fukuda, M.; Mochizuki, W.; Yamauchi, Y.; Nozaki, K.; Yui, S.; Nemoto, Y.; Nagaishi, T.; et al. Real-Time Analysis of P-Glycoprotein-Mediated Drug Transport across Primary Intestinal Epithelium Three-Dimensionally Cultured in Vitro. Biochem. Biophys. Res. Commun. 2012, 419, 238–243. [Google Scholar] [CrossRef]
- Kakni, P.; López-Iglesias, C.; Truckenmüller, R.; Habibović, P.; Giselbrecht, S. Reversing Epithelial Polarity in Pluripotent Stem Cell-Derived Intestinal Organoids. Front. Bioeng. Biotechnol. 2022, 10, 879024. [Google Scholar] [CrossRef]
- Co, J.Y.; Margalef-Català, M.; Monack, D.M.; Amieva, M.R. Controlling the Polarity of Human Gastrointestinal Organoids to Investigate Epithelial Biology and Infectious Diseases. Nat. Prot. 2021, 16, 5171–5192. [Google Scholar] [CrossRef]
- Costa, J.; Ahluwalia, A. Advances and Current Challenges in Intestinal in Vitro Model Engineering: A Digest. Front. Bioeng. Biotechnol. 2019, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Snyder, J.; Wang, C.-M.; Zhang, A.Q.; Li, Y.; Luchan, J.; Hosic, S.; Koppes, R.; Carrier, R.L.; Koppes, A. Materials and Microenvironments for Engineering the Intestinal Epithelium. Ann. Biomed. Eng. 2020, 48, 1916–1940. [Google Scholar] [CrossRef] [PubMed]
- Costello, C.M.; Hongpeng, J.; Shaffiey, S.; Yu, J.; Jain, N.K.; Hackam, D.; March, J.C. Synthetic Small Intestinal Scaffolds for Improved Studies of Intestinal Differentiation. Biotechnol. Bioeng. 2014, 111, 1222–1232. [Google Scholar] [CrossRef] [PubMed]
- Devriese, S.; Van den Bossche, L.; Van Welden, S.; Holvoet, T.; Pinheiro, I.; Hindryckx, P.; De Vos, M.; Laukens, D. T84 Monolayers Are Superior to Caco-2 as a Model System of Colonocytes. Histochem. Cell Biol. 2017, 148, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Gjorevski, N.; Sachs, N.; Manfrin, A.; Giger, S.; Bragina, M.E.; Ordóñez-Morán, P.; Clevers, H.; Lutolf, M.P. Designer Matrices for Intestinal Stem Cell and Organoid Culture. Nature 2016, 539, 560–564. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, Y.; Davis, K.M.; Wang, Q.; Rnjak-Kovacina, J.; Li, C.; Isberg, R.R.; Kumamoto, C.A.; Mecsas, J.; Kaplan, D.L. Robust Bioengineered 3D Functional Human Intestinal Epithelium. Sci. Rep. 2015, 5, 13708. [Google Scholar] [CrossRef]
- Pusch, J.; Votteler, M.; Göhler, S.; Engl, J.; Hampel, M.; Walles, H.; Schenke-Layland, K. The Physiological Performance of a Three-Dimensional Model That Mimics the Microenvironment of the Small Intestine. Biomaterials 2011, 32, 7469–7478. [Google Scholar] [CrossRef]
- Wang, L.; Murthy, S.K.; Barabino, G.A.; Carrier, R.L. Synergic Effects of Crypt-like Topography and ECM Proteins on Intestinal Cell Behavior in Collagen Based Membranes. Biomaterials 2010, 31, 7586–7598. [Google Scholar] [CrossRef]
- Andrée, B.; Bär, A.; Haverich, A.; Hilfiker, A. Small Intestinal Submucosa Segments as Matrix for Tissue Engineering: Review. Tissue Eng. Part. B Rev. 2013, 19, 279–291. [Google Scholar] [CrossRef]
- Schweinlin, M.; Wilhelm, S.; Schwedhelm, I.; Hansmann, J.; Rietscher, R.; Jurowich, C.; Walles, H.; Metzger, M. Development of an Advanced Primary Human In Vitro Model of the Small Intestine. Tissue Eng. Part. C Methods 2016, 22, 873–883. [Google Scholar] [CrossRef]
- Koppes, A.N.; Kamath, M.; Pfluger, C.A.; Burkey, D.D.; Dokmeci, M.; Wang, L.; Carrier, R.L. Complex, Multi-Scale Small Intestinal Topography Replicated in Cellular Growth Substrates Fabricated via Chemical Vapor Deposition of Parylene C. Biofabrication 2016, 8, 035011. [Google Scholar] [CrossRef]
- Kitano, K.; Schwartz, D.M.; Zhou, H.; Gilpin, S.E.; Wojtkiewicz, G.R.; Ren, X.; Sommer, C.A.; Capilla, A.V.; Mathisen, D.J.; Goldstein, A.M.; et al. Bioengineering of Functional Human Induced Pluripotent Stem Cell-Derived Intestinal Grafts. Nat. Commun. 2017, 8, 765. [Google Scholar] [CrossRef]
- Moroni, L.; Burdick, J.A.; Highley, C.; Lee, S.J.; Morimoto, Y.; Takeuchi, S.; Yoo, J.J. Biofabrication Strategies for 3D in Vitro Models and Regenerative Medicine. Nat. Rev. Mater. 2018, 3, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, S.E.; Longo, B.N.; Tse, M.W.; Houchin, M.R.; Shokoufandeh, M.M.; Chen, Y.; Kaplan, D.L. Crypt-Villus Scaffold Architecture for Bioengineering Functional Human Intestinal Epithelium. ACS Biomater. Sci. Eng. 2022, 8, 4942–4955. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, M.; Mitrofanova, O.; Broguiere, N.; Geraldo, S.; Dutta, D.; Tabata, Y.; Elci, B.; Brandenberg, N.; Kolotuev, I.; Gjorevski, N.; et al. Homeostatic Mini-Intestines through Scaffold-Guided Organoid Morphogenesis. Nature 2020, 585, 574–578. [Google Scholar] [CrossRef]
- Wang, Y.; Gunasekara, D.B.; Reed, M.I.; DiSalvo, M.; Bultman, S.J.; Sims, C.E.; Magness, S.T.; Allbritton, N.L. A Microengineered Collagen Scaffold for Generating a Polarized Crypt-Villus Architecture of Human Small Intestinal Epithelium. Biomaterials 2017, 128, 44–55. [Google Scholar] [CrossRef]
- El-Khateeb, E.; Burkhill, S.; Murby, S.; Amirat, H.; Rostami-Hodjegan, A.; Ahmad, A. Physiological-based Pharmacokinetic Modeling Trends in Pharmaceutical Drug Development over the Last 20-years; In-depth Analysis of Applications, Organizations, and Platforms. Biopharm. Drug Dispos. 2021, 42, 107–117. [Google Scholar] [CrossRef]
- Collander, R. The Permeability of Nitella Cells to Non-Eleetrolytes. Physiol. Plant. 1954, 7, 420–445. [Google Scholar] [CrossRef]
- Palm, K.; Luthman, K.; Unge, A.-L.; Strandlund, G.; Artursson, P. Correlation of Drug Absorption with Molecular Surface Properties. J. Pharm. Sci. 1996, 85, 32–39. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Palm, K.; Luthman, K.; Ungell, A.-L.; Strandlund, G.; Beigi, F.; Lundahl, P.; Artursson, P. Evaluation of Dynamic Polar Molecular Surface Area as Predictor of Drug Absorption: Comparison with Other Computational and Experimental Predictors. J. Med. Chem. 1998, 41, 5382–5392. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Over, B.; Matsson, P.; Tyrchan, C.; Artursson, P.; Doak, B.C.; Foley, M.A.; Hilgendorf, C.; Johnston, S.E.; Lee, M.D.; Lewis, R.J.; et al. Structural and Conformational Determinants of Macrocycle Cell Permeability. Nat. Chem. Biol. 2016, 12, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Rossi Sebastiano, M.; Doak, B.C.; Backlund, M.; Poongavanam, V.; Over, B.; Ermondi, G.; Caron, G.; Matsson, P.; Kihlberg, J. Impact of Dynamically Exposed Polarity on Permeability and Solubility of Chameleonic Drugs Beyond the Rule of 5. J. Med. Chem. 2018, 61, 4189–4202. [Google Scholar] [CrossRef] [PubMed]
- Matsson, P.; Bergström, C.A.S.; Nagahara, N.; Tavelin, S.; Norinder, U.; Artursson, P. Exploring the Role of Different Drug Transport Routes in Permeability Screening. J. Med. Chem. 2005, 48, 604–613. [Google Scholar] [CrossRef]
- Pereira, T.; Abbasi, M.; Oliveira, J.L.; Ribeiro, B.; Arrais, J. Optimizing Blood–Brain Barrier Permeation through Deep Reinforcement Learning for de Novo Drug Design. Bioinformatics 2021, 37, i84–i92. [Google Scholar] [CrossRef]
- Schlessinger, A.; Welch, M.A.; van Vlijmen, H.; Korzekwa, K.; Swaan, P.W.; Matsson, P. Molecular Modeling of Drug-Transporter Interactions-An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 2018, 104, 818–835. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors. Methods and Principles in Medicinal Chemistry; Wiley-VCH: Weinheim, Germany, 2008; ISBN 9783527313686. [Google Scholar]
- O’Hagan, S.; Kell, D.B. The Apparent Permeabilities of Caco-2 Cells to Marketed Drugs: Magnitude, and Independence from Both Biophysical Properties and Endogenite Similarities. PeerJ 2015, 3, e1405. [Google Scholar] [CrossRef]
- Venable, R.M.; Krämer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef]
- Dickson, C.J.; Hornak, V.; Pearlstein, R.A.; Duca, J.S. Structure–Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc. 2017, 139, 442–452. [Google Scholar] [CrossRef]
- Sethio, D.; Poongavanam, V.; Xiong, R.; Tyagi, M.; Duy Vo, D.; Lindh, R.; Kihlberg, J. Simulation Reveals the Chameleonic Behavior of Macrocycles. J. Chem. Inf. Model. 2023, 63, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Linker, S.M.; Schellhaas, C.; Kamenik, A.S.; Veldhuizen, M.M.; Waibl, F.; Roth, H.-J.; Fouché, M.; Rodde, S.; Riniker, S. Lessons for Oral Bioavailability: How Conformationally Flexible Cyclic Peptides Enter and Cross Lipid Membranes. J. Med. Chem. 2023, 66, 2773–2788. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Lennernas, H.; Welage, L.S.; Barnett, J.L.; Landowski, C.P.; Foster, D.; Fleisher, D.; Lee, K.-D.; Amidon, G.L. Comparison of Human Duodenum and Caco-2 Gene Expression Profiles for 12,000 Gene Sequences Tags and Correlation with Permeability of 26 Drugs. Pharm. Res. 2002, 19, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- Franco, Y.L.; Da Silva, L.; Cristofoletti, R. Navigating Through Cell-Based in Vitro Models Available for Prediction of Intestinal Permeability and Metabolism: Are We Ready for 3D? AAPS J. 2021, 24, 2. [Google Scholar] [CrossRef]
- Lennernäs, H. Human Jejunal Effective Permeability and Its Correlation with Preclinical Drug Absorption Models. J. Pharm. Pharmacol. 2011, 49, 627–638. [Google Scholar] [CrossRef]
- Markopoulos, C.; Thoenen, F.; Preisig, D.; Symillides, M.; Vertzoni, M.; Parrott, N.; Reppas, C.; Imanidis, G. Biorelevant Media for Transport Experiments in the Caco-2 Model to Evaluate Drug Absorption in the Fasted and the Fed State and Their Usefulness. Eur. J. Pharm. Biopharm. 2014, 86, 438–448. [Google Scholar] [CrossRef]
- Lee, J.B.; Zgair, A.; Taha, D.A.; Zang, X.; Kagan, L.; Kim, T.H.; Kim, M.G.; Yun, H.; Fischer, P.M.; Gershkovich, P. Quantitative Analysis of Lab-to-Lab Variability in Caco-2 Permeability Assays. Eur. J. Pharm. Biopharm. 2017, 114, 38–42. [Google Scholar] [CrossRef]
- Sugano, K. Computational Oral Absorption Simulation for Low-Solubility Compounds. Chem. Biodivers. 2009, 6, 2014–2029. [Google Scholar] [CrossRef]
- Roffey, S.J.; Obach, R.S.; Gedge, J.I.; Smith, D.A. What Is the Objective of the Mass Balance Study? A Retrospective Analysis of Data in Animal and Human Excretion Studies Employing Radiolabeled Drugs. Drug Metab. Rev. 2007, 39, 17–43. [Google Scholar] [CrossRef]
- Sjögren, E.; Abrahamsson, B.; Augustijns, P.; Becker, D.; Bolger, M.B.; Brewster, M.; Brouwers, J.; Flanagan, T.; Harwood, M.; Heinen, C.; et al. In Vivo Methods for Drug Absorption—Comparative Physiologies, Model Selection, Correlations with In Vitro Methods (IVIVC), and Applications for Formulation/API/Excipient Characterization Including Food Effects. Eur. J. Pharm. Sci. 2014, 57, 99–151. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Zur, M.; Beig, A.; Fine, N.; Cohen, Y.; González-Álvarez, M.; Merino-Sanjuán, M.; González-Álvarez, I.; Bermejo, M.; Dahan, A. Segmental-Dependent Permeability throughout the Small Intestine Following Oral Drug Administration: Single-Pass vs. Doluisio Approach to in-Situ Rat Perfusion. Int. J. Pharm. 2016, 515, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Dubbelboer, I.R.; Dahlgren, D.; Sjögren, E.; Lennernäs, H. Rat Intestinal Drug Permeability: A Status Report and Summary of Repeated Determinations. Eur. J. Pharm. Biopharm. 2019, 142, 364–376. [Google Scholar] [CrossRef]
- Sjögren, E.; Dahlgren, D.; Roos, C.; Lennernäs, H. Human in Vivo Regional Intestinal Permeability: Quantitation Using Site-Specific Drug Absorption Data. Mol. Pharm. 2015, 12, 2026–2039. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Roos, C.; Peters, K.; Lundqvist, A.; Tannergren, C.; Sjögren, E.; Sjöblom, M.; Lennernäs, H. Evaluation of Drug Permeability Calculation Based on Luminal Disappearance and Plasma Appearance in the Rat Single-Pass Intestinal Perfusion Model. Eur. J. Pharm. Biopharm. 2019, 142, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Cummins, C.L.; Salphati, L.; Reid, M.J.; Benet, L.Z. In Vivo Modulation of Intestinal CYP3A Metabolism by P-Glycoprotein: Studies Using the Rat Single-Pass Intestinal Perfusion Model. J. Pharmacol. Exp. Ther. 2003, 305, 306–314. [Google Scholar] [CrossRef]
- Song, N.-N.; Li, Q.-S.; Liu, C.-X. Intestinal Permeability of Metformin Using Single-Pass Intestinal Perfusion in Rats. World J. Gastroenterol. 2006, 12, 4064–4070. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M.; Hilfinger, J.M.; Yamashita, S.; Yu, L.X.; Lennernäs, H.; Amidon, G.L. High-Permeability Criterion for BCS Classification: Segmental/PH Dependent Permeability Considerations. Mol. Pharm. 2010, 7, 1827–1834. [Google Scholar] [CrossRef]
- Ho, Y.-F.; Lai, M.-Y.; Yu, H.-Y.; Huang, D.-K.; Hsueh, W.-C.; Tsai, T.-H.; Lin, C.-C. Application of Rat In Situ Single-Pass Intestinal Perfusion in the Evaluation of Presystemic Extraction of Indinavir Under Different Perfusion Rates. J. Fomosan Med. Assoc. 2008, 107, 37–45. [Google Scholar] [CrossRef]
- Braeckmans, M.; Augustijns, P.; Mols, R.; Servais, C.; Brouwers, J. Investigating the Mechanisms behind the Positive Food Effect of Abiraterone Acetate: In Vitro and Rat In Situ Studies. Pharmaceutics 2022, 14, 952. [Google Scholar] [CrossRef]
- Singh, G.; Pai, R.S. Trans -Resveratrol Self-Nano-Emulsifying Drug Delivery System (SNEDDS) with Enhanced Bioavailability Potential: Optimization, Pharmacokinetics and in Situ Single Pass Intestinal Perfusion (SPIP) Studies. Drug Deliv. 2015, 22, 522–530. [Google Scholar] [CrossRef]
- Lozoya-Agullo, I.; Gonzalez-Alvarez, I.; Zur, M.; Fine-Shamir, N.; Cohen, Y.; Markovic, M.; Garrigues, T.M.; Dahan, A.; Gonzalez-Alvarez, M.; Merino-Sanjuán, M.; et al. Closed-Loop Doluisio (Colon, Small Intestine) and Single-Pass Intestinal Perfusion (Colon, Jejunum) in Rat—Biophysical Model and Predictions Based on Caco-2. Pharm. Res. 2018, 35, 2. [Google Scholar] [CrossRef] [PubMed]
- Nylander, O.; Kvietys, P.; Granger, D.N. Effects of Hydrochloric Acid on Duodenal and Jejunal Mucosal Permeability in the Rat. Am. J. Physiol. Gastrointest. Liver Physiol. 1989, 257, G653–G660. [Google Scholar] [CrossRef] [PubMed]
- Sjöberg, Å.; Lutz, M.; Tannergren, C.; Wingolf, C.; Borde, A.; Ungell, A.L. Comprehensive Study on Regional Human Intestinal Permeability and Prediction of Fraction Absorbed of Drugs Using the Ussing Chamber Technique. Eur. J. Pharm. Sci. 2013, 48, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Incecayir, T.; Tsume, Y.; Amidon, G.L. Comparison of the Permeability of Metoprolol and Labetalol in Rat, Mouse, and Caco-2 Cells: Use as a Reference Standard for BCS Classification. Mol. Pharm. 2013, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Roos, C.; Johansson, P.; Tannergren, C.; Lundqvist, A.; Langguth, P.; Sjöblom, M.; Sjögren, E.; Lennernäs, H. The Effects of Three Absorption-Modifying Critical Excipients on the in Vivo Intestinal Absorption of Six Model Compounds in Rats and Dogs. Int. J. Pharm. 2018, 547, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Roos, C.; Lundqvist, A.; Abrahamsson, B.; Tannergren, C.; Hellström, P.M.; Sjögren, E.; Lennernäs, H. Regional Intestinal Permeability of Three Model Drugs in Human. Mol. Pharm. 2016, 13, 3013–3021. [Google Scholar] [CrossRef]
- Sutton, S.C.; Evans, L.A.; Fortner, J.H.; McCarthy, J.M.; Sweeney, K. Dog Colonoscopy Model for Predicting Human Colon Absorption. Pharm. Res. 2006, 23, 1554–1563. [Google Scholar] [CrossRef]
- Wilsson-Rahmberg, M.; Jonsson, O. Method for Long-Term Intestinal Access in the Dog. Lab. Anim. 1997, 31, 231–240. [Google Scholar] [CrossRef]
- van Nuland, M.; Rosing, H.; Huitema, A.D.R.; Beijnen, J.H. Predictive Value of Microdose Pharmacokinetics. Clin. Pharmacokinet. 2019, 58, 1221–1236. [Google Scholar] [CrossRef]
- Burt, T.; Young, G.; Lee, W.; Kusuhara, H.; Langer, O.; Rowland, M.; Sugiyama, Y. Phase 0/Microdosing Approaches: Time for Mainstream Application in Drug Development? Nat. Rev. Drug Discov. 2020, 19, 801–818. [Google Scholar] [CrossRef]
- Lennernäs, H.; Aarons, L.; Augustijns, P.; Beato, S.; Bolger, M.; Box, K.; Brewster, M.; Butler, J.; Dressman, J.; Holm, R.; et al. Oral Biopharmaceutics Tools—Time for a New Initiative—An Introduction to the IMI Project OrBiTo. Eur. J. Pharm. Sci. 2014, 57, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Senekowitsch, S.; Schick, P.; Abrahamsson, B.; Augustijns, P.; Gießmann, T.; Lennernäs, H.; Matthys, C.; Marciani, L.; Pepin, X.; Perkins, A.; et al. Application of In Vivo Imaging Techniques and Diagnostic Tools in Oral Drug Delivery Research. Pharmaceutics 2022, 14, 801. [Google Scholar] [CrossRef] [PubMed]
- Heidelberg Medical Product History. Available online: https://www.phcapsule.com/welcome/about-heidelberg-medical/ (accessed on 2 February 2023).
- Youngberg, C.A.; Berardi, R.R.; Howatt, W.F.; Hyneck, M.L.; Amidon, G.L.; Meyer, J.H.; Dressman, J.B. Comparison of Gastrointestinal PH in Cystic Fibrosis and Healthy Subjects. Dig. Dis. Sci. 1987, 32, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Dressman, J.B.; Berardi, R.R.; Dermentzoglou, L.C.; Russell, T.L.; Schmaltz, S.P.; Barnett, J.L.; Jarvenpaa, K.M. Upper Gastrointestinal (GI) PH in Young, Healthy Men and Women. Pharm. Res. 1990, 7, 756–761. [Google Scholar] [CrossRef]
- Russell, T.L.; Berardi, R.R.; Barnett, J.L.; Dermentzoglou, L.C.; Jarvenpaa, K.M.; Schmaltz, S.P.; Dressman, J.B. Upper Gastrointestinal PH in Seventy-Nine Healthy, Elderly, North American Men and Women. Pharm. Res. 1993, 10, 187–196. [Google Scholar] [CrossRef]
- Mojaverian, P. Evaluation of Gastrointestinal PH and Gastric Residence Time via the Heidelberg Radiotelemetry Capsule: Pharmaceutical Application. Drug Dev. Res. 1996, 38, 73–85. [Google Scholar] [CrossRef]
- SmartPill Product Information. Available online: https://www.medtronic.com/covidien/de-de/products/motility-testing/smartpill-motility-testing-system.html# (accessed on 2 February 2023).
- Koziolek, M.; Schneider, F.; Grimm, M.; Modeβ, C.; Seekamp, A.; Roustom, T.; Siegmund, W.; Weitschies, W. Intragastric PH and Pressure Profiles after Intake of the High-Caloric, High-Fat Meal as Used for Food Effect Studies. J. Control. Release 2015, 220, 71–78. [Google Scholar] [CrossRef]
- Weitschies, W.; Müller, L.; Grimm, M.; Koziolek, M. Ingestible Devices for Studying the Gastrointestinal Physiology and Their Application in Oral Biopharmaceutics. Adv. Drug Deliv. Rev. 2021, 176, 113853. [Google Scholar] [CrossRef]
- Söderlind, E.; Abrahamsson, B.; Erlandsson, F.; Wanke, C.; Iordanov, V.; von Corswant, C. Validation of the IntelliCap® System as a Tool to Evaluate Extended Release Profiles in Human GI Tract Using Metoprolol as Model Drug. J. Control. Release 2015, 217, 300–307. [Google Scholar] [CrossRef]
- Wilding, I.R.; Coupe, A.J.; Davis, S.S. The Role of γ-Scintigraphy in Oral Drug Delivery. Adv. Drug Deliv. Rev. 2001, 46, 103–124. [Google Scholar] [CrossRef]
- Becker, D.; Zhang, J.; Heimbach, T.; Penland, R.C.; Wanke, C.; Shimizu, J.; Kulmatycki, K. Novel Orally Swallowable IntelliCap® Device to Quantify Regional Drug Absorption in Human GI Tract Using Diltiazem as Model Drug. AAPS PharmSciTech 2014, 15, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, L.; Månsson, W.; Abrahamsson, B.; Seidegård, J.; Borgå, O. A Convenient Method for Local Drug Administration at Predefined Sites in the Entire Gastrointestinal Tract: Experiences from 13 Phase I Studies. Eur. J. Pharm. Sci. 2007, 30, 432–440. [Google Scholar] [CrossRef]
- Dahlgren, D.; Roos, C.; Johansson, P.; Lundqvist, A.; Tannergren, C.; Abrahamsson, B.; Sjögren, E.; Lennernäs, H. Regional Intestinal Permeability in Dogs: Biopharmaceutical Aspects for Development of Oral Modified-Release Dosage Forms. Mol. Pharm. 2016, 13, 3022–3033. [Google Scholar] [CrossRef]
- Hofmann, M.; Thieringer, F.; Nguyen, M.A.; Månsson, W.; Galle, P.R.; Langguth, P. A Novel Technique for Intraduodenal Administration of Drug Suspensions/Solutions with Concurrent PH Monitoring Applied to Ibuprofen Formulations. Eur. J. Pharm. Biopharm. 2019, 136, 192–202. [Google Scholar] [CrossRef]
- Fraunhofer-Institute for Translational Medicine and Pharmacology. Press Release “e-Pille”. Available online: https://www.itmp.fraunhofer.de/en/press/e-Pille.html (accessed on 2 February 2023).
- Vaithianathan, S.; Haidar, S.H.; Zhang, X.; Jiang, W.; Avon, C.; Dowling, T.C.; Shao, C.; Kane, M.; Hoag, S.W.; Flasar, M.H.; et al. Reply to “On the Effect of Common Excipients on the Oral Absorption of Class 3 Drugs”. J. Pharm. Sci. 2016, 105, 1355–1357. [Google Scholar] [CrossRef] [PubMed]
- Vaithianathan, S.; Haidar, S.H.; Zhang, X.; Jiang, W.; Avon, C.; Dowling, T.C.; Shao, C.; Kane, M.; Hoag, S.W.; Flasar, M.H.; et al. Effect of Common Excipients on the Oral Drug Absorption of Biopharmaceutics Classification System Class 3 Drugs Cimetidine and Acyclovir. J. Pharm. Sci. 2016, 105, 996–1005. [Google Scholar] [CrossRef]
- Polli, J.E. In Vitro Studies Are Sometimes Better than Conventional Human Pharmacokinetic In Vivo Studies in Assessing Bioequivalence of Immediate-Release Solid Oral Dosage Forms. AAPS J. 2008, 10, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.E.; McLean, A.M. Novel Direct Curve Comparison Metrics for Bioequivalence. Pharm. Res. 2001, 18, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Metry, M.; Krug, S.A.; Karra, V.K.; Ekins, S.; Hoag, S.W.; Kane, M.A.; Fink, J.C.; Polli, J.E. Lack of an Effect of Polysorbate 80 on Intestinal Drug Permeability in Humans. Pharm. Res. 2022, 39, 1881–1890. [Google Scholar] [CrossRef]
- Rege, B.D.; Kao, J.P.Y.; Polli, J.E. Effects of Nonionic Surfactants on Membrane Transporters in Caco-2 Cell Monolayers. Eur. J. Pharm. Sci. 2002, 16, 237–246. [Google Scholar] [CrossRef]
- Parr, A.; Hidalgo, I.J.; Bode, C.; Brown, W.; Yazdanian, M.; Gonzalez, M.A.; Sagawa, K.; Miller, K.; Jiang, W.; Stippler, E.S. The Effect of Excipients on the Permeability of BCS Class III Compounds and Implications for Biowaivers. Pharm. Res. 2016, 33, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Zizzari, A.T.; Pliatsika, D.; Gall, F.M.; Fischer, T.; Riedl, R. New Perspectives in Oral Peptide Delivery. Drug Discov. Today 2021, 26, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic Delivery of Peptides by the Oral Route: Formulation and Medicinal Chemistry Approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Peptide Delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Anderson, S.L.; Beutel, T.R.; Trujillo, J.M. Oral Semaglutide in Type 2 Diabetes. J. Diabetes Complicat. 2020, 34, 107520. [Google Scholar] [CrossRef]
- Rybelsus. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/Rybelsus (accessed on 30 September 2022).
- Buckley, S.T.; Bækdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Rønne, J.; Madsen, K.G.; Schéele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular Stomach Absorption of a Derivatized Glucagon-Like Peptide-1 Receptor Agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef] [PubMed]
- Pechenov, S.; Revell, J.; Will, S.; Naylor, J.; Tyagi, P.; Patel, C.; Liang, L.; Tseng, L.; Huang, Y.; Rosenbaum, A.I.; et al. Development of an Orally Delivered GLP-1 Receptor Agonist through Peptide Engineering and Drug Delivery to Treat Chronic Disease. Sci. Rep. 2021, 11, 22521. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Patel, P.J.; Aburub, A.; Sperry, A.; Estwick, S.; ElSayed, M.E.H.; Mannan, A.D. Identification of a Multi-Component Formulation for Intestinal Delivery of a GLP-1/Glucagon Co-Agonist Peptide. Pharm. Res. 2022, 39, 2555–2567. [Google Scholar] [CrossRef]
- Mycapssa. Available online: https://Www.Ema.Europa.Eu/En/Medicines/Human/EPAR/Mycapssa#authorisation-Details-Section (accessed on 30 October 2022).
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A Novel Suspension Formulation Enhances Intestinal Absorption of Macromolecules Via Transient and Reversible Transport Mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef]
- Stern, W.; Mehta, N.; Carl, S.M. Peptide Delivery—Oral Delivery of Peptides by Peptelligence Technology. Drug Dev. Deliv. 2013, in press.
- A Study of Pharmacokinetic/Pharmacodynamic Profile of Orally Administered Leuprolide in Healthy Female Volunteers. Available online: https://Clinicaltrials.Gov/Ct2/Show/NCT02807363?Term=ovarest&draw=1&rank=2 (accessed on 30 October 2022).
- Shangold, G.A.; Rubin, A.; Daggs, T.; Vrettos, J.; Rasums, A.; Consalvo, A.; Skeet, N.; Variam, S.P.; Ramakrishnan, K.; Shields, P. Pharmacokinetic (PK) Study of Oral Leuprolide Delivery with Ovarest® Achieves Drug Levels Exceeding Those of Approved Injectable Products. Fertil. Steril. 2022, 118, e102. [Google Scholar] [CrossRef]
- Study to Evaluate the Pharmacodynamics and Efficacy of Leuprolide Tablets (Ovarest®) in Women with Endometriosis. Available online: https://Clinicaltrials.Gov/Ct2/Show/NCT05096065?Term=ovarest&draw=2&rank=1 (accessed on 30 October 2022).
- McCartney, F.; Jannin, V.; Chevrier, S.; Boulghobra, H.; Hristov, D.R.; Ritter, N.; Miolane, C.; Chavant, Y.; Demarne, F.; Brayden, D.J. Labrasol® is an Efficacious Intestinal Permeation Enhancer across Rat Intestine: Ex Vivo and In Vivo Rat Studies. J. Control. Release 2019, 310, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.J.; Embrey, M.W.; Alleyne, C.; Amin, R.P.; Bass, A.; Bhatt, B.; Bianchi, E.; Branca, D.; Bueters, T.; Buist, N.; et al. A Series of Novel, Highly Potent, and Orally Bioavailable Next-Generation Tricyclic Peptide PCSK9 Inhibitors. J. Med. Chem. 2021, 64, 16770–16800. [Google Scholar] [CrossRef] [PubMed]
- Hristov, D.; McCartney, F.; Beirne, J.; Mahon, E.; Reid, S.; Bhattacharjee, S.; Penarier, G.; Werner, U.; Bazile, D.; Brayden, D.J. Silica-Coated Nanoparticles with a Core of Zinc, l-Arginine, and a Peptide Designed for Oral Delivery. ACS Appl. Mater. Interfaces 2020, 12, 1257–1269. [Google Scholar] [CrossRef] [PubMed]
- Lamson, N.G.; Berger, A.; Fein, K.C.; Whitehead, K.A. Anionic Nanoparticles Enable the Oral Delivery of Proteins by Enhancing Intestinal Permeability. Nat. Biomed. Eng. 2019, 4, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.; Uggla, T.; Antonsson, M.; Nunes, S.F.; Englund, M.; Rosengren, L.; Fahraj, M.; Wu, X.; Govender, R.; Söderberg, M.; et al. Evaluation in Pig of an Intestinal Administration Device for Oral Peptide Delivery. J. Control. Release 2023, 353, 792–801. [Google Scholar] [CrossRef]
- Maher, S.; Brayden, D.J. Formulation Strategies to Improve the Efficacy of Intestinal Permeation Enhancers. Adv. Drug Deliv. Rev. 2021, 177, 113925. [Google Scholar] [CrossRef]
- Gleeson, J.P.; McCartney, F. Striving Towards the Perfect In Vitro Oral Drug Absorption Model. Trends Pharmacol. Sci. 2019, 40, 720–724. [Google Scholar] [CrossRef]
- Martinez, M.N.; Wu, F.; Sinko, B.; Brayden, D.J.; Grass, M.; Kesisoglou, F.; Stewart, A.; Sugano, K. A Critical Overview of the Biological Effects of Excipients (Part II): Scientific Considerations and Tools for Oral Product Development. AAPS J. 2022, 24, 61. [Google Scholar] [CrossRef]
- McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics 2019, 11, 565. [Google Scholar] [CrossRef]
- von Erlach, T.; Saxton, S.; Shi, Y.; Minahan, D.; Reker, D.; Javid, F.; Lee, Y.-A.A.L.; Schoellhammer, C.; Esfandiary, T.; Cleveland, C.; et al. Robotically Handled Whole-Tissue Culture System for the Screening of Oral Drug Formulations. Nat. Biomed. Eng. 2020, 4, 544–559. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.; Aihara, E.; Mohammed, F.A.; Qu, H.; Riley, A.; Su, Y.; Lai, X.; Huang, S.; Aburub, A.; Chen, J.J.H.; et al. In Vivo Mechanism of Action of Sodium Caprate for Improving the Intestinal Absorption of a GLP1/GIP Coagonist Peptide. Mol. Pharm. 2023, 20, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Reppas, C.; Karatza, E.; Goumas, C.; Markopoulos, C.; Vertzoni, M. Characterization of Contents of Distal Ileum and Cecum to Which Drugs/Drug Products Are Exposed during Bioavailability/Bioequivalence Studies in Healthy Adults. Pharm. Res. 2015, 32, 3338–3349. [Google Scholar] [CrossRef] [PubMed]
- Schiller, C.; Fröhlich, C.P.; Giessmann, T.; Siegmund, W.; Mönnikes, H.; Hosten, N.; Weitschies, W. Intestinal Fluid Volumes and Transit of Dosage Forms as Assessed by Magnetic Resonance Imaging. Aliment. Pharmacol. Ther. 2005, 22, 971–979. [Google Scholar] [CrossRef]
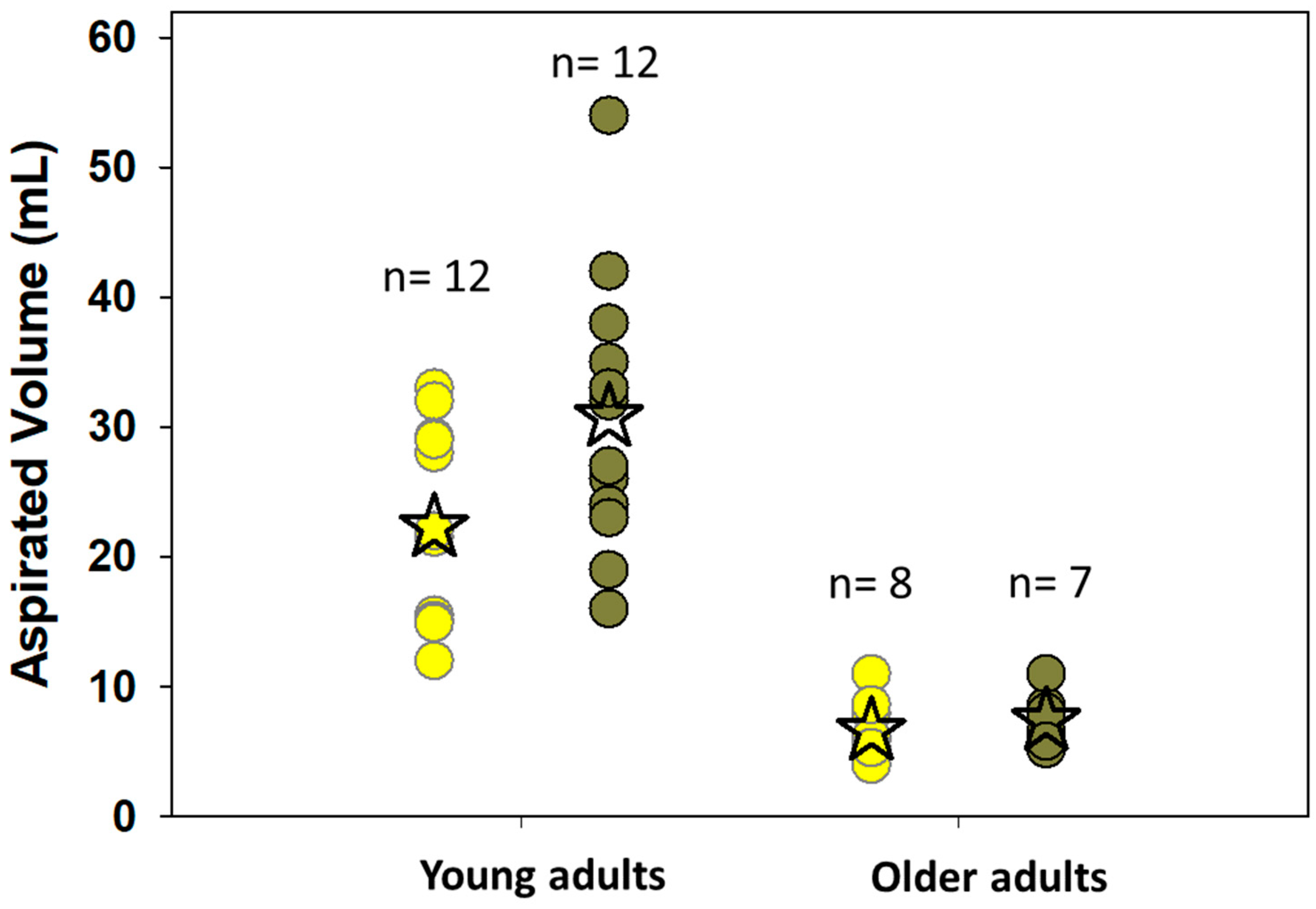
- Vertzoni, M.; Sulaiman, S.; Goumas, K.; Kersten, E.; Anlahr, J.; Muenster, U.; Reppas, C. Characteristics of Contents of Lower Intestine in the 65–74 Years of Age Range Could Impact the Performance of Safe and Efficacious Modified Release Products. J. Pharm. Sci. 2021, 110, 251–258. [Google Scholar] [CrossRef]
- Diakidou, A.; Vertzoni, M.; Goumas, K.; Söderlind, E.; Abrahamsson, B.; Dressman, J.; Reppas, C. Characterization of the Contents of Ascending Colon to Which Drugs Are Exposed after Oral Administration to Healthy Adults. Pharm. Res. 2009, 26, 2141–2151. [Google Scholar] [CrossRef]
- Wilson, C.G. The Transit of Dosage Forms through the Colon. Int. J. Pharm. 2010, 395, 17–25. [Google Scholar] [CrossRef]
- Georgaka, D.; Butler, J.; Kesisoglou, F.; Reppas, C.; Vertzoni, M. Evaluation of Dissolution in the Lower Intestine and Its Impact on the Absorption Process of High Dose Low Solubility Drugs. Mol. Pharm. 2017, 14, 4181–4191. [Google Scholar] [CrossRef]
- Markopoulos, C.; Andreas, C.J.; Vertzoni, M.; Dressman, J.B.; Reppas, C. In-Vitro Simulation of Luminal Conditions for Evaluation of Performance of Oral Drug Products: Choosing the Appropriate Test Media. Eur. J. Pharm. Biopharm. 2015, 93, 173–182. [Google Scholar] [CrossRef]
- Sousa, T.; Paterson, R.; Moore, V.; Carlsson, A.; Abrahamsson, B.; Basit, A.W. The Gastrointestinal Microbiota as a Site for the Biotransformation of Drugs. Int. J. Pharm. 2008, 363, 1–25. [Google Scholar] [CrossRef]
- Beeck, R.; Glöckl, G.; Krause, J.; Schick, P.; Weitschies, W. Mimicking the Dynamic Colonic Microbiota in Vitro to Gain a Better Understanding on the in Vivo Metabolism of Xenobiotics: Degradation of Sulfasalazine. Int. J. Pharm. 2021, 603, 120704. [Google Scholar] [CrossRef]
- Vertzoni, M.; Kersten, E.; van der Mey, D.; Muenster, U.; Reppas, C. Evaluating the Clinical Importance of Bacterial Degradation of Therapeutic Agents in the Lower Intestine of Adults Using Adult Fecal Material. Eur. J. Pharm. Sci. 2018, 125, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Vertzoni, M.; Carlsson, A.; Abrahamsson, B.; Goumas, K.; Reppas, C. Degradation Kinetics of Metronidazole and Olsalazine by Bacteria in Ascending Colon and in Feces of Healthy Adults. Int. J. Pharm. 2011, 413, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Karatza, E.; Goumas, C.; Muenster, U.; Reppas, C.; Vertzoni, M. Ex Vivo Evaluation of Degradation Rates of Metronidazole and Olsalazine in Distal Ileum and in Cecum: The Impact of Prandial State. Int. J. Pharm. 2017, 534, 237–241. [Google Scholar] [CrossRef]
- Kostantini, C.; Arora, S.; Söderlind, E.; Ceulemans, J.; Reppas, C.; Vertzoni, M. Usefulness of Optimized Human Fecal Material in Simulating the Bacterial Degradation of Sulindac and Sulfinpyrazone in the Lower Intestine. Mol. Pharm. 2022, 19, 2542–2548. [Google Scholar] [CrossRef]
- Tannergren, C.; Bergendal, A.; Lennernäs, H.; Abrahamsson, B. Toward an Increased Understanding of the Barriers to Colonic Drug Absorption in Humans: Implications for Early Controlled Release Candidate Assessment. Mol. Pharm. 2009, 6, 60–73. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
Discovery (Pre-CN) |
Development (Post-CN) | Clinical | ||
|---|---|---|---|---|
| Drug Substance | Drug Substance | Drug Product | Drug Substance/Drug Product | |
| Physicochemical parameters | Lipinski rule of 5 (MW, LogP/LogD, HBD, HBA), PSA aromatic rings, number of rotatable bonds, etc.; bRo5 requires additional property evaluations | |||
| In vitro/ex vivo tools (Papp, Peff) | PAMPA, cellular transport screening | Passive cellular permeability and active transport | Dissolution–permeation systems (e.g., biphasic dissolution, µFlux) | |
| In vivo models (F, fa) | Rodent PK | PK in higher preclinical species and food effect studies, extended release and regional absorption recorded in dogs | Human PK (Fabs), bioequivalence, microdosing (Fabs), perfusion studies | |
| Modeling (from Papp, Peff, F to kA, fA, fG, fH, predicted dose, human PK) | Static predictions: in silico tools, multiparametric scores (AB-), machine learning, artificial intelligence [34] | Dynamic models: PBPK modeling | ||
| Model | Advantages | Limitations | Applications |
|---|---|---|---|
| SPIP | Defined intestinal segment and surface area Allows direct drug permeability determination from luminal disappearance or indirect determination from plasma appearance Physiological regulation of gut functions is maintained Controlled luminal conditions Physiologically relevant permeability values | Slightly more labor intensive than the intestinal bolus model Tubing could cause non-specific binding of APIs | Determination of permeability independent of other absorption mechanisms Investigations of luminal, physiological and pharmaceutical effects on drug permeability Drug permeability can be studied at different physio-logical and pathophysiological conditions |
| Intraintestinal dosing | Very simple and efficient setup Minimal material use takes away risk of non-specific binding Can be used to investigate the entire absorption process, including dissolution, precipitation, gastric emptying kinetics, transit, etc. Useful for comparative formulation assessment | Permeability determination requires more assumptions than in the SPIP model Absolute Peff calculation less accurate due to lack of defined segment (relative assessment) | More generalized and formulation-related approach Investigation of absorption mechanisms as opposed to isolated permeability testing By comparing different formulation groups, different processes can be assessed more isolated (early preclinical formulation development) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koziolek, M.; Augustijns, P.; Berger, C.; Cristofoletti, R.; Dahlgren, D.; Keemink, J.; Matsson, P.; McCartney, F.; Metzger, M.; Mezler, M.; et al. Challenges in Permeability Assessment for Oral Drug Product Development. Pharmaceutics 2023, 15, 2397. https://doi.org/10.3390/pharmaceutics15102397
Koziolek M, Augustijns P, Berger C, Cristofoletti R, Dahlgren D, Keemink J, Matsson P, McCartney F, Metzger M, Mezler M, et al. Challenges in Permeability Assessment for Oral Drug Product Development. Pharmaceutics. 2023; 15(10):2397. https://doi.org/10.3390/pharmaceutics15102397
Chicago/Turabian StyleKoziolek, Mirko, Patrick Augustijns, Constantin Berger, Rodrigo Cristofoletti, David Dahlgren, Janneke Keemink, Pär Matsson, Fiona McCartney, Marco Metzger, Mario Mezler, and et al. 2023. "Challenges in Permeability Assessment for Oral Drug Product Development" Pharmaceutics 15, no. 10: 2397. https://doi.org/10.3390/pharmaceutics15102397
APA StyleKoziolek, M., Augustijns, P., Berger, C., Cristofoletti, R., Dahlgren, D., Keemink, J., Matsson, P., McCartney, F., Metzger, M., Mezler, M., Niessen, J., Polli, J. E., Vertzoni, M., Weitschies, W., & Dressman, J. (2023). Challenges in Permeability Assessment for Oral Drug Product Development. Pharmaceutics, 15(10), 2397. https://doi.org/10.3390/pharmaceutics15102397