Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases


Abstract

1. Introduction
2. Adeno-Associated Virus (AAV) Background
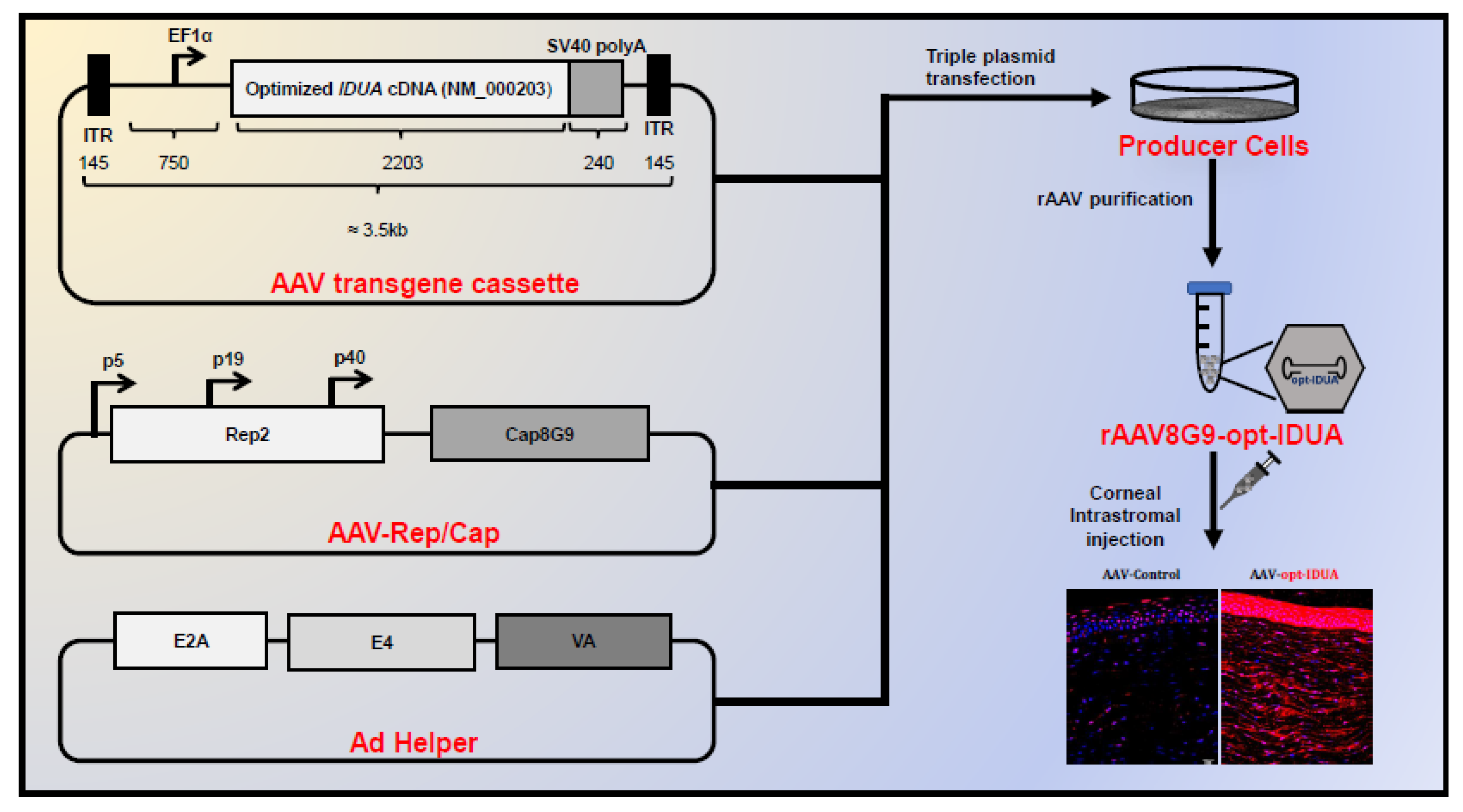
3. Developing AAV as a Vector for In Vivo Gene Delivery
4. Targeting Disease with AAV Gene Therapy
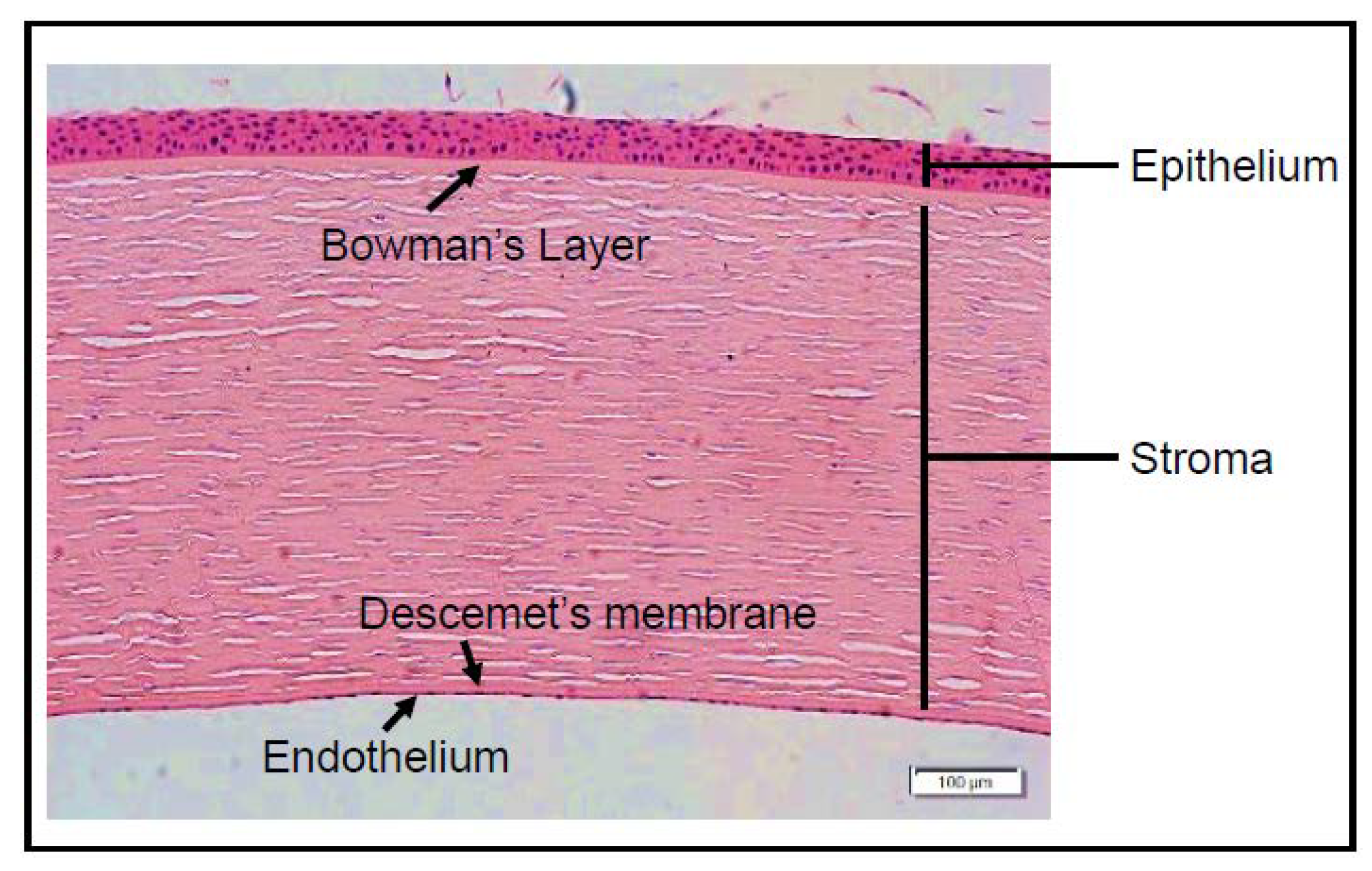
5. Cornea: Structure and Function
6. Corneal Disorders
6.1. Mechanical Injuries and Chemical Burns
6.2. Infectious Keratitis
6.3. Dry Eye Disease
6.4. Corneal Dystrophies
6.5. Fuchs Endothelial Corneal Dystrophy
6.6. Corneal Opacity Associated with Mucopolysaccharidoses
6.7. Corneal Graft Rejection
6.8. Corneal Neovascularization
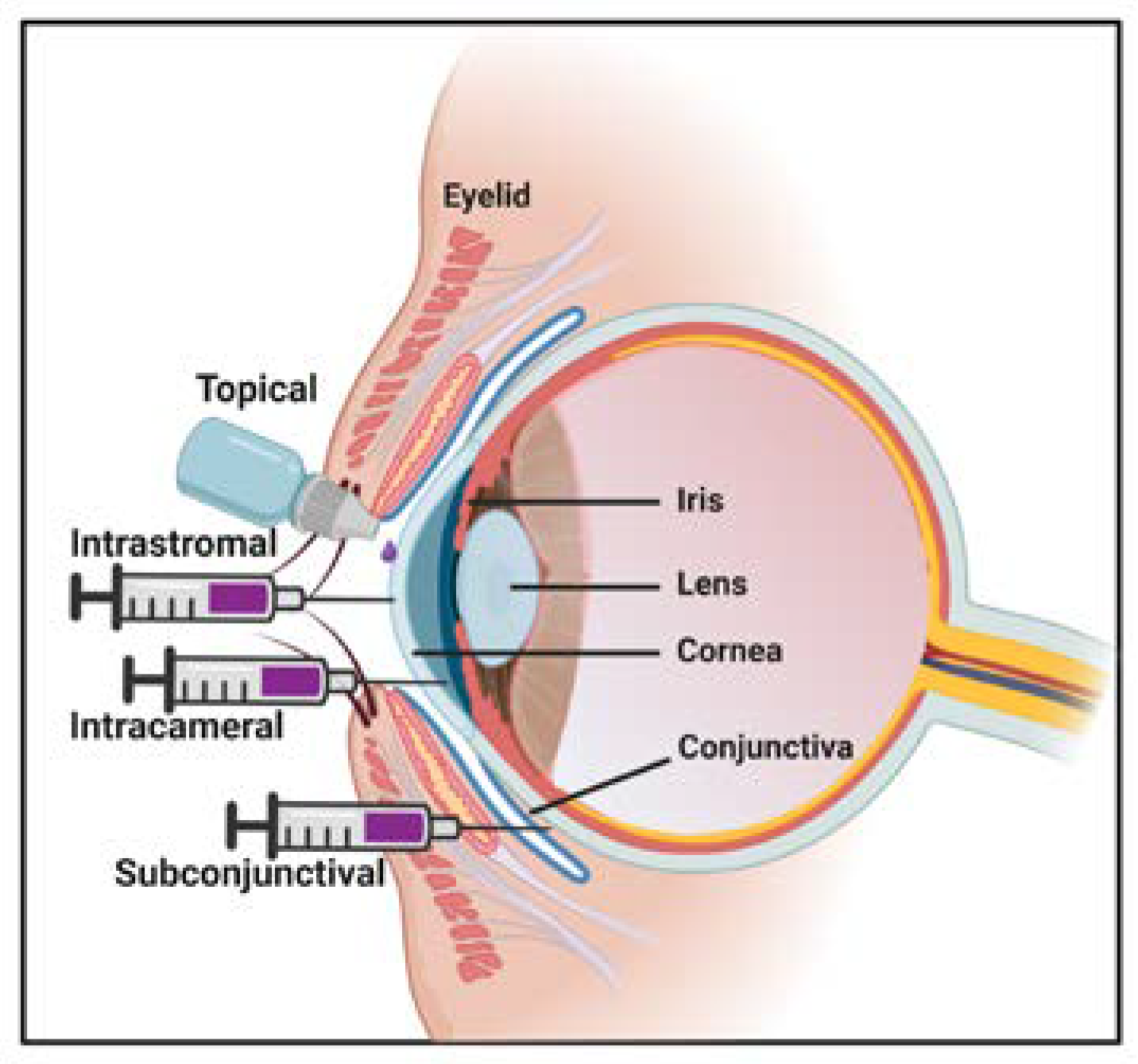
7. Route of Administration for AAV Gene Therapy
8. Immune Response Following rAAV Gene Therapy
9. Unanswered Questions in AAV Corneal Gene Therapy
10. Challenges and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bastola, P.; Oien, D.B.; Cooley, M.; Chien, J. Emerging Cancer Therapeutic Targets in Protein Homeostasis. Aaps. J. 2018, 20, 94. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic. Acids. Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.L.; High, K.A. Gene therapy for haemophilia. Br. J. Haematol. 2008, 140, 479–487. [Google Scholar] [CrossRef]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef]
- Lai, L.J.; Xiao, X.; Wu, J.H. Inhibition of corneal neovascularization with endostatin delivered by adeno-associated viral (AAV) vector in a mouse corneal injury model. J. Biomed. Sci. 2007, 14, 313–322. [Google Scholar] [CrossRef]
- Hodges, C.A.; Conlon, R.A. Delivering on the promise of gene editing for cystic fibrosis. Genes. Dis. 2018, 6, 97–108. [Google Scholar] [CrossRef]
- Al-Dosari, M.S.; Gao, X. Nonviral gene delivery: Principle, limitations, and recent progress. Aaps. J. 2009, 11, 671–681. [Google Scholar] [CrossRef]
- Ramamoorth, M.; Narvekar, A. Non viral vectors in gene therapy- an overview. J. Clin. Diagn. Res. 2015, 9, GE01–GE06. [Google Scholar] [CrossRef]
- Sung, Y.K.; Kim, S.W. Recent advances in the development of gene delivery systems. Biomater. Res. 2019, 23, 8. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359. [Google Scholar] [CrossRef]
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef] [PubMed]
- Organisation, W.H. Blindness and Vision Impairment Prevention. Priority Eye Diseases. Available online: https://www.who.int/blindness/causes/priority/en/index8.html. (accessed on 29 May 2020).
- Miller, W.L. Cornea: Fundamentals, Diagnosis and Management. In Optometry and Vision Science, 4th ed.; Lippincott Williams & Wilkins Company: Philadelphia, PA, USA, 2018; Volume 95. [Google Scholar]
- Atchison, R.W.; Casto, B.C.; Hammon, W.M. Adenovirus-Associated Defective Virus Particles. Science 1965, 149, 754–756. [Google Scholar] [CrossRef]
- Buller, R.M.; Janik, J.E.; Sebring, E.D.; Rose, J.A. Herpes simplex virus types 1 and 2 completely help adenovirus-associated virus replication. J. Virol. 1981, 40, 241–247. [Google Scholar] [CrossRef]
- Ogston, P.; Raj, K.; Beard, P. Productive replication of adeno-associated virus can occur in human papillomavirus type 16 (HPV-16) episome-containing keratinocytes and is augmented by the HPV-16 E2 protein. J. Virol. 2000, 74, 3494–3504. [Google Scholar] [CrossRef]
- Yalkinoglu, A.O.; Heilbronn, R.; Burkle, A.; Schlehofer, J.R.; zur Hausen, H. DNA amplification of adeno-associated virus as a response to cellular genotoxic stress. Cancer Res. 1988, 48, 3123–3129. [Google Scholar]
- Yakobson, B.; Hrynko, T.A.; Peak, M.J.; Winocour, E. Replication of adeno-associated virus in cells irradiated with UV light at 254 nm. J. Virol. 1989, 63, 1023–1030. [Google Scholar] [CrossRef]
- Yakobson, B.; Koch, T.; Winocour, E. Replication of adeno-associated virus in synchronized cells without the addition of a helper virus. J. Virol. 1987, 61, 972–981. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Wolf, S.J.; Samulski, R.J. Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors. Methods Mol. Biol. 2016, 1382, 21–39. [Google Scholar] [CrossRef]
- Naso, M.F.; Tomkowicz, B.; Perry, W.L.; Strohl, W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs 2017, 31, 317–334. [Google Scholar] [CrossRef]
- Sonntag, F.; Schmidt, K.; Kleinschmidt, J.A. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc. Natl. Acad. Sci. USA 2010, 107, 10220–10225. [Google Scholar] [CrossRef]
- Ogden, P.J.; Kelsic, E.D.; Sinai, S.; Church, G.M. Comprehensive AAV capsid fitness landscape reveals a viral gene and enables machine-guided design. Science 2019, 366, 1139–1143. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Tian, W.; Liu, C.; Lian, Z.; Dong, X.; Wu, X. Deletion of the B-B’ and C-C’ regions of inverted terminal repeats reduces rAAV productivity but increases transgene expression. Sci. Rep. 2017, 7, 5432. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Prasad, K.-M.R.; Trempe, J.P. The Adeno-Associated Virus Rep78 Protein Is Covalently Linked to Viral DNA in a Preformed Virion. Virology 1995, 214, 360–370. [Google Scholar] [CrossRef][Green Version]
- Prasad, K.M.R.; Zhou, C.; Trempe, J.P. Characterization of the Rep78/Adeno-Associated Virus Complex. Virology 1997, 229, 183–192. [Google Scholar] [CrossRef]
- Bleker, S.; Pawlita, M.; Kleinschmidt, J.A. Impact of capsid conformation and Rep-capsid interactions on adeno-associated virus type 2 genome packaging. J. Virol. 2006, 80, 810–820. [Google Scholar] [CrossRef]
- Maurer, A.C.; Weitzman, M.D. Adeno-Associated Virus Genome Interactions Important for Vector Production and Transduction. Hum. Gene Ther. 2020, 31, 499–511. [Google Scholar] [CrossRef]
- Berns, K.I.; Muzyczka, N. AAV: An Overview of Unanswered Questions. Hum. Gene Ther. 2017, 28, 308–313. [Google Scholar] [CrossRef]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef]
- Li, C.; Bowles, D.E.; van Dyke, T.; Samulski, R.J. Adeno-associated virus vectors: Potential applications for cancer gene therapy. Cancer Gene 2005, 12, 913–925. [Google Scholar] [CrossRef]
- Stilwell, J.L.; Samulski, R.J. Adeno-associated virus vectors for therapeutic gene transfer. Biotechniques 2003, 34, 148–150, 152, 154. [Google Scholar] [CrossRef]
- Carter, P.J.; Samulski, R.J. Adeno-associated viral vectors as gene delivery vehicles. Int. J. Mol. Med. 2000, 6, 17–27. [Google Scholar] [CrossRef]
- Colella, P.; Ronzitti, G.; Mingozzi, F. Emerging Issues in AAV-Mediated In Vivo Gene Therapy. Mol. Methods Clin. Dev. 2018, 8, 87–104. [Google Scholar] [CrossRef]
- Nidetz, N.F.; McGee, M.C.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharm. Ther. 2020, 207, 107453. [Google Scholar] [CrossRef]
- Samulski, R.J.; Berns, K.I.; Tan, M.; Muzyczka, N. Cloning of adeno-associated virus into pBR322: Rescue of intact virus from the recombinant plasmid in human cells. Proc. Natl. Acad. Sci. USA 1982, 79, 2077–2081. [Google Scholar] [CrossRef]
- Hermonat, P.L.; Muzyczka, N. Use of adeno-associated virus as a mammalian DNA cloning vector: Transduction of neomycin resistance into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA 1984, 81, 6466–6470. [Google Scholar] [CrossRef]
- Tratschin, J.D.; West, M.H.; Sandbank, T.; Carter, B.J. A human parvovirus, adeno-associated virus, as a eucaryotic vector: Transient expression and encapsidation of the procaryotic gene for chloramphenicol acetyltransferase. Mol. Cell Biol. 1984, 4, 2072–2081. [Google Scholar] [CrossRef]
- Samulski, R.J.; Chang, L.S.; Shenk, T. Helper-free stocks of recombinant adeno-associated viruses: Normal integration does not require viral gene expression. J. Virol. 1989, 63, 3822–3828. [Google Scholar] [CrossRef]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [CrossRef]
- Alroy, J.; Haskins, M.; Birk, D.E. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp. Eye Res. 1999, 68, 523–530. [Google Scholar] [CrossRef]
- Huang, Y.; Bron, A.J.; Meek, K.M.; Vellodi, A.; McDonald, B. Ultrastructural study of the cornea in a bone marrow-transplanted Hurler syndrome patient. Exp. Eye Res. 1996, 62, 377–387. [Google Scholar] [CrossRef]
- Vance, M.; Llanga, T.; Bennett, W.; Woodard, K.; Murlidharan, G.; Chungfat, N.; Asokan, A.; Gilger, B.; Kurtzberg, J.; Samulski, R.J.; et al. AAV Gene Therapy for MPS1-associated Corneal Blindness. Sci. Rep. 2016, 6, 22131. [Google Scholar] [CrossRef]
- Hippert, C.; Ibanes, S.; Serratrice, N.; Court, F.; Malecaze, F.; Kremer, E.J.; Kalatzis, V. Corneal transduction by intra-stromal injection of AAV vectors in vivo in the mouse and ex vivo in human explants. PLoS ONE 2012, 7, e35318. [Google Scholar] [CrossRef]
- Sharma, A.; Tovey, J.C.; Ghosh, A.; Mohan, R.R. AAV serotype influences gene transfer in corneal stroma in vivo. Exp. Eye Res. 2010, 91, 440–448. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xie, J.; Mao, Q.; Tai, P.W.L.; He, R.; Ai, J.; Su, Q.; Zhu, Y.; Ma, H.; Li, J.; Gong, S.; et al. Short DNA Hairpins Compromise Recombinant Adeno-Associated Virus Genome Homogeneity. Mol. Ther. 2017, 25, 1363–1374. [Google Scholar] [CrossRef]
- Miyadera, K.; Conatser, L.; Llanga, T.A.; Carlin, K.; O’Donnell, P.; Bagel, J.; Song, L.; Kurtzberg, J.; Samulski, R.J.; Gilger, B.; et al. Intrastromal Gene Therapy Prevents and Reverses Advanced Corneal Clouding in a Canine Model of Mucopolysaccharidosis I. Mol. Ther. 2020. [Google Scholar] [CrossRef]
- Ferrari, F.K.; Samulski, T.; Shenk, T.; Samulski, R.J. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J. Virol. 1996, 70, 3227–3234. [Google Scholar] [CrossRef]
- McCarty, D.M.; Monahan, P.E.; Samulski, R.J. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8, 1248–1254. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, H.I.; Li, J.; Sun, L.; Zhang, J.; Xiao, X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003, 10, 2105–2111. [Google Scholar] [CrossRef]
- Gray, S.J.; Choi, V.W.; Asokan, A.; Haberman, R.A.; McCown, T.J.; Samulski, R.J. Production of recombinant adeno-associated viral vectors and use in in vitro and in vivo administration. In Current Protocols in Neuroscience; Wiley-Liss Inc.: Hoboken, NJ, USA, 2011; Chapter 4, Unit4.17–14.17. [Google Scholar] [CrossRef]
- Grieger, J.C.; Choi, V.W.; Samulski, R.J. Production and characterization of adeno-associated viral vectors. Nat. Protoc. 2006, 1, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bower, J.J.; Llanga, T.; Salmon, J.H.; Hirsch, M.L.; Gilger, B.C. Ocular Tolerability and Immune Response to Corneal Intrastromal AAV-IDUA Gene Therapy in New Zealand White Rabbits. Mol. Ther. Methods Clin. Dev. 2020, 18, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Linden, R.M. Adeno-associated virus biology. Methods Mol. Biol. 2011, 807, 1–23. [Google Scholar] [CrossRef]
- Sridhar, M.S. Anatomy of cornea and ocular surface. Indian J. Ophthalmol. 2018, 66, 190–194. [Google Scholar] [CrossRef]
- Teichmann, J.; Valtink, M.; Nitschke, M.; Gramm, S.; Funk, R.H.W.; Engelmann, K.; Werner, C. Tissue engineering of the corneal endothelium: A review of carrier materials. J. Funct. Biomater. 2013, 4, 178–208. [Google Scholar] [CrossRef]
- Eghrari, A.O.; Riazuddin, S.A.; Gottsch, J.D. Overview of the Cornea: Structure, Function, and Development. Prog. Mol. Biol. Transl. Sci. 2015, 134, 7–23. [Google Scholar] [CrossRef]
- Quantock, A.J.; Young, R.D. Development of the corneal stroma, and the collagen-proteoglycan associations that help define its structure and function. Dev. Dyn. 2008, 237, 2607–2621. [Google Scholar] [CrossRef]
- Cholkar, K.; Patel, S.P.; Vadlapudi, A.D.; Mitra, A.K. Novel strategies for anterior segment ocular drug delivery. J. Ocul. Pharm. Ther. 2013, 29, 106–123. [Google Scholar] [CrossRef]
- Mantelli, F.; Argueso, P. Functions of ocular surface mucins in health and disease. Curr. Opin. Allergy Clin. Immunol. 2008, 8, 477–483. [Google Scholar] [CrossRef]
- McDermott, A.M. Antimicrobial compounds in tears. Exp. Eye Res. 2013, 117, 53–61. [Google Scholar] [CrossRef]
- Panda, A.; Vanathi, M.; Kumar, A.; Dash, Y.; Priya, S. Corneal graft rejection. Surv. Ophthalmol. 2007, 52, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.P.; Nichols, K.K.; Akpek, E.K.; Caffery, B.; Dua, H.S.; Joo, C.K.; Liu, Z.; Nelson, J.D.; Nichols, J.J.; Tsubota, K.; et al. TFOS DEWS II Definition and Classification Report. Ocul Surf. 2017, 15, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Farrand, K.F.; Fridman, M.; Stillman, I.O.; Schaumberg, D.A. Prevalence of Diagnosed Dry Eye Disease in the United States Among Adults Aged 18 Years and Older. Am. J. Ophthalmol. 2017, 182, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Musch, D.C.; Niziol, L.M.; Stein, J.D.; Kamyar, R.M.; Sugar, A. Prevalence of corneal dystrophies in the United States: Estimates from claims data. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6959–6963. [Google Scholar] [CrossRef] [PubMed]
- Mohan, R.R.; Tandon, A.; Sharma, A.; Cowden, J.W.; Tovey, J.C. Significant inhibition of corneal scarring in vivo with tissue-selective, targeted AAV5 decorin gene therapy. Invest. Ophthalmol. Vis. Sci. 2011, 52, 4833–4841. [Google Scholar] [CrossRef]
- Mohan, R.R.; Tovey, J.C.; Sharma, A.; Schultz, G.S.; Cowden, J.W.; Tandon, A. Targeted decorin gene therapy delivered with adeno-associated virus effectively retards corneal neovascularization in vivo. PLoS ONE 2011, 6, e26432. [Google Scholar] [CrossRef]
- Gupta, S.; Rodier, J.T.; Sharma, A.; Giuliano, E.A.; Sinha, P.R.; Hesemann, N.P.; Ghosh, A.; Mohan, R.R. Targeted AAV5-Smad7 gene therapy inhibits corneal scarring in vivo. PLoS ONE 2017, 12, e0172928. [Google Scholar] [CrossRef]
- Saika, S.; Yamanaka, O.; Okada, Y.; Miyamoto, T.; Kitano, A.; Flanders, K.C.; Ohnishi, Y.; Nakajima, Y.; Kao, W.W.; Ikeda, K. Effect of overexpression of PPARgamma on the healing process of corneal alkali burn in mice. Am. J. Physiol. Cell Physiol. 2007, 293, C75–C86. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Conatser, L.M.; Smith, S.M.; Salmon, J.H.; Wu, J.; Buglak, N.E.; Davis, R.; Gilger, B.C. AAV vector-meditated expression of HLA-G reduces injury-induced corneal vascularization, immune cell infiltration, and fibrosis. Sci. Rep. 2017, 7, 17840. [Google Scholar] [CrossRef]
- Robaei, D.; Carnt, N.; Watson, S. Established and emerging ancillary techniques in management of microbial keratitis: A review. Br. J. Ophthalmol. 2016, 100, 1163–1170. [Google Scholar] [CrossRef]
- Roy, P.; Das, S.; Singh, N.P.; Saha, R.; Kajla, G.; Snehaa, K.; Gupta, V.P. Changing trends in fungal and bacterial profile of infectious keratitis at a tertiary care hospital: A six-year study. Clin. Epidemiol. Glob. Health 2017, 5, 40–45. [Google Scholar] [CrossRef]
- Williams, K.A.; Klebe, S. Gene therapy for corneal dystrophies and disease, where are we? Curr. Opin. Ophthalmol. 2012, 23, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Farooq, A.V.; Shukla, D. Herpes simplex epithelial and stromal keratitis: An epidemiologic update. Surv. Ophthalmol. 2012, 57, 448–462. [Google Scholar] [CrossRef] [PubMed]
- Asbell, P.A. Valacyclovir for the prevention of recurrent herpes simplex virus eye disease after excimer laser photokeratectomy. Trans. Am. Ophthalmol. Soc. 2000, 98, 285–303. [Google Scholar]
- Poole, C.L.; James, S.H. Antiviral Therapies for Herpesviruses: Current Agents and New Directions. Clin. Ther. 2018, 40, 1282–1298. [Google Scholar] [CrossRef]
- Watson, Z.L.; Washington, S.D.; Phelan, D.M.; Lewin, A.S.; Tuli, S.S.; Schultz, G.S.; Neumann, D.M.; Bloom, D.C. In Vivo Knockdown of the Herpes Simplex Virus 1 Latency-Associated Transcript Reduces Reactivation from Latency. J. Virol. 2018, 92, e00812–e00818. [Google Scholar] [CrossRef] [PubMed]
- van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.G.; Bruggeling, C.E.; Schürch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.H.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef]
- Roehm, P.C.; Shekarabi, M.; Wollebo, H.S.; Bellizzi, A.; He, L.; Salkind, J.; Khalili, K. Inhibition of HSV-1 Replication by Gene Editing Strategy. Sci. Rep. 2016, 6, 23146. [Google Scholar] [CrossRef]
- Oh, H.S.; Neuhausser, W.M.; Eggan, P.; Angelova, M.; Kirchner, R.; Eggan, K.C.; Knipe, D.M. Herpesviral lytic gene functions render the viral genome susceptible to novel editing by CRISPR/Cas9. eLife 2019, 8, e51662. [Google Scholar] [CrossRef]
- Rolinski, J.; Hus, I. Immunological aspects of acute and recurrent herpes simplex keratitis. J. Immunol. Res. 2014, 2014, 513560. [Google Scholar] [CrossRef] [PubMed]
- Bainbridge, J.W.; Mistry, A.; De Alwis, M.; Paleolog, E.; Baker, A.; Thrasher, A.J.; Ali, R.R. Inhibition of retinal neovascularisation by gene transfer of soluble VEGF receptor sFlt-1. Gene Ther. 2002, 9, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.C.; Yeh, S.I.; Tsao, Y.P.; Kuo, P.C. Subconjunctival injection of recombinant AAV-angiostatin ameliorates alkali burn induced corneal angiogenesis. Mol. Vis. 2007, 13, 2344–2352. [Google Scholar] [PubMed]
- Lai, C.M.; Estcourt, M.J.; Wikstrom, M.; Himbeck, R.P.; Barnett, N.L.; Brankov, M.; Tee, L.B.; Dunlop, S.A.; Degli-Esposti, M.A.; Rakoczy, E.P. rAAV.sFlt-1 gene therapy achieves lasting reversal of retinal neovascularization in the absence of a strong immune response to the viral vector. Invest. Ophthalmol. Vis. Sci. 2009, 50, 4279–4287. [Google Scholar] [CrossRef] [PubMed]
- Coursey, T.G.; de Paiva, C.S. Managing Sjogren’s Syndrome and non-Sjogren Syndrome dry eye with anti-inflammatory therapy. Clin. Ophthalmol. 2014, 8, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Huynh, K.; Grubbs, J., Jr.; Davis, R.M. Autoimmunity in the pathogenesis and treatment of keratoconjunctivitis sicca. Curr. Allergy Asthma. Rep. 2014, 14, 403. [Google Scholar] [CrossRef]
- Erdelyi, B.; Kraak, R.; Zhivov, A.; Guthoff, R.; Nemeth, J. In vivo confocal laser scanning microscopy of the cornea in dry eye. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 39–44. [Google Scholar] [CrossRef]
- Blecha, C.; Wolff, D.; Holler, B.; Holler, E.; Weber, D.; Vogt, R.; Helbig, H.; Dietrich-Ntoukas, T. Verification of the new grading scale for ocular chronic graft-versus-host disease developed by the German-Austrian-Swiss consensus conference on chronic GVHD. Ann. Hematol. 2016, 95, 493–499. [Google Scholar] [CrossRef]
- Kam, K.W.; Chen, L.J.; Wat, N.; Young, A.L. Topical Olopatadine in the Treatment of Allergic Conjunctivitis: A Systematic Review and Meta-analysis. Ocul. Immunol. Inflamm. 2017, 25, 663–677. [Google Scholar] [CrossRef]
- Zhu, Z.; Stevenson, D.; Schechter, J.E.; Mircheff, A.K.; Ritter, T.; Labree, L.; Trousdale, M.D. Prophylactic effect of IL-10 gene transfer on induced autoimmune dacryoadenitis. Invest. Ophthalmol. Vis. Sci. 2004, 45, 1375–1381. [Google Scholar] [CrossRef][Green Version]
- Trousdale, M.D.; Zhu, Z.; Stevenson, D.; Schechter, J.E.; Ritter, T.; Mircheff, A.K. Expression of TNF inhibitor gene in the lacrimal gland promotes recovery of tear production and tear stability and reduced immunopathology in rabbits with induced autoimmune dacryoadenitis. J. Autoimmune Dis. 2005, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; Cotrim, A.P.; Zheng, C.; Riveros, P.P.; Baum, B.J.; Chiorini, J.A. Recovery of radiation-induced dry eye and corneal damage by pretreatment with adenoviral vector-mediated transfer of erythropoietin to the salivary glands in mice. Hum. Gene Ther. 2013, 24, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Yin, H.; Cabrera-Pérez, J.; Guimaro, M.C.; Afione, S.; Michael, D.G.; Glenton, P.; Patel, A.; Swaim, W.D.; Zheng, C.; et al. Aquaporin gene therapy corrects Sjögren’s syndrome phenotype in mice. Proc. Natl. Acad. Sci. USA 2016, 113, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Sirk, S.J.; Shui, S.-L.; Liu, J. Genome-Editing Technologies: Principles and Applications. Cold Spring Harb. Perspect. Biol. 2016, 8, a023754. [Google Scholar] [CrossRef]
- Pâques, F.; Duchateau, P. Meganucleases and DNA double-strand break-induced recombination: Perspectives for gene therapy. Curr. Gene Ther. 2007, 7, 49–66. [Google Scholar] [CrossRef]
- Komor, A.C.; Badran, A.H.; Liu, D.R. CRISPR-Based Technologies for the Manipulation of Eukaryotic Genomes. Cell 2017, 168, 20–36. [Google Scholar] [CrossRef]
- Bastola, P.; Bilkis, R.; De Souza, C.; Minn, K.; Chien, J. Heterozygous mutations in valosin-containing protein (VCP) and resistance to VCP inhibitors. Sci. Rep. 2019, 9, 11002. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Marzec, M.; Brąszewska-Zalewska, A.; Hensel, G. Prime Editing: A New Way for Genome Editing. Trends Cell Biol. 2020, 30, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Aarts, M.; te Riele, H. Progress and prospects: Oligonucleotide-directed gene modification in mouse embryonic stem cells: A route to therapeutic application. Gene Ther. 2011, 18, 213–219. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Courtney, D.G.; Moore, J.E.; Atkinson, S.D.; Maurizi, E.; Allen, E.H.A.; Pedrioli, D.M.L.; McLean, W.H.I.; Nesbit, M.A.; Moore, C.B.T. CRISPR/Cas9 DNA cleavage at SNP-derived PAM enables both in vitro and in vivo KRT12 mutation-specific targeting. Gene Ther. 2016, 23, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Christie, K.A.; Courtney, D.G.; DeDionisio, L.A.; Shern, C.C.; De Majumdar, S.; Mairs, L.C.; Nesbit, M.A.; Moore, C.B.T. Towards personalised allele-specific CRISPR gene editing to treat autosomal dominant disorders. Sci. Rep. 2017, 7, 16174. [Google Scholar] [CrossRef]
- Taketani, Y.; Kitamoto, K.; Sakisaka, T.; Kimakura, M.; Toyono, T.; Yamagami, S.; Amano, S.; Kuroda, M.; Moore, T.; Usui, T.; et al. Repair of the TGFBI gene in human corneal keratocytes derived from a granular corneal dystrophy patient via CRISPR/Cas9-induced homology-directed repair. Sci. Rep. 2017, 7, 16713. [Google Scholar] [CrossRef]
- Wang, L.; Yang, Y.; Breton, C.; Bell, P.; Li, M.; Zhang, J.; Che, Y.; Saveliev, A.; He, Z.; White, J.; et al. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020, 6, eaax5701. [Google Scholar] [CrossRef]
- Kamata, Y.; Okuyama, T.; Kosuga, M.; O’Hira, A.; Kanaji, A.; Sasaki, K.; Yamada, M.; Azuma, N. Adenovirus-mediated gene therapy for corneal clouding in mice with mucopolysaccharidosis type VII. Mol. Ther. 2001, 4, 307–312. [Google Scholar] [CrossRef]
- Serratrice, N.; Cubizolle, A.; Ibanes, S.; Mestre-Francés, N.; Bayo-Puxan, N.; Creyssels, S.; Gennetier, A.; Bernex, F.; Verdier, J.-M.; Haskins, M.E.; et al. Corrective GUSB transfer to the canine mucopolysaccharidosis VII cornea using a helper-dependent canine adenovirus vector. J. Control. Release 2014, 181, 22–31. [Google Scholar] [CrossRef]
- Cotugno, G.; Annunziata, P.; Tessitore, A.; O’Malley, T.; Capalbo, A.; Faella, A.; Bartolomeo, R.; O’Donnell, P.; Wang, P.; Russo, F.; et al. Long-term amelioration of feline Mucopolysaccharidosis VI after AAV-mediated liver gene transfer. Mol. Ther. J. Am. Soc. Gene Ther. 2011, 19, 461–469. [Google Scholar] [CrossRef]
- Okumura, N.; Kinoshita, S.; Koizumi, N. Application of Rho Kinase Inhibitors for the Treatment of Corneal Endothelial Diseases. J. Ophthalmol. 2017, 2017, 2646904. [Google Scholar] [CrossRef]
- Hinderer, C.; Bell, P.; Gurda, B.L.; Wang, Q.; Louboutin, J.P.; Zhu, Y.; Bagel, J.; O’Donnell, P.; Sikora, T.; Ruane, T.; et al. Intrathecal gene therapy corrects CNS pathology in a feline model of mucopolysaccharidosis I. Mol. Ther. 2014, 22, 2018–2027. [Google Scholar] [CrossRef] [PubMed]
- Duman, F.; Kosker, M.; Suri, K.; Reddy, J.C.; Ma, J.F.; Hammersmith, K.M.; Nagra, P.K.; Rapuano, C.J. Indications and outcomes of corneal transplantation in geriatric patients. Am. J. Ophthalmol. 2013, 156, 600–607.e602. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Ponzin, D.; Ashworth, J.L.; Fahnehjelm, K.T.; Summers, C.G.; Harmatz, P.R.; Scarpa, M. Diagnosis and management of ophthalmological features in patients with mucopolysaccharidosis. Br. J. Ophthalmol. 2011, 95, 613–619. [Google Scholar] [CrossRef]
- Gullingsrud, E.O.; Krivit, W.; Summers, C.G. Ocular abnormalities in the mucopolysaccharidoses after bone marrow transplantation. Longer follow-up. Ophthalmology 1998, 105, 1099–1105. [Google Scholar] [CrossRef]
- Prasad, V.K.; Kurtzberg, J. Transplant outcomes in mucopolysaccharidoses. Semin. Hematol. 2010, 47, 59–69. [Google Scholar] [CrossRef]
- Bothun, E.D.; Decanini, A.; Summers, C.G.; Orchard, P.J.; Tolar, J. Outcome of penetrating keratoplasty for mucopolysaccharidoses. Arch. Ophthalmol. 2011, 129, 138–144. [Google Scholar] [CrossRef][Green Version]
- Fuchsluger, T.A.; Jurkunas, U.; Kazlauskas, A.; Dana, R. Anti-apoptotic gene therapy prolongs survival of corneal endothelial cells during storage. Gene Ther. 2011, 18, 778–787. [Google Scholar] [CrossRef]
- Thomas, P.B.; Samant, D.M.; Selvam, S.; Wei, R.H.; Wang, Y.; Stevenson, D.; Schechter, J.E.; Apparailly, F.; Mircheff, A.K.; Trousdale, M.D. Adeno-associated virus-mediated IL-10 gene transfer suppresses lacrimal gland immunopathology in a rabbit model of autoimmune dacryoadenitis. Invest. Ophthalmol. Vis. Sci. 2010, 51, 5137–5144. [Google Scholar] [CrossRef][Green Version]
- Chang, J.H.; Gabison, E.E.; Kato, T.; Azar, D.T. Corneal neovascularization. Curr. Opin. Ophthalmol. 2001, 12, 242–249. [Google Scholar] [CrossRef]
- Feizi, S.; Azari, A.A.; Safapour, S. Therapeutic approaches for corneal neovascularization. Eye Vis. 2017, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.M.; Brankov, M.; Zaknich, T.; Lai, Y.K.; Shen, W.Y.; Constable, I.J.; Kovesdi, I.; Rakoczy, P.E. Inhibition of angiogenesis by adenovirus-mediated sFlt-1 expression in a rat model of corneal neovascularization. Hum. Gene Ther. 2001, 12, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tai, P.W.L.; Ai, J.; Gessler, D.J.; Su, Q.; Yao, X.; Zheng, Q.; Zamore, P.D.; Xu, X.; Gao, G. Transcriptome Profiling of Neovascularized Corneas Reveals miR-204 as a Multi-target Biotherapy Deliverable by rAAVs. Mol. Ther. Nucleic Acids 2018, 10, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Llanga, T.; Conatser, L.M.; Zaric, V.; Gilger, B.C.; Hirsch, M.L. Serotype survey of AAV gene delivery via subconjunctival injection in mice. Gene Ther. 2018, 25, 402–414. [Google Scholar] [CrossRef]
- Basche, M.; Kampik, D.; Kawasaki, S.; Branch, M.J.; Robinson, M.; Larkin, D.F.; Smith, A.J.; Ali, R.R. Sustained and Widespread Gene Delivery to the Corneal Epithelium via In Situ Transduction of Limbal Epithelial Stem Cells, Using Lentiviral and Adeno-Associated Viral Vectors. Hum. Gene Ther. 2018, 29, 1140–1152. [Google Scholar] [CrossRef]
- Tsai, M.L.; Chen, S.L.; Chou, P.I.; Wen, L.Y.; Tsai, R.J.; Tsao, Y.P. Inducible adeno-associated virus vector-delivered transgene expression in corneal endothelium. Invest. Ophthalmol. Vis. Sci. 2002, 43, 751–757. [Google Scholar]
- Buie, L.K.; Rasmussen, C.A.; Porterfield, E.C.; Ramgolam, V.S.; Choi, V.W.; Markovic-Plese, S.; Samulski, R.J.; Kaufman, P.L.; Borras, T. Self-complementary AAV virus (scAAV) safe and long-term gene transfer in the trabecular meshwork of living rats and monkeys. Invest. Ophthalmol Vis. Sci. 2010, 51, 236–248. [Google Scholar] [CrossRef]
- Bogner, B.; Boye, S.L.; Min, S.H.; Peterson, J.J.; Ruan, Q.; Zhang, Z.; Reitsamer, H.A.; Hauswirth, W.W.; Boye, S.E. Capsid Mutated Adeno-Associated Virus Delivered to the Anterior Chamber Results in Efficient Transduction of Trabecular Meshwork in Mouse and Rat. PLoS ONE 2015, 10, e0128759. [Google Scholar] [CrossRef]
- O’Callaghan, J.; Crosbie, D.E.; Cassidy, P.S.; Sherwood, J.M.; Flugel-Koch, C.; Lutjen-Drecoll, E.; Humphries, M.M.; Reina-Torres, E.; Wallace, D.; Kiang, A.S.; et al. Therapeutic potential of AAV-mediated MMP-3 secretion from corneal endothelium in treating glaucoma. Hum. Mol. Genet. 2017, 26, 1230–1246. [Google Scholar] [CrossRef]
- Igarashi, T.; Miyake, K.; Suzuki, N.; Kato, K.; Takahashi, H.; Ohara, K.; Shimada, T. New strategy for in vivo transgene expression in corneal epithelial progenitor cells. Curr. Eye Res. 2002, 24, 46–50. [Google Scholar] [CrossRef]
- Mohan, R.R.; Schultz, G.S.; Hong, J.W.; Mohan, R.R.; Wilson, S.E. Gene transfer into rabbit keratocytes using AAV and lipid-mediated plasmid DNA vectors with a lamellar flap for stromal access. Exp. Eye Res. 2003, 76, 373–383. [Google Scholar] [CrossRef]
- Liu, J.; Saghizadeh, M.; Tuli, S.S.; Kramerov, A.A.; Lewin, A.S.; Bloom, D.C.; Hauswirth, W.W.; Castro, M.G.; Schultz, G.S.; Ljubimov, A.V. Different tropism of adenoviruses and adeno-associated viruses to corneal cells: Implications for corneal gene therapy. Mol. Vis. 2008, 14, 2087–2096. [Google Scholar] [PubMed]
- Mohan, R.R.; Sharma, A.; Netto, M.V.; Sinha, S.; Wilson, S.E. Gene therapy in the cornea. Prog. Retin. Eye Res. 2005, 24, 537–559. [Google Scholar] [CrossRef]
- Mohan, R.R.; Sharma, A.; Cebulko, T.C.; Tandon, A. Vector delivery technique affects gene transfer in the cornea in vivo. Mol. Vis. 2010, 16, 2494–2501. [Google Scholar] [PubMed]
- Mohan, R.R.; Sinha, S.; Tandon, A.; Gupta, R.; Tovey, J.C.; Sharma, A. Efficacious and safe tissue-selective controlled gene therapy approaches for the cornea. PLoS ONE 2011, 6, e18771. [Google Scholar] [CrossRef] [PubMed]
- Cone, R.E.; Pais, R. Anterior Chamber-Associated Immune Deviation (ACAID): An Acute Response to Ocular Insult Protects from Future Immune-Mediated Damage? Ophthalmol. Eye Dis. 2009, 1, 33–40. [Google Scholar] [CrossRef]
- Hewitt, F.C.; Li, C.; Gray, S.J.; Cockrell, S.; Washburn, M.; Samulski, R.J. Reducing the risk of adeno-associated virus (AAV) vector mobilization with AAV type 5 vectors. J. Virol. 2009, 83, 3919–3929. [Google Scholar] [CrossRef]
- Song, L.; Samulski, R.J.; Hirsch, M.L. Wild type AAV, recombinant AAV, and Adenovirus super infection impact on AAV vector mobilization. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deyle, D.R.; Russell, D.W. Adeno-associated virus vector integration. Curr. Opin. Mol. Ther. 2009, 11, 442–447. [Google Scholar]
- West-Mays, J.A.; Dwivedi, D.J. The keratocyte: Corneal stromal cell with variable repair phenotypes. Int. J. Biochem. Cell Biol. 2006, 38, 1625–1631. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorders | Genes/Chr Location | Inheritance Pattern |
|---|---|---|
| CD, Avellino Type | TGFBI | autosomal dominant |
| CD, Congenital Endothelial 1 | 20p11.2–q11.2 locus | autosomal dominant |
| CD, Congenital Stromal | DCN | autosomal dominant |
| CD, Epithelial Basement Membrane | TGFBI | autosomal dominant |
| CD, Fleck | PIKFYVE | autosomal dominant |
| CD, Fuchs Endothelial, Early Onset | COL8A2 | autosomal dominant |
| CD, Fuchs Endothelial, Late Onset | ZEB1 | autosomal dominant |
| CD, Fuchs Endothelial, Late Onset 2 | TCF4 | autosomal dominant |
| CD, Granular | TGFBI | autosomal dominant |
| CD, Lattice Type I | TGFBI | autosomal dominant |
| CD, Lattice Type II | GSN | autosomal dominant |
| CD, Meesmann | KRT12, KRT3 | autosomal dominant |
| CD, Posterior Amorphous | 12q21.33 deletion | autosomal dominant |
| CD, Posterior Polymorphous 1 | OVOL2 | autosomal dominant |
| CD, Posterior Polymorphous 2 | COL8A2 | autosomal dominant |
| CD, Posterior Polymorphous 3 | ZEB1 | autosomal dominant |
| CD, Posterior Polymorphous 4 | GRHL2 | autosomal dominant |
| CD, Recurrent Epithelial Erosions | unknown | autosomal dominant |
| CD, Reis-Bücklers | TGFBI | autosomal dominant |
| CD, Schnyder | UBIAD1 | autosomal dominant |
| CD, Stocker-Holt | KRT12 | autosomal dominant |
| CD, Subepithelial Mucinous | unknown | autosomal dominant |
| CD, Thiel-Behnke | TGFBI | autosomal dominant |
| CD, Band-Shaped | unknown | unknown |
| CD, Congenital Endothelial 2 | SLC4A11 | autosomal recessive |
| CD, Gelatinous Drop-like | M1S1 (TACSTD2) | autosomal recessive |
| CD, Macular | CHST6 | autosomal recessive |
| CD, Lisch Epithelial | unknown | X-linked dominant |
| CD, Endothelial X-Linked | Xq25 locus | X-linked unclear* |
| Transduced Cell/Section | Route of Administration | Species (Model) | Dose/Volume | Serotype | Promoter | Transgene | Ref |
|---|---|---|---|---|---|---|---|
| Stroma | Intrastromal | C57BL/6 mouse | 1 × 109 vg/2 µL | AAV1, 2, 5, 8 | CMV | EGFP | [46] |
| Stroma | Intrastromal | Human (ex vivo) | 5 × 1010 vg/300 µL | AAV1, 2, 8 | CMV | EGFP | [46] |
| Entire cornea | Intrastromal | Human (ex vivo) | 1 × 1010 vg/50 µL | AAV8G9 | CMV | GFP, IDUA | [45] |
| Entire cornea | Intrastromal | MPS I dogs | 5–8 × 1010 vg/50–80 µL | AAV8G9 | CMV | GFP, IDUA | [49] |
| Entire cornea | Intrastromal | Male New Zealand white rabbit | 5 × 1010 vg/50 µL | scAAV8G9 | JET | GFP, HLA-G | [73] |
| Stroma & Epithelium | Intrastromal | C57BL/6J mouse | 1.4 × 1011 vg/2 µL | AAV2/5, 2/8, 2/9, 2/8Y733F, AAV[ShH10], AAV[Anc80] | CMV | EGFP | [128] |
| Entire cornea | Intraperitoneal | Ai9 mouse | 7.2 × 1011 vg/10 µL | AAV9 | CMV | Cre | [128] |
| Endothelium | Intracameral | New Zealand white rabbits | 1 × 107 vg/25 µL | AAV2 | CMV | LacZ | [129] |
| Endothelium | Intracameral | Brown Norway & Wistar Rat | 3 × 109–6 × 109 VP/3–5 µL | ssAAV2 | CMV | GFP | [130] |
| Endothelium | Intracameral | Cynomolgus monkey | 3 × 1010 VP/30 µL | scAAV2 | CMV | GFP | [130] |
| Endothelium | Intracameral | C57BL/6 mouse | 9 × 108 vg–3 × 109 vg/1 µL | scAAV2, scAAV2 (variants), scAAV8(variants) | CMV-CBA | GFP | [131] |
| Endothelium | Intracameral | Sprague Dawley rat | 1.8 × 109 vg–6 × 109 vg/2 µL | scAAV2, scAAV2 (variants), scAAV8 (variants) | CMV-CBA | GFP | [131] |
| Endothelium | Intracameral | C57BL6 mouse | 4 × 109 vg/2 µL or 1 × 1011 vg/2 µL | AAV9 | CMV, Tet | EGFP, MMP-3 | [132] |
| Epithelium | Topical a | Rat | Unknown/20 µL | AAV2 | CAG | EGFP | [133] |
| Keratocyte | Topical b | New Zealand white rabbit | 5 × 1011 VP/25 µL or 1 × 1011 VP/10 µL | Not specified | CMV | β-gal, CAT | [134] |
| Entire cornea | Topical c | New Zealand white rabbit | 2 × 1011 vg | AAV1, 2, 5, 7, 8 | CMV-CBA | GFP | [135] |
| Entire cornea | Topical c | Human cornea (ex vivo) | 1.2–7.8 × 1010 vg | AAV1, 2, 5, 7, 8 | CMV-CBA | GFP | [135] |
| Keratocytes | Topical d or microinjection | Mouse | Not specified | AAV2, 5 | CMV-CBA | EGFP | [136] |
| Keratocytes | Topical e | C57 Mouse | 2 × 109 vg/2 µL | AAV6, 8, 9 | RSV | Alkaline phosphatase | [47] |
| Stroma | Topical f | Female C57 black mouse | 2.2 × 108 vg/2 µL | AAV8 | RSV | Alkaline phosphatase | [137] |
| Stroma | Topical f | New Zealand white rabbit | 5 × 1011 vg/100 µL | AAV5 | CMV-CBA | GFP, Decorin | [70] |
| Keratocyte | Topical g | New Zealand white rabbit | 6.5 × 1011 vg/100 µL | AAV5 | CMV-CBA | GFP | [138] |
| Stroma | Topical h | New Zealand white Rabbit | 2.0 × 1012/75 µL | AAV5 | CMV-CBA | Smad7 | [71] |
| Epithelium | Subconjunctival | CD-1 mouse | 2.5 × 107 VP/5 µL | AAV2 | CMV | EGFP, Endostatin | [5] |
| Endothelium | Subconjunctival | C57BL/6J mouse | 7 × 109 vg/14 µL | AAV6 | CMV | GFP | [127] |
| Stroma | Subconjunctival | C57BL/6J mouse | 7 × 109 vg/14 µL | AAV8 | CMV | GFP | [127] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastola, P.; Song, L.; Gilger, B.C.; Hirsch, M.L. Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases. Pharmaceutics 2020, 12, 767. https://doi.org/10.3390/pharmaceutics12080767
Bastola P, Song L, Gilger BC, Hirsch ML. Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases. Pharmaceutics. 2020; 12(8):767. https://doi.org/10.3390/pharmaceutics12080767
Chicago/Turabian StyleBastola, Prabhakar, Liujiang Song, Brian C. Gilger, and Matthew L. Hirsch. 2020. "Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases" Pharmaceutics 12, no. 8: 767. https://doi.org/10.3390/pharmaceutics12080767
APA StyleBastola, P., Song, L., Gilger, B. C., & Hirsch, M. L. (2020). Adeno-Associated Virus Mediated Gene Therapy for Corneal Diseases. Pharmaceutics, 12(8), 767. https://doi.org/10.3390/pharmaceutics12080767