
Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs




Abstract
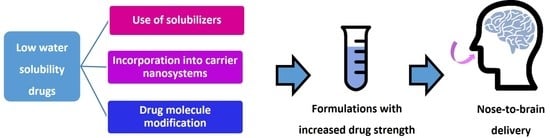
1. Introduction
2. Overview of Intranasal Formulation Strategies for Drugs with Low Aqueous Solubility
3. Use of Excipients for Enhanced Aqueous Solubility
3.1. Adjustment of the pH
3.2. Cyclodextrins
3.3. Cosolvents and Surfactants
4. Nanometric Dispersions
4.1. Nanosuspensions
4.2. Polymeric Carrier Nanosystems
4.3. Solid Lipid Nanoparticles and Nanostructured Lipid Carriers
4.4. Liposomes and Liposome-Related Vesicular Nanosystems
4.5. Nanometric Emulsions
4.6. Polymer-Coated Nanometric Emulsions
5. Drug Molecule Modification
6. Final Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, A.; Pandey, A.N.; Jain, S.K. Nasal-Nanotechnology: Revolution for Efficient Therapeutics Delivery. Drug Deliv. 2016, 23, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Cloyd, J.C.; Siegel, R.A. A Review of Intranasal Formulations for the Treatment of Seizure Emergencies. J. Control. Release 2016, 237, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Erdó, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of Intranasal Delivery Route of Drug Administration for Brain Targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Melo, D.; Santos, A.O. Intranasal Delivery of Antiseizure Drugs. In Drug Delivery Devices and Therapeutic Systems; Academic Press: Cambridge, MA, USA, 2021; pp. 623–646. ISBN 978-0-12-819838-4. [Google Scholar]
- Robinson, A.; Wermeling, D.P. Intranasal Naloxone Administration for Treatment of Opioid Overdose. Am. J. Health-Syst. Pharm. 2014, 71, 2129–2135. [Google Scholar] [CrossRef]
- Djupesland, P.G.; Messina, J.C.; Mahmoud, R.A. The Nasal Approach to Delivering Treatment for Brain Diseases: An Anatomic, Physiologic, and Delivery Technology Overview. Ther. Deliv. 2014, 5, 709–733. [Google Scholar] [CrossRef]
- Costa, C.; Moreira, J.N.; Amaral, M.H.; Lobo, J.M.S.; Silva, A.C. Nose-to-Brain Delivery of Lipid-Based Nanosystems for Epileptic Seizures and Anxiety Crisis. J. Control. Release 2019, 295, 187–200. [Google Scholar] [CrossRef]
- Katare, Y.K.; Piazza, J.E.; Bhandari, J.; Daya, R.P.; Akilan, K.; Simpson, M.J.; Hoare, T.; Mishra, R.K. Intranasal Delivery of Antipsychotic Drugs. Schizophr. Res. 2016, 184, 2–13. [Google Scholar] [CrossRef]
- Zieglmayer, P.; Schmutz, R.; Lemell, P.; Unger-Manhart, N.; Nakowitsch, S.; Goessl, A.; Savli, M.; Zieglmayer, R.; Prieschl-Grassauer, E. Fast Effectiveness of a Solubilized Low-Dose Budesonide Nasal Spray in Allergic Rhinitis. Clin. Exp. Allergy 2020, 50, 1065–1077. [Google Scholar] [CrossRef]
- Marinomed Biotech AG Marinosolv®—The Technology Platform for Aqueous Formulations. Available online: https://www.marinosolv.com/en (accessed on 31 August 2021).
- Pozzoli, M.; Traini, D.; Young, P.M.; Sukkar, M.B.; Sonvico, F. Development of a Soluplus Budesonide Freeze-Dried Powder for Nasal Drug Delivery. Drug Dev. Ind. Pharm. 2017, 43, 1510–1518. [Google Scholar] [CrossRef]
- Racaniello, G.F.; Laquintana, V.; Summonte, S.; Lopedota, A.; Cutrignelli, A.; Lopalco, A.; Franco, M.; Bernkop-Schnürch, A.; Denora, N. Spray-Dried Mucoadhesive Microparticles Based on S-Protected Thiolated Hydroxypropyl-β-Cyclodextrin for Budesonide Nasal Delivery. Int. J. Pharm. 2021, 603, 120728. [Google Scholar] [CrossRef]
- Shibuya, K.; Morikawa, S.; Miyamoto, M.; Ogawa, S.I.; Tsunenari, Y.; Urano, Y.; Noguchi, N. Brain Targeting of Acyl-CoA:Cholesterol O-Acyltransferase-1 Inhibitor K-604 via the Intranasal Route Using a Hydroxycarboxylic Acid Solution. ACS Omega 2019, 4, 16943–16955. [Google Scholar] [CrossRef] [PubMed]
- Zolkowska, D.; Wu, C.Y.; Rogawski, M.A. Intranasal Allopregnanolone Confers Rapid Seizure Protection: Evidence for Direct Nose-to-Brain Delivery. Neurotherapeutics 2021, 18, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.; Wong, L.R.; Xie, H.; Ho, P.C.L. In Vitro and in Vivo Comparison of Curcumin-Encapsulated Chitosan-Coated Poly (Lactic-Co-Glycolic Acid) Nanoparticles and Curcumin/Hydroxypropyl-β-Cyclodextrin Inclusion Complexes Administered Intranasally as Therapeutic Strategies for Alzheimer’s Disease. Mol. Pharm. 2020, 17, 4256–4269. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorso, A.; Gigliobianco, M.R.; Pellitteri, R.; Santonocito, D.; Carbone, C.; Di Martino, P.; Puglisi, G.; Musumeci, T. Optimization of Curcumin Nanocrystals as Promising Strategy for Nose-to-Brain Delivery Application. Pharmaceutics 2020, 12, 476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.; Huang, L.; Ho, P.C.L. Poly (Ethylene Glycol)-Block-Poly (D, L-Lactide) (PEG-PLA) Micelles for Brain Delivery of Baicalein through Nasal Route for Potential Treatment of Neurodegenerative Diseases Due to Oxidative Stress and Inflammation: An in Vitro and in Vivo Study. Int. J. Pharm. 2020, 591, 119981. [Google Scholar] [CrossRef] [PubMed]
- Madane, R.G.; Mahajan, H.S. Curcumin-Loaded Nanostructured Lipid Carriers (NLCs) for Nasal Administration: Design, Characterization, and in Vivo Study. Drug Deliv. 2016, 23, 1326–1334. [Google Scholar] [CrossRef]
- De Oliveira, E.R., Jr.; Truzzi, E.; Ferraro, L.; Fogagnolo, M.; Pavan, B.; Beggiato, S.; Rustichelli, C.; Maretti, E.; Lima, E.M.; Leo, E.; et al. Nasal Administration of Nanoencapsulated Geraniol/Ursodeoxycholic Acid Conjugate: Towards a New Approach for the Management of Parkinson’s Disease. J. Control. Release 2020, 321, 540–552. [Google Scholar] [CrossRef]
- Esposito, E.; Ravani, L.; Drechsler, M.; Mariani, P.; Contado, C.; Ruokolainen, J.; Ratano, P.; Campolongo, P.; Trezza, V.; Nastruzzi, C.; et al. Cannabinoid Antagonist in Nanostructured Lipid Carriers (NLCs): Design, Characterization and in Vivo Study. Mater. Sci. Eng. C 2015, 48, 328–336. [Google Scholar] [CrossRef]
- Ahmed, O.A.A.; Fahmy, U.A.; Badr-Eldin, S.M.; Aldawsari, H.M.; Awan, Z.A.; Asfour, H.Z.; Kammoun, A.K.; Caruso, G.; Caraci, F.; Alfarsi, A.; et al. Application of Nanopharmaceutics for Flibanserin Brain Delivery Augmentation via the Nasal Route. Nanomaterials 2020, 10, 1270. [Google Scholar] [CrossRef]
- Salem, H.F.; Kharshoum, R.M.; Abou-Taleb, H.A.; Naguib, D.M. Nanosized Transferosome-Based Intranasal in Situ Gel for Brain Targeting of Resveratrol: Formulation, Optimization, in Vitro Evaluation, and in Vivo Pharmacokinetic Study. AAPS PharmSciTech 2019, 20, 181. [Google Scholar] [CrossRef]
- Shinde, R.L.; Devarajan, P.V. Docosahexaenoic Acid–Mediated, Targeted and Sustained Brain Delivery of Curcumin Microemulsion. Drug Deliv. 2017, 24, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Sintov, A.C. AmyloLipid Nanovesicles: A Self-Assembled Lipid-Modified Starch Hybrid System Constructed for Direct Nose-to-Brain Delivery of Curcumin. Int. J. Pharm. 2020, 588, 119725. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Santos, L.T.; Rodrigues, M.; Alves, G.; Santos, A.O. Intranasal Fosphenytoin: The Promise of Phosphate Esters in Nose-to-Brain Delivery of Poorly Soluble Drugs. Int. J. Pharm. 2021, 592, 120040. [Google Scholar] [CrossRef] [PubMed]
- Rautiola, D.; Maglalang, P.D.; Cheryala, N.; Nelson, K.M.; Georg, G.I.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A.; Hanson, L.R.; Mishra, U.; et al. Intranasal Coadministration of a Diazepam Prodrug with a Converting Enzyme Results in Rapid Absorption of Diazepam in Rats. J. Pharmacol. Exp. Ther. 2019, 370, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Gao, H. Progress and Perspectives on Targeting Nanoparticles for Brain Drug Delivery. Acta Pharm. Sin. B 2016, 6, 268–286. [Google Scholar] [CrossRef]
- Pires, P.C.; Santos, A.O. Nanosystems in Nose-to-Brain Drug Delivery: A Review of Non-Clinical Brain Targeting Studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Gangurde, P.K.; Ajitkumar, B.N.; Kumar, L. Lamotrigine Lipid Nanoparticles for Effective Treatment of Epilepsy: A Focus on Brain Targeting via Nasal Route. J. Pharm. Innov. 2019, 14, 91–111. [Google Scholar] [CrossRef]
- Lopez-Toledano, M.A.; Saxena, V.; Legassie, J.D.; Liu, H.; Ghanta, A.; Riseman, S.; Cocilova, C.; Daak, A.; Thorsteinsson, T.; Rabinowicz, A.L.; et al. Advanced Lipid Technologies® (ALT®): A Proven Formulation Platform to Enhance the Bioavailability of Lipophilic Compounds. J. Drug Deliv. 2019, 2019, 1957360. [Google Scholar] [CrossRef]
- Karavasili, C.; Fatouros, D.G. Smart Materials: In Situ Gel-Forming Systems for Nasal Delivery. Drug Discov. Today 2016, 21, 157–166. [Google Scholar] [CrossRef]
- Shaikh, R.; Singh, T.; Garland, M.J.; Woolfson, A.D.; Donnelly, R.F. Mucoadhesive Drug Delivery Systems. J. Pharm. Bioallied Sci. 2011, 3, 89–100. [Google Scholar] [CrossRef]
- Ohmoto, T.; Nishitsuji, K.; Yoshitani, N.; Mizuguchi, M.; Yanagisawa, Y.; Saito, H.; Sakashita, N. K604, a Specific Acyl-CoA:Cholesterol Acyltransferase 1 Inhibitor, Suppresses Proliferation of U251-MG Glioblastoma Cells. Mol. Med. Rep. 2015, 12, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Wermeling, D.P. Intranasal Delivery of Antiepileptic Medications for Treatment of Seizures. Neurother. J. Am. Soc. Exp. NeuroTher. 2009, 6, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Zelcer, M.; Goldman, R.D. Intranasal Midazolam for Seizure Cessation in the Community Setting. Can. Fam. Physician 2016, 62, 559–561. [Google Scholar] [PubMed]
- Brigo, F.; Nardone, R.; Tezzon, F.; Trinka, E. Nonintravenous Midazolam versus Intravenous or Rectal Diazepam for the Treatment of Early Status Epilepticus: A Systematic Review with Meta-Analysis. Epilepsy Behav. 2015, 49, 325–336. [Google Scholar] [CrossRef]
- Haut, S.R.; Seinfeld, S.; Pellock, J. Benzodiazepine Use in Seizure Emergencies: A Systematic Review. Epilepsy Behav. 2016, 63, 109–117. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Midazolam. Available online: https://go.drugbank.com/drugs/DB00683 (accessed on 24 July 2021).
- Andersin, R. Solubility and Acid-Base Behaviour of Midazolam in Media of Different PH, Studied by Ultraviolet Spectrophotometry with Multicomponent Software. J. Pharm. Biomed. Anal. 1991, 9, 451–455. [Google Scholar] [CrossRef]
- Knoester, P.D.; Jonker, D.M.; van der Hoeven, R.T.M.; Vermeij, T.A.C.; Edelbroek, P.M. Pharmacokinetics and Pharmacodynamics of Midazolam Administered as a Concentrated Intranasal Spray. A Study in Healthy Volunteers. J. Clin. Pharmacol. 2002, 53, 501–507. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Brexanolone. Available online: https://go.drugbank.com/drugs/DB11859 (accessed on 24 July 2021).
- Jia, F.; Chibhabha, F.; Yang, Y.; Kuang, Y.; Zhang, Q.; Ullah, S.; Liang, Z.; Xie, M.; Li, F. Detection and Monitoring of the Neuroprotective Behavior of Curcumin Micelles Based on an AIEgen Probe. J. Mater. Chem. B 2021, 9, 731–745. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Curcumin. Available online: https://go.drugbank.com/drugs/DB11672 (accessed on 10 June 2021).
- Muankaew, C.; Loftsson, T. Cyclodextrin-Based Formulations: A Non-Invasive Platform for Targeted Drug Delivery. Basic Clin. Pharmacol. Toxicol. 2018, 122, 46–55. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Nayzilam—New Drug Application. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=211321 (accessed on 20 February 2020).
- U.S. Food and Drug Administration Valtoco—New Drug Application. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/211635s000lbl.pdf (accessed on 12 June 2021).
- Cartt, S.; Medeiros, D.; Gwozdz, G.T.; Maggio, A.; Mark, L.; David, M.; Hale, E.T. Administration of Benzodiazepine Compositions. US 9,763,876 B2, 19 September 2017. [Google Scholar]
- Boddu, S.H.S.; Kumari, S. A Short Review on the Intranasal Delivery of Diazepam for Treating Acute Repetitive Seizures. Pharmaceutics 2020, 12, 1167. [Google Scholar] [CrossRef]
- Wermeling, D.P.; Record, K.A.; Kelly, T.H.; Archer, S.M.; Clinch, T.; Rudy, A.C. Pharmacokinetics and Pharmacodynamics of a New Intranasal Midazolam Formulation in Healthy Volunteers. Anesth. Analg. 2006, 103, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Suter, K.; Hofmann, S.; Witschi, R.; Fröhlich, J.; Imanidis, G.; Drewe, J.; Briellmann, T.A.; Dussy, F.E.; Krähenbühl, S.; et al. Pharmacokinetics and Pharmacodynamics of Nasally Delivered Midazolam: Intranasal Delivery of Midazolam. Br. J. Clin. Pharmacol. 2010, 69, 607–616. [Google Scholar] [CrossRef]
- Lindhardt, K.; Gizurarson, S.; Stefánsson, S.B.; Òlafsson, D.R.; Bechgaard, E. Electroencephalographic Effects and Serum Concentrations after Intranasal and Intravenous Administration of Diazepam to Healthy Volunteers. Br. J. Clin. Pharmacol. 2001, 52, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Kriel, R.L.; Brundage, R.C.; Ivaturi, V.D.; Cloyd, J.C. A Pilot Study Assessing the Bioavailability and Pharmacokinetics of Diazepam after Intranasal and Intravenous Administration in Healthy Volunteers. Epilepsy Res. 2013, 105, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Hogan, R.E.; Tarquinio, D.; Sperling, M.R.; Klein, P.; Miller, I.; Segal, E.B.; Rabinowicz, A.L.; Carrazana, E. Pharmacokinetics and Safety of VALTOCO (NRL-1; Diazepam Nasal Spray) in Patients with Epilepsy during Seizure (Ictal/Peri-Ictal) and Nonseizure (Interictal) Conditions: A Phase 1, Open-Label Study. Epilepsia 2020, 61, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Sachdeva, M.; Agarwal, V. Nanosuspension Technology: Recent Patents on Drug Delivery and Their Characterizations. Recent Pat. Drug Deliv. Formul. 2019, 13, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Ryan, K.M.; Padrela, L. Production and Isolation of Pharmaceutical Drug Nanoparticles. Int. J. Pharm. 2021, 603, 120708. [Google Scholar] [CrossRef]
- Patel, H.P.; Chaudhari, P.S.; Gandhi, P.A.; Desai, B.V.; Desai, D.T.; Dedhiya, P.P.; Vyas, B.A.; Maulvi, F.A. Nose to Brain Delivery of Tailored Clozapine Nanosuspension Stabilized Using (+)-Alpha-Tocopherol Polyethylene Glycol 1000 Succinate: Optimization and in Vivo Pharmacokinetic Studies. Int. J. Pharm. 2021, 600, 120474. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Clozapine. Available online: https://go.drugbank.com/drugs/DB00363 (accessed on 10 June 2021).
- Abdelbary, G.A.; Tadros, M.I. Brain Targeting of Olanzapine via Intranasal Delivery of Core-Shell Difunctional Block Copolymer Mixed Nanomicellar Carriers: In Vitro Characterization, Ex Vivo Estimation of Nasal Toxicity and in Vivo Biodistribution Studies. Int. J. Pharm. 2013, 452, 300–310. [Google Scholar] [CrossRef]
- Nour, S.A.; Abdelmalak, N.S.; Naguib, M.J.; Rashed, H.M.; Ibrahim, A.B. Intranasal Brain-Targeted Clonazepam Polymeric Micelles for Immediate Control of Status Epilepticus: In Vitro Optimization, Ex Vivo Determination of Cytotoxicity, in Vivo Biodistribution and Pharmacodynamics Studies. Drug Deliv. 2016, 23, 3681–3695. [Google Scholar] [CrossRef]
- Rashed, H.M.; Shamma, R.N.; Basalious, E.B. Contribution of Both Olfactory and Systemic Pathways for Brain Targeting of Nimodipine-Loaded Lipo-Pluronics Micelles: In Vitro Characterization and in Vivo Biodistribution Study after Intranasal and Intravenous Delivery. Drug Deliv. 2017, 24, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Rui, W.; Li, S.; Xiao, H.; Xiao, M.; Shi, J. Baicalein Attenuates Neuroinflammation by Inhibiting NLRP3/Caspase-1/GSDMD Pathway in MPTP-Induced Mice Model of Parkinson’s Disease. Int. J. Neuropsychopharmacol. 2020, 23, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.K.; Uversky, V.N.; Chinnathambi, S. Baicalein Inhibits Heparin-Induced Tau Aggregation by Initializing Non-Toxic Tau Oligomer Formation. Cell Commun. Signal. 2021, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- OMx Personal Health Analytics Inc. Baicalein. Available online: https://go.drugbank.com/drugs/DB16101 (accessed on 10 June 2021).
- Singh, A.P.; Saraf, S.K.; Saraf, S.A. SLN Approach for Nose-to-Brain Delivery of Alprazolam. Drug Deliv. Transl. Res. 2012, 2, 498–507. [Google Scholar] [CrossRef]
- Patel, S.; Chavhan, S.; Soni, H.; Babbar, A.K.; Mathur, R.; Mishra, A.K.; Sawant, K. Brain Targeting of Risperidone-Loaded Solid Lipid Nanoparticles by Intranasal Route. J. Drug Target. 2011, 19, 468–474. [Google Scholar] [CrossRef]
- Jain, K.; Sood, S.; Gowthamarajan, K. Optimization of Artemether-Loaded NLC for Intranasal Delivery Using Central Composite Design. Drug Deliv. 2015, 22, 940–954. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Rimonabant. Available online: https://go.drugbank.com/drugs/DB06155 (accessed on 10 June 2021).
- Salama, H.A.; Mahmoud, A.A.; Kamel, A.O.; Hady, M.A.; Awad, G.A.S. Brain Delivery of Olanzapine by Intranasal Administration of Transfersomal Vesicles. J. Liposome Res. 2012, 22, 336–345. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Flibanserin. Available online: https://go.drugbank.com/drugs/DB04908 (accessed on 10 June 2021).
- Yang, A.J.T.; Bagit, A.; Macpherson, R.E.K. Resveratrol, Metabolic Dysregulation, and Alzheimer’s Disease: Considerations for Neurogenerative Disease. Int. J. Mol. Sci. 2021, 22, 4628. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Resveratrol. Available online: https://go.drugbank.com/drugs/DB02709 (accessed on 12 June 2021).
- Patel, R.B.; Patel, M.R.; Bhatt, K.K.; Patel, B.G.; Gaikwad, R. V Microemulsion-Based Drug Delivery System for Transnasal Delivery of Carbamazepine: Preliminary Brain-Targeting Study. Drug Deliv. 2016, 23, 207–213. [Google Scholar] [CrossRef]
- Patel, M.R.; Patel, R.B.; Bhatt, K.K.; Patel, B.G.; Gaikwad, V.; Patel, M.R.; Patel, R.B.; Bhatt, K.K.; Patel, B.G.; Gaikwad, R. V Paliperidone Microemulsion for Nose-to-Brain Targeted Drug Delivery System: Pharmacodynamic and Pharmacokinetic Evaluation. Drug Deliv. 2016, 23, 346–354. [Google Scholar] [CrossRef]
- Lalani, J.; Baradia, D.; Lalani, R.; Misra, A. Brain Targeted Intranasal Delivery of Tramadol: Comparative Study of Microemulsion and Nanoemulsion. Pharm. Dev. Technol. 2014, 20, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.; Sharma, S.; Mustafa, G.; Ali, J.; Baboota, S. Vitamin E Loaded Resveratrol Nanoemulsion for Brain Targeting for the Treatment of Parkinson’s Disease by Reducing Oxidative Stress. Nanotechnology 2014, 25, 485102. [Google Scholar] [CrossRef] [PubMed]
- El-setouhy, D.A.; Ibrahim, A.B.; Amin, M.M.; Khowessah, O.M.; Elzanfaly, E.S. Intranasal Haloperidol-Loaded Miniemulsions for Brain Targeting: Evaluation of Locomotor Suppression and in-Vivo Biodistribution. Eur. J. Pharm. Sci. 2016, 92, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Peixoto, D.; Teixeira, I.; Rodrigues, M.; Alves, G.; Santos, A.O. Nanoemulsions and Thermosensitive Nanoemulgels of Phenytoin and Fosphenytoin for Intranasal Administration: Formulation Development and in Vitro Characterization. Eur. J. Pharm. Sci. 2020, 141, 105099. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Salt Formation to Improve Drug Solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Breijyeh, Z.; Karaman, R. Enzyme Models—From Catalysis to Prodrugs. Molecules 2021, 26, 3248. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Phenytoin. Available online: https://go.drugbank.com/drugs/DB00252 (accessed on 12 June 2021).
- Antunes Viegas, D.; Rodrigues, M.; Francisco, J.; Falcão, A.; Alves, G.; Santos, A.O. Development and Application of an Ex Vivo Fosphenytoin Nasal Bioconversion/Permeability Evaluation Method. Eur. J. Pharm. Sci. 2016, 89, 61–72. [Google Scholar] [CrossRef]
- OMx Personal Health Analytics Inc. Diazepam. Available online: https://go.drugbank.com/drugs/DB00829 (accessed on 12 June 2021).


{kind=link}
{kind=link}
| Global Strategy | Formulation Strategy | Drug | Approximate Water Solubility (mg/mL) | Achieved Drug Strength 1 (mg/mL) | Drug Product or Bibliographic Reference |
|---|---|---|---|---|---|
| Use of solubilizers | Change in pH (acidification) | K-604 | 0.05 | 10.8 | [13] |
| Midazolam | 0.01 | 5 | Midazolam injection USP | ||
| Complexation (cyclodextrins) | Allopregnanolone | 0.001 | 16 | [14] | |
| Curcumin | 0.006 | ~3 | [15] | ||
| Cosolvents and surfactants | Midazolam | 0.01 | 50 | Nayzilam® | |
| Diazepam | 0.05 | 50–100 | ValtocoTM | ||
| Nanosuspensions and incorporation into carrier nanosystems | Nanosuspensions | Curcumin | 0.006 | 3.42 | [16] |
| Polymeric nanosystems | Curcumin | 0.006 | ~1.5 | [15] | |
| Baicalein | 0.2 | 0.8 2 | [17] | ||
| Solid lipid nanoparticles and nanostructured lipid carriers | Curcumin | 0.006 | 500 | [18] | |
| Geraniol- ursodeoxycholic acid conjugate | 0.0002 | ~4.5 | [19] | ||
| Rimonabant | 0.002 | ~2 | [20] | ||
| Liposomes and related vesicular nanosystems | Flibanserin | 0.2 | 10 | [21] | |
| Resveratrol | 0.07 | NR | [22] | ||
| Nanometric emulsions 3 | Curcumin | 0.006 | 5 | [23] | |
| Polymer-coated nanometric emulsions 4 | Curcumin | 0.006 | 1.9 | [24] | |
| Drug molecule modification | Salts and hydrophilic prodrugs | Phenytoin (used as fosphenytoin) | 0.07 | 34.8 (equivalent to 50 mg/mL fosphenytoin) | [25] |
| Diazepam (avizafone) | 0.05 | Up to the equivalent of ~13.5 mg/mL of diazepam | [26] |
| Global Strategy | Formulation Strategy | Advantages | Limitations |
|---|---|---|---|
| Use of solubilizers | Change in pH (acidification) | Increased drug solubility | Irritation of the nose and upper respiratory tract |
| Complexation (cyclodextrins) | Increased drug solubility, protection, and permeation | Safety is dependent on the type of cyclodextrin, their concentration, and the administration route | |
| Cosolvents and surfactants | Increased drug solubility and permeation | Irritation of the nose and upper respiratory tract | |
| Nanosuspensions and incorporation into carrier nanosystems | Nanosuspensions | Increased drug strength, simplicity of preparation, controlled drug release, and reduced toxicity | Physical instability and drug precipitation |
| Polymeric nanosystems | Increased drug strength, controlled drug release, targeted drug delivery, and prolonged therapeutic effect | Physical instability, low drug loading, and excipients not biocompatible | |
| Solid lipid nanoparticles and nanostructured lipid carriers | Increased drug strength, high safety (biocompatible), and controlled release profile | Physical instability and low drug loading | |
| Liposomes and related vesicular nanosystems | Increased drug strength, high safety (biocompatible), and enhanced permeation | Physical instability and low drug loading | |
| Nanometric emulsions | Increased drug solubilization, easy preparation (some), and enhanced permeability | Physical instability and low drug solubilization | |
| Drug molecule modification | Salts and hydrophilic prodrugs | Increased drug solubility and safety | Might not be enough to increase drug strength (has to be joined by other strategies) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pires, P.C.; Rodrigues, M.; Alves, G.; Santos, A.O. Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics 2022, 14, 588. https://doi.org/10.3390/pharmaceutics14030588
Pires PC, Rodrigues M, Alves G, Santos AO. Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics. 2022; 14(3):588. https://doi.org/10.3390/pharmaceutics14030588
Chicago/Turabian StylePires, Patrícia C., Márcio Rodrigues, Gilberto Alves, and Adriana O. Santos. 2022. "Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs" Pharmaceutics 14, no. 3: 588. https://doi.org/10.3390/pharmaceutics14030588
APA StylePires, P. C., Rodrigues, M., Alves, G., & Santos, A. O. (2022). Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics, 14(3), 588. https://doi.org/10.3390/pharmaceutics14030588